

Abstract
Bistability is a fundamental phenomenon in nature. In biology, a number of fine properties of bistability have been identified. However, these properties are only consequences of bistability at the physiological level, which do not explain why it had to emerge during evolution. Using optimal homeostasis as the first principle, I find that bistability emerges as an indispensable control mechanism. It is the only solution to a dilemma in glucose homeostasis: high insulin efficiency is required to confer rapidness in plasma glucose clearance, whereas an insulin sparing state is required to guarantee the brain's safety during fasting. The optimality consideration renders a clear correspondence between the molecular and physiological levels. This new perspective can illuminate studies on the twin epidemics of obesity and diabetes and the corresponding intervening strategies. For example, overnutrition and sedentary lifestyle may represent sudden environmental changes that cause the lose of optimality, which may contribute to the marked rise of obesity and diabetes in our generation. Because this bistability result is independent of the parameters of the mathematical model (for which the result is quite general), some other biological systems may also use bistability to control homeostasis.
Keywords: bistability, homeostasis, optimal control, glucose–insulin feedback system, insulin signalling pathway, AKT
1. Introduction
Systems theory becomes increasingly important in the post-genomic era in which the object of study is a complex network of interactions among many kinds of biomolecules. A central theme in systems biology is to reveal the intricate relationship among network structure, dynamical behaviour and physiology. As the intermediate between structure and physiology, dynamical behaviour is of particular importance [1]. One ubiquitous dynamical behaviour is bistability [2–10]. Instead of a graded response to the stimulus, a bistable molecule toggles between active and inactive states (all-or-none), but the threshold concentration of the stimulus for activation significantly differs from that for deactivation (hysteresis). The red lines in figure 1c constitute a bistable switch. Biological examples of bistability include Escherichia coli lac operon [2,3], mitogen-activated protein kinase cascades [4], cell cycle circuits [5,11,12], insulin signalling pathway [13–15] and synthetic gene switches [16]. A comprehensive review of bistability was made in [17]. Bistability corresponds to multiple fine properties: it produces a ‘memory’ of a transient stimulus, which is important for cell differentiation and cell cycle progression [5,18–20]; it enables cell's robustness to noises while responding sensitively to signals [21–23]; it is a mechanism through which small non-coding RNAs can mediate gene regulation [24]; it confers adaptivity by adjusting hysteresis [14]; it helps one to achieve biological rhythms with widely tunable frequency and near-constant amplitude [25]. On the other hand, disturbances to bistability often lead to complex diseases such as cancer and diabetes [13,14,26].
Figure 1.

The set of locally optimal controls whose utilization rates are within the range η ± Δη, where Δη = 5 × 10−5. (a) The distribution of control counts over the T-values. (b) Three u(t) are illustrated: the worst control (grey, T = 373), an average control (blue, T = 290) and the globally optimal control (red, T = 203.8). (c) The three controls in the form of u(I). They are obtained by following the conversion procedure in figure 2. The dashed lines are for illustration purpose only; they are not part of the controls.
However, these fine properties do not explain why bistability had to emerge from evolution. First, the wide variety of miscellaneous properties appear too special to explain ubiquity. Occam's razor (the principle that plurality should not be postulated unless absolutely necessary) argues for a simple and general explanation. Second, bistability may not be unique in conferring these properties. Other dynamics can possibly realize the same physiological properties as well.
The first concern can be addressed by seeking an overarching principle that involves only general notions. The second concern can be addressed by demonstrating that the overarching principle necessitates bistability. One starts from the principle and then deduces how the organism should be organized. If bistability emerges as an indispensable mechanism, then bistability proves to be necessary for the overarching principle, and the generality of the principle can explain the ubiquity of bistability.
Optimal homeostasis is such an overarching principle. Darwin's theory of natural selection provided an obvious mechanism for optimization in biology: more optimized individuals are more likely to survive [27]. After many rounds of natural selection, a species is optimally adapted to the environment it evolved in. Homeostasis, first defined by Claude Bernard and later by Walter Bradford Cannon [28,29], has become a central concept in biology. Primarily through negative feedback control, homeostasis allows an organism to maintain a stable parameter such as temperature. The negative feedback control must accommodate competing or even conflicting internal requirements and possibly wide environmental variations. Those that achieve optimal compromise are advantageous to survive natural selection. Optimal homeostatic regulation appears to be an overarching principle acquired through evolution.
Glucose homeostasis was an example frequently used by W. B. Cannon to elucidate homeostasis [29]. During the process, a balanced distribution of glucose is achieved between the brain and the other tissues. Neurons, relying absolutely on glucose as their energy source, can uptake glucose without any help from insulin. The other cells each possess a molecular pathway that, in response to insulin signalling, assimilates glucose from the blood. The insulin signalling pathway is sitting on the horns of a dilemma: high insulin efficiency is desired for fast removal of glucose after a meal to avoid hyperglycemia and the consequential toxicity; on the other hand, an insulin sparing state is required to guarantee the brain's safety during fasting (although insulin reduces to the baseline level during fasting, the majority of tissue cells, greatly outnumbering neurons, could still seize the limited endogenous glucose that should be reserved for the brain; thus, insulin sparing is required) [30–33]. Indeed, insulin therapy (to treat diabetes) has demonstrated risks of hypoglycemia and brain damages [33,34]. The dilemma was especially acute for ancient hunter–gatherers who had to deal with sporadic food availability—a sudden surfeit of food followed by a prolonged starvation [35]. Apparently, an optimal strategy has to be evolved to solve the dilemma.
In this paper, I use a mathematical model to describe the glucose–insulin system. The plasma glucose and insulin concentrations are controlled by the insulin signalling pathway. It is a negative feedback control because the rising glucose and insulin levels activate the control, but the effect of the control is to reduce the glucose and insulin levels. I first assume that the control is optimal in solving the earlier-mentioned dilemma. By minimizing an objective function, I find that the optimal control has to be a bistable switch (optimality ⇒ bistability). Therefore, the best compromise is not a median insulin response, but a timely, hysteretic switching between full responsiveness and unresponsiveness. Note that the optimal control is obtained entirely from physiological considerations, without using any information from the molecular level (the insulin signalling pathway).
This optimal solution is in line with a previous discovery that the actual control provided by the insulin signalling pathway manifests bistability, which was obtained entirely from analysing the insulin signalling pathway, without using any information from the physiological level [13,14]. By adjusting the parameters of the insulin signalling pathway, I show in this paper that an exact match can be achieved between the actual control and the optimal control. The matching also reveals the structure–function relationship of the insulin signalling pathway. It turns out that the pathway can be divided into three components, each playing a distinct role in modulating bistability.
This paper uses a physiology-driven approach, which starts from physiological properties to deduce how network dynamics and structure are constrained (physiology ⇒ dynamics ⇒ structure). Together with the fact that positive feedback is necessary for bistability [6,36], this paper establishes a chain of necessities: optimality ⇒ bistability ⇒ positive feedback. Although less explored, physiology-driven approaches have received more and more attention recently [37–41]. In comparison, the majority of works in systems biology are network-based. They start from a particular network to deduce higher level properties (structure ⇒ dynamics ⇒ physiology).
2. Model system
2.1. Glucose–insulin feedback system
During fasting, the glucose and insulin levels in our blood are maintained within a narrow range: 65–105 mg dl–1 for glucose [42] and 5–10 µU ml–1 for insulin [43]. Prolonged deviations from the range can cause serious health problems. Hyperglycemia can damage certain tissues and is the hallmark of diabetes. Hypoglycemia can damage the brain, which depends critically on glucose. Hyperinsulinemia increases the risks of many diseases. The regulation of glucose and insulin must be tight to keep them within the normal range.
Glucose homeostasis is achieved by negative feedback. Upon a sudden glucose increase following meal ingestion, the pancreatic beta cells are stimulated to secrete more insulin. The rising insulin level stimulates tissue cells (primarily in the muscles, adipose tissues and the liver) to uptake glucose through a molecular circuit known as insulin signalling pathway, whereby the plasma glucose level is lowered. The insulin level falls accordingly because the beta cells become less stimulated. Finally, both glucose and insulin return to their baseline levels.
Besides the insulin-mediated glucose utilization, there is always a small but critical stream of glucose to the brain. Unlike other tissue cells, neurons need to consume glucose all the time, owing to their inability to convert glucose into glycogen for storage. This is not usually a problem while exogenous glucose is abundant. During fasting, the liver generates glucose to sustain the brain's needs. The glucose uptake by neurons is insulin-independent.
2.2. Mathematical model
The dynamics of the glucose–insulin system can be described by
| 2.1 |
and
| 2.2 |
where t is the time; G(t) is the plasma glucose concentration; I(t) is the plasma insulin concentration; s is the basal rate of glucose required by the brain; during fasting s is exactly the rate of glucose generated by the liver; m(t) is the rate of glucose supplied by the meal; during fasting, one has m(t) = 0; λ is the rate of insulin-independent glucose utilization (by the brain) per unit glucose concentration; u(I) is the rate of insulin-dependent glucose utilization (by the majority of tissue cells) per unit glucose concentration, it is the main factor controlling plasma glucose clearance; k is the rate of insulin clearance; f(G) denotes the rate of pancreatic insulin secretion in response to glucose, it is a monotonically increasing function of the glucose concentration [44–47].
It is natural to think that u(I) is a continuous and monotonically increasing function (the more insulin, the faster glucose uptake), which has been assumed by others [47–51].
The present paper does not assume the function form of u(I). Instead, it aims to determine u(I) that renders optimal glucose homeostasis, based solely on equations (2.1) and (2.2).
3. Methods
Following the standard approach in optimal control theory [52,53], the determination of an optimal control u(I) consists of the following four steps:
— determine a time function u(t) that is optimal, without considering its practicality;
— numerically solve equations (2.1) and (2.2) with the optimal u(t) to obtain the corresponding G(t) and I(t);
— construct the optimal control u(I) from the optimal u(t) and the corresponding I(t); and
— determine whether or not the obtained optimal control u(I) is practical, by examining whether or not the insulin signalling pathway can realize the desired u(I). If yes, then the optimal control u(I) is practical. If not, then one has to repeat the whole process with a relaxed optimization criterion.
3.1. Determination of the optimal u(t)
The objective function for optimization is designed as follows. First, the efficiency of insulin action can be represented by T, the time when homeostasis is restored upon an initial perturbation (e.g. by meal ingestion) beginning at time t = 0. The smaller the T is, the more efficient the insulin action. Therefore, the optimization should be in terms of the minimization of T. Second, the optimization is constrained by a fixed average: 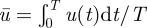 . By setting
. By setting 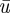 significantly smaller than umax (the upper bound of u(I)), the overall action of insulin is limited, which is necessary for brain safety. I actually use
significantly smaller than umax (the upper bound of u(I)), the overall action of insulin is limited, which is necessary for brain safety. I actually use  , termed the utilization rate, to set the constraint. The utilization rate η of normal cells should be small. A much larger utilization rate may be adopted by cancer cells, which must consume large amounts of glucose for them to become rapidly dividing. In summary, the aim is to find the control u(t) that restores homeostasis in the shortest time, whose average is fixed at ηumax for some small η. That is,
, termed the utilization rate, to set the constraint. The utilization rate η of normal cells should be small. A much larger utilization rate may be adopted by cancer cells, which must consume large amounts of glucose for them to become rapidly dividing. In summary, the aim is to find the control u(t) that restores homeostasis in the shortest time, whose average is fixed at ηumax for some small η. That is,
 |
3.1 |
On the basis of the earlier-mentioned objective of optimization, one first finds those u(t) that are locally optimal, by employing Pontryagin's maximum principle [52] (electronic supplementary material, §2). One then searches for the globally optimal u(t) among the local optima (electronic supplementary material, §3).
3.2. Determination of G(t) and I(t) corresponding to the optimal u(t)
With the optimal control u(t), one integrates equations (2.1) and (2.2) to obtain the glucose dynamics G(t) and the insulin dynamics I(t).
3.3. Determination of the optimal control u(I)
Since it is insulin that mediates the massive glucose utilization, the optimal control has to be in the form of u(I), which can be obtained by synthesizing the optimal u(t) and the corresponding I(t). That is, the optimal control u(I) is a trajectory on the I versus u plane whose coordinates at time t is (I(t), u(t)). For example, if u(t) = 1 + sin(t) and I(t) = 1 + cos(t), then the control u(I) is along the unit circle (I − 1)2 + (u − 1)2 = 1 rotating counterclockwise.
For the present problem, figures 2 and 4, and electronic supplementary material, figure S3 give three examples of the determination procedure.
Figure 2.

A computer simulation of the glucose–insulin system. (a) The control u(t) is taken from the red control in figure 1b. The ‘on’ phase of the control is re-coloured in green. (b) The controlled (red and green) and uncontrolled (grey) insulin dynamics. (c) The controlled and uncontrolled glucose dynamics. (d) The control in the form of u(I). (e) Twenty-four hour profile of plasma insulin concentration averaged from 14 normal (red circles with solid lines) and 15 obese (yellow circles with dashed lines) subjects. There are three spikes, caused by the three meals at 09.00, 13.00 and 18.00. (f) Twenty-four hour profile of plasma glucose concentration. Normal person denotes red circles with solid lines and obese person denotes yellow circles with dashed lines.
Figure 4.

A computer simulation of equations (4.3) and (4.4). (a) The control u(t) with umax = 1, ton = 2 and toff = 5. (b) The controlled (black) and uncontrolled (grey) insulin dynamics. (c) The controlled and uncontrolled glucose dynamics. (d) The control in the form of u(I). (e) The function m(t) is a triangular function centring on t = 2 and with height 10.
3.4. Practicality of the optimal control u(I)
To justify its practicality, the obtained optimal control u(I) should be compared with the actual control, which is generated by the insulin signalling pathway, whose mathematical description can be found in electronic supplementary material, §4. At the centre of the insulin signalling pathway is the protein kinase AKT, which is a master regulator of cell growth, proliferation and survival [54,55]. In particular, the activation of AKT leads to glucose uptake. Therefore, the actual control can be represented by the function A(I), where A represents the concentration of the activated AKT and I represents the insulin level. The value of A(I) is bounded between 0 and Amax, where Amax represents the total concentration of AKT. The protein AKT is stable and Amax can be assumed constant [54,56]. The actual control A(I) is constrained by equation (S40) (see also eqn (A2) of [14]).
4. Results
4.1. Optimal homeostasis necessitates all-or-none
By applying Pontryagin's maximum principle, I find that a locally optimal control u(t) must be of a ‘bang–bang’ type—switching abruptly between two extreme values 0 and umax, in a similar way as a residential thermostat in response to the temperature change. This optimality then imposes an all-or-none requirement at the molecular level: either no AKT activation or full AKT activation.
All-or-none is a recurring phenomenon in biology, which was noticed as early as 1871 by Henry Pickering Bowditch while he was studying the contraction of heart muscle [57]. However, the advantages of all-or-none remain largely unclear. Here, I demonstrate that all-or-none at the molecular level is necessary for the optimal regulation at the physiological level.
The result is independent of the parameters in equations (2.1) and (2.2). The computations have been symbolic only. I did not assign values for the parameters, nor assume the functional forms for f(G) and m(t) in the model. The model thus represents a general homeostatic system. Because homeostasis and optimality are fundamental properties in biology, all-or-none must be a ubiquitous property and its emergence is inevitable.
4.2. Optimal control as a time function
The mathematical model has two ending time points: t = 0 is the time when the perturbation (meal ingestion) begins, and t = T is the time when homeostasis is restored. At both time points, the system is close to the fasting state and the control must be off: u(0) = u(T) = 0. This equality implies that an optimal u(t) switches between 0 and umax only an even number of times during 0 < t < T. If u(t) switched an odd number of times, then one would have u(0) ≠ u(T). Further analysis reveals that an optimal control only switches twice (electronic supplementary material, §2.4). That is, an optimal control must have the following form:
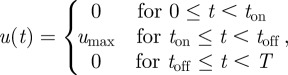 |
4.1 |
where ton and toff are the time points at which u(t) switches. In the following, a control satisfying equation (4.1) is often abbreviated by 0-umax-0. Figure 1b illustrates three examples of 0-umax-0 controls.
This result is also independent of the parameters and functional forms in the mathematical model. The obtained local optimality is likely to apply to the homeostasis of all such systems using negative feedback to control concentrations.
Equation (4.1) is also sufficient for u(t) to be optimal, provided that a parametric condition (electronic supplementary material, expression (S33)) is satisfied. As explained in the electronic supplementary material, expression (S33) should always hold when the model parameters are reasonable. For the 8.3 × 105 controls tested, I find electronic supplementary material, expression (S33) always holds. Therefore, an arbitrary 0-umax-0 control is almost certainly an optimal one. This implies that local optimality is easy to maintain (the change of ton and toff will not remove optimality). This would allow the system to easily resettle into another optimum when metabolic conditions change.
It should be noted that the utilization rate of a 0-umax-0 control reduces to the duty cycle: η = (toff – ton)/T.
4.3. Optimal homeostasis necessitates bistability
The optimal control u(I) can be determined from u(t) and I(t). Figure 2 shows an example of the determination. The to-be-converted u(t) is illustrated in figure 2a. It is also illustrated in figure 1b as the red control (the one with ton = 70.5 and toff = 90.95). With this u(t), I run the model to obtain G(t) and I(t). The values for the model parameters are obtained from the literature (electronic supplementary material, table S1). As shown in figure 2b,c, glucose and insulin concentrations have a spike-like change before returning to the baseline levels G0 and I0, respectively. The results are realistic as demonstrated by the comparison with clinical data (figure 2e,f, which are adapted from [58]). By synthesizing u(t) and I(t), the control u(I) is traced out in figure 2d. It is also presented as the red control in figure 1c.
The obtained u(I) is a bistable switch characterized by all-or-none and hysteresis. As I increases, the glucose uptake switches on fully, once I exceeds the threshold Ion = I(ton) = 43.70. As I decreases, the glucose uptake switches off fully, once I drops below the threshold Ioff = I(toff) = 12.23. Interestingly, hysteresis emerges during the switching: glucose uptake is triggered (terminated) by a larger (smaller) insulin concentration (Ion > Ioff).
The emergence of hysteresis is explained as follows. Once the control switches on at ton, the glucose utilization is so massive that the plasma glucose and insulin concentrations drop almost immediately. In this event, Ion is either the peak of I(t) (figure 2b) or is close to the peak (electronic supplementary material, figure S3b). Therefore, Ion > Ioff is almost inevitable.
Although intuitively natural, assumptions about u(I) (i.e. continuity, monotonicity and even the property of being a function in previous studies [47–51] are challenged by the locally optimal solution obtained here. First, all-or-none implies discontinuity. Second, hysteresis defies monotonicity. Finally, an optimal control is not a strict function: in the range Ioff < I < Ion, one preimage corresponds to two images.
4.4. The globally optimal control
The globally optimal control can be obtained by enumerating the local optima, which are characterized by only two parameters ton and toff. In the ton versus toff plane, the area of all the meaningful points is illustrated in electronic supplementary material, figure S5a. I enumerate all the 8.3 × 105 points in the area, up to a grid size 0.05 min for both axes. For each point, the homeostasis time T and the utilization rate η are calculated (electronic supplementary material, §3.2).
The globally optimal control is in terms of a given utilization rate. Therefore, it should be searched among all the points whose utilization rates are within a narrow range η ± Δη, where the tolerance Δη = 5 × 10−5 accounts for the fact that few, if any, controls have a utilization rate exactly η.
I first study the set with η = 0.1, which contains 1048 points whose T-values are heterogeneous (figure 1a). Figure 1b shows u(t) of three points in the set: the grey, blue and red controls achieve the slowest (T = 373 min), an average (T = 290 min) and the fastest (T = 203.8 min) homeostasis, respectively. The red control is therefore the globally optimal control for η = 0.1.
Figure 2 was produced by using the globally optimal control for η = 0.1 (i.e. the red control in figure 1b). As a comparison, the uncontrolled dynamics 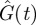 and
and 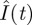 were also obtained (the grey curves in figure 2). One sees that ton = 70.5 min happens to be the time when
were also obtained (the grey curves in figure 2). One sees that ton = 70.5 min happens to be the time when 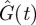 and
and 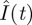 are at their peaks, which confers two respective advantages. The first advantage is rapidity. The peak of
are at their peaks, which confers two respective advantages. The first advantage is rapidity. The peak of 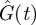 corresponds to the largest possible plasma glucose concentration (Gon ≈ 133 mg dl–1). Thus, absorbing glucose around ton = 70.5 min would yield the maximum efficiency (because the rate umaxGon is the largest possible). The homeostasis time T = 203.8 min is realistic because it typically takes about four hours to restore homeostasis upon a regular meal [42,49,58]. The second advantage is brain safety. From figure 2b, one sees that Ion is about 43.7 µU ml–1, the peak of
corresponds to the largest possible plasma glucose concentration (Gon ≈ 133 mg dl–1). Thus, absorbing glucose around ton = 70.5 min would yield the maximum efficiency (because the rate umaxGon is the largest possible). The homeostasis time T = 203.8 min is realistic because it typically takes about four hours to restore homeostasis upon a regular meal [42,49,58]. The second advantage is brain safety. From figure 2b, one sees that Ion is about 43.7 µU ml–1, the peak of  . This large threshold would prevent fortuitous glucose uptake triggered by small insulin levels, thus sparing glucose for the brain. Interestingly, although the globally optimal control was selected according to rapidity, it is also optimal in terms of its opposite—brain safety.
. This large threshold would prevent fortuitous glucose uptake triggered by small insulin levels, thus sparing glucose for the brain. Interestingly, although the globally optimal control was selected according to rapidity, it is also optimal in terms of its opposite—brain safety.
Electronic supplementary material, figure S3 shows the dynamics resulting from the sub-optimal blue control in figure 1. Because the control turns on early (ton = 22.25), the spikes are suppressed. However, this marked early suppression does not bring a long-term benefit: T = 290 > 203.8. Moreover, Ion = 13.9 µU ml–1 is a little too small. It may be fine for a normal person whose basal insulin level is around I0 = 10 µU ml–1 [58]. But a typical obese person has I0 around 20 µU ml–1 [58], which has exceeded 13.9 µU ml–1. This implies a massive glucose uptake during the fasting state of that obese person, which is dangerous to the brain.
The globally optimal controls for other small η-values may also have Ion around 43.7 µU ml–1, because Ion is around the peak of 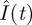 , which is not affected by the control. To test this idea, I determine u(I) for 10 different η-values: 0.01, 0.02, 0.03, 0.04, 0.05, 0.10, 0.15, 0.20, 0.25 and 0.30 (six of them are shown in figure 3a). The first eight cases all have Ion ≈ 43.7. Therefore, Ion is indeed independent of η as long as η is not too large. For η = 0.25 and 0.30, Ion deviates to 41.8 and 37.7, respectively. Now consider the threshold Ioff. As η increases, Ioff apparently decreases. The delayed switching off leads to a shorter homeostasis time. As η increases from 0.05 to 0.30, T decreases from 363.5 to 79.3 min.
, which is not affected by the control. To test this idea, I determine u(I) for 10 different η-values: 0.01, 0.02, 0.03, 0.04, 0.05, 0.10, 0.15, 0.20, 0.25 and 0.30 (six of them are shown in figure 3a). The first eight cases all have Ion ≈ 43.7. Therefore, Ion is indeed independent of η as long as η is not too large. For η = 0.25 and 0.30, Ion deviates to 41.8 and 37.7, respectively. Now consider the threshold Ioff. As η increases, Ioff apparently decreases. The delayed switching off leads to a shorter homeostasis time. As η increases from 0.05 to 0.30, T decreases from 363.5 to 79.3 min.
Figure 3.

(a) The globally optimal control u(I) for six η-values. The dashed lines are for illustration purpose only. (b) The actual controls A(I) with fixed β- and six α-values, under the limit condition K = 0. The α-values are specially chosen to render an exact match with (a). The solid and dashed lines indicate stable and unstable branches, respectively. (c) The same controls but with K = 10−4. (d) The same controls but with K = 10−2. (e) Under the condition K = 0, the solutions to electronic supplementary material, equation (S40) reduce to three straight lines, which constitute a bistable switch with Ion = cβ−1 and ΔI = cα. (f) Under the condition K = 0, the actual controls A(I) with fixed α- and four β-values.
4.5. Optimal controls are realizable
To prove that optimal controls are realizable, one needs only to show that the insulin signalling pathway can generate bistability, which is indeed true [13–15]. Moreover, the insulin signalling pathway is well organized to generate bistability (electronic supplementary material, §4). The pathway has three components, which play uncorrelated roles in modulating the shape of the actual control A(I): the input component (green colour in electronic supplementary material, figure S7) sets the threshold Ion; the output component (blue colour in electronic supplementary material, figure S7) makes A(I) an ideal step function that switches at Ion (electronic supplementary material, figure S8a); the positive feedback (red colour in electronic supplementary material, figure S7) deforms the step function into a bistable switch (electronic supplementary material, figure S8b).
Note that electronic supplementary material, figure S8 corresponds to the ideal condition K = 0, where K = Km/Amax [14]. Here Km is the Michaelis constant of the AKT phosphorylation and dephosphorylation cycle. The parameter K affects the shape of the actual control through electronic supplementary material, equation (S40). Figure 3b–d illustrate actual controls (generated by the pathway) with K = 0, 10−4 and 10−2, respectively. Under the ideal condition K = 0, the actual controls (figure 3b) match exactly with the optimal controls (figure 3a). As K becomes larger, the actual controls become less sensitive and more deviated from the optimal controls. The smallness of K (i.e. the largeness of Amax) is thus crucial to confer sensitivity.
This result concurs with a previous discovery that enzyme saturation is an important mechanism of ultrasensitivity [59–61]. When in high concentration, AKT, as the substrate of enzyme reaction, saturates the surface of the converter enzymes. Enzyme saturation alone can achieve ultrasensitivity in the absence of allosteric cooperativity [59]. Because the saturation portion of the enzyme reaction curve has a slope of nearly zero, this type of sensitivity was called zeroth-order ultrasensitivity [59].
4.6. The actual control is adaptive
The globally optimal control is subject to changes in metabolic conditions (starvation, overnutrition, pregnancy, inflammation, etc.). This necessitates the adaptivity of the actual control so that the global optimum can be tracked as much as possible. Mathematical analysis of the insulin signalling pathway reveals that the actual control is parametrized by K, α and β (electronic supplementary material, equation (S40)), which characterize the output, feedback and the input components of the pathway, respectively. It turns out that the three parameters play orthogonal roles in adjusting the actual control. The parameter K is to produce all-or-none: the smaller the K is, the more sensitive the control [14,59–61]. For simplicity, one can assume K = 0, which renders simple solutions to electronic supplementary material, equation (S40):
 |
where c is a scaling constant. In figure 3e, the solutions are represented by three lines, which constitute a bistable switch with Ion = cβ−1 and Ioff = cβ−1 − cα. Define ΔI = Ion − Ioff = cα to be the hysteresis width. It now becomes clear that α and β play uncorrelated roles: α adjusts the hysteresis width ΔI without changing the threshold Ion; β adjusts the threshold Ion without changing the hysteresis width ΔI.
To better understand the roles played by α, six actual controls are generated with K = 0, a fixed β and six α (figure 3b). The α-values are so selected that the resultant controls match exactly the globally optimal ones in figure 3a. For example, the α of the red A(I) is determined as follows: by using the relation ΔI/Ion = cα/(cβ−1) = 24.5/43.7 = 0.56, one obtains α = 0.56β−1. The exact match between figure 3a,b implies that the insulin signalling pathway is ideal if K → 0. Moreover, the correspondence becomes clear between the molecular parameter α and the physiologic parameter η. As α or η increases, ΔI increases but Ion remains the same. The parameter α negatively correlates with the intracellular nutrient level Ψ (sensed by the protein complex mTOR). Because nutritional conditions can range from starvation to overnutrition [62–64], α is highly tunable. A larger α (i.e. a smaller intracellular nutrient level Ψ) means that the cell is more ‘hungry’, which necessitates a larger η to pump more glucose into the cell. In this way, the adaptive mTOR signalling translates to the adaptive regulation of the glucose–insulin system.
To better understand the roles played by β, four actual controls are generated with K = 0, a fixed α and four β (figure 3f). As β decreases, both Ion and Ioff increase, but ΔI is fixed. That is, the bistable switch drifts along the I-axis without any distortions. This drift may well be an adaptation to some systemic change of insulin levels. For example, the overall insulin levels (including the baseline I0) of a typical obese person are twice as large as those of a normal person (figure 2e). This necessitates a corresponding rise of Ion, the threshold for the insulin signalling pathway to switch on glucose uptake. If otherwise, then the plasma insulin level could easily exceed Ion because the baseline level I0 is too high, causing fortuitous glucose uptake that is adverse to brain safety. The raise of Ion is achieved as follows. The parameter β reflects PI3-kinase activity, which is negatively regulated by many factors such as free fatty acid [65–67]. As a person becomes obese, the free fatty acid level becomes elevated, which weakens PI3-kinase activity and makes β smaller. Consequently, Ion is raised because it is inversely proportional to β.
To better understand the roles played by K, figure 3b is reproduced with non-zero K-values. For K = 10−4, the controls are deformed only slightly (figure 3c). Even for K as large as 10−2, the actual controls are still close to optima (figure 3d). The smallness of K can be realized by the condition ‘enzyme saturation’, a well-known mechanism of ultrasensitivity, first discovered in [59].
4.7. Generality
With the parameter values in electronic supplementary material, table S1, the mathematical model (equations (2.1) and (2.2)) is just for glucose–insulin homeostasis. Without assigning any parameter values, equations (2.1) and (2.2) represent a general negative feedback system in biology.
To demonstrate the generality of equations (2.1) and (2.2), I start from a simple model:
| 4.2 |
which stabilizes at G0 = 1. With a transient stimulation m(t) added to the right-hand side of equation (4.2), the state point leaves, but will finally return to, G0. To expedite homeostasis, a control −u is used, which turns the model into
If u is a constant, then the control is open-loop (non-feedback). To be a feedback control, u must change adaptively with a signal reflecting the level of G. The signal can be G itself. For example, u = γG is a proportional control that leads to the dynamics 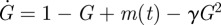 . However, it is common that G is impossible or inappropriate for signal transduction. In this event, another entity I, which tracks the change of G (equation (4.4)), serves as the feedback signal. The model can be described by
. However, it is common that G is impossible or inappropriate for signal transduction. In this event, another entity I, which tracks the change of G (equation (4.4)), serves as the feedback signal. The model can be described by
| 4.3 |
and
| 4.4 |
Being negative, the control −u(I) is a negative feedback one.
Equations (4.3) and (4.4) represent a general homeostatic system achieved by negative feedback. This general model, however, is only a special case of equations (2.1) and (2.2) (with s = λ = k = 1, and f(G) ≡ G), which implies that equations (2.1) and (2.2) are even more general.
My analysis on equations (2.1) and (2.2) has shown that locally optimal u(t) must be 0-umax-0 (equation (4.1)), which certainly applies to the model described by equations (4.3) and (4.4). An intuitive explanation of this result is as follows.
Without any constraints, it is easy to see that the optimal control is ‘all’ (i.e. u(t) ≡ umax). By consistently applying the maximum control umax, the homeostasis time is certainly minimal. But this ‘all’ solution might be unacceptable owing to many biological constraints: a great deal of energy would be wasted; the constitutive activation of a molecule might lead to cancer; potential conflicts with other physiological requirements; etc.
With the constraint η < 1, the optimal control cannot be ‘all’. My analysis has revealed that it is still simple: ‘all-or-none’. The control turns on fully at time ton and turns off fully at time toff. This result implies that a bell-shaped u(t) (increases gradually and decreases gradually) cannot be an optimal control.
Figure 4a illustrates a locally optimal u(t). By integrating equations (4.3) and (4.4) with this control, one obtains G(t) and I(t), illustrated in figure 4b,c, respectively. One then synthesizes u(t) and I(t) to obtain u(I), which is again a bistable switch (figure 4d). Note that the parameters here (s = λ = k = 1) significantly differ from those of the glucose–insulin system (s = 0.425, λ = 0.005 and k = 0.3). The function m(t) here (figure 4e) is also different from the one used for the glucose–insulin system (electronic supplementary material, figure S4). Here f(G) is just the linear function G, while in the glucose–insulin system f(G) is a nonlinear, Hill function (electronic supplementary material, equation (S36)). Despite all these differences, the same bistability feature was obtained. This is in line with the theoretical results that the optimal control is largely independent of the values of the parameters in equations (2.1) and (2.2).
5. Summary
The ubiquity of bistability now has a reason. As organisms become more complex, homeostatic regulation of the internal environment becomes more difficult owing to the increasing number of competing requirements that have to be accommodated simultaneously. This paper has shown that bistability is necessary to confer optimal compromise in homeostatic regulation. Although the mathematical model was initially for glucose homeostasis, the bistability solution is independent of the parameters in the mathematical model. The model can thus represent a general homeostatic system beyond merely glucose homeostasis. Bistability is thus required for the optimal homeostasis of all such systems using negative feedback to control concentrations.
Glucose homeostasis is a critical process, in which hyperglycemia and hypoglycemia are both unacceptable. To ensure the priority of the brain in receiving glucose, the insulin signalling pathway works like an overflow valve. During the fasting state, the valves are closed firmly so that the limited glucose all goes to the brain. The opening of the valves is significantly delayed to wait until the plasma glucose concentration builds up (in order that the subsequent glucose transportation is more efficient) and until the insulin level exceeds a preset threshold Ion. The other tissue cells can then enjoy the surplus glucose. The valves close and close fully when the insulin level drops below another threshold Ioff, which is smaller than Ion. The valve is thus hysteretic. This optimal strategy has been confirmed by mathematical and computational analysis. We now realize that what is required for the optimal regulation at the physiologic level is exactly what is provided by the insulin signalling pathway at the molecular level.
The valve is tunable to track the varying metabolic conditions. As a person becomes obese, his/her insulin levels increase, which is balanced by the corresponding increase in the threshold Ion. However, this adaptivity is not without limitations—excessive production of insulin may damage the pancreas and finally lead to insulin shortage and the emergence of diabetes.
In the evolutionary time scale, abundance of food and sedentary lifestyle are only recent things, which constitute acute environmental changes that may challenge the evolved optimality that worked well for our ancestors. The twin epidemics of obesity and diabetes have raised an alarm about maladaptation. It is the time to adjust our lifestyle to restore the optimality and to avoid being eliminated by natural selection.
Acknowledgements
The author thanks Prof. William C. Parke for his critical reading and suggestions. This work was supported by National Science Foundation grant CDI-0941228, and an Institutional Research grant (IRG-08-091-01) from the American Cancer Society to George Washington University Cancer Institute.
References
- 1.Sherman A. 2011. Dynamical systems theory in physiology. J. Gen. Physiol. 138, 13–19 10.1085/jgp.201110668 (doi:10.1085/jgp.201110668) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Novick A., Weiner M. 1957. Enzyme induction as an all-or-none phenomenon. Proc. Natl Acad. Sci. USA 43, 553–566 10.1073/pnas.43.7.553 (doi:10.1073/pnas.43.7.553) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santillán M., Mackey M. C. 2008. Quantitative approaches to the study of bistability in the lac operon of Escherichia coli. J. R. Soc. Interface 5(Suppl. 1), S29–S39 10.1098/rsif.2008.0086.focus (doi:10.1098/rsif.2008.0086.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferrell J. E., Jr, Machleder E. M. 1998. The biochemical basis of an all-or-none cell fate switch in Xenopus oocytes. Science 280, 895–898 10.1126/science.280.5365.895 (doi:10.1126/science.280.5365.895) [DOI] [PubMed] [Google Scholar]
- 5.Sha W., Moore J., Chen K., Lassaletta A. D., Yi C.-S., Tyson J. J., Sible J. C. 2003. Hysteresis drives cell-cycle transitions in Xenopus laevis egg extracts. Proc. Natl Acad. Sci. USA 100, 975–980 10.1073/pnas.0235349100 (doi:10.1073/pnas.0235349100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Angeli D., Ferrell J. E., Jr, Sontag E. D. 2004. Detection of multistability, bifurcations, and hysteresis in a large class of biological positive-feedback systems. Proc. Natl Acad. Sci. USA 101, 1822–1827 10.1073/pnas.0308265100 (doi:10.1073/pnas.0308265100) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ge H., Qian H. 2011. Non-equilibrium phase transition in mesoscopic biochemical systems: from stochastic to nonlinear dynamics and beyond. J. R. Soc. Interface 8, 107–116 10.1098/rsif.2010.0202 (doi:10.1098/rsif.2010.0202) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Qian H., Shi P.-Z., Xing J. 2009. Stochastic bifurcation, slow fluctuations, and bistability as an origin of biochemical complexity. Phys. Chem. Chem. Phys. 11, 4861–4870 10.1039/b900335p (doi:10.1039/b900335p) [DOI] [PubMed] [Google Scholar]
- 9.Bishop L. M., Qian H. 2010. Stochastic bistability and bifurcation in a mesoscopic signaling system with autocatalytic kinase. Biophys. J. 98, 1–11 10.1016/j.bpj.2009.09.055 (doi:10.1016/j.bpj.2009.09.055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vellela M., Qian H. 2009. Stochastic dynamics and non-equilibrium thermodynamics of a bistable chemical system: the Schlögl model revisited. J. R. Soc. Interface 6, 925–940 10.1098/rsif.2008.0476 (doi:10.1098/rsif.2008.0476) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pomerening J. R., Sontag E. D., Ferrell J. E., Jr 2003. Building a cell cycle oscillator: hysteresis and bistability in the activation of Cdc2. Nat. Cell Biol. 5, 346–351 10.1038/ncb954 (doi:10.1038/ncb954) [DOI] [PubMed] [Google Scholar]
- 12.Cross F. R., Archambault V., Miller M., Klovstad M. 2002. Testing a mathematical model of the yeast cell cycle. Mol. Biol. Cell 13, 52–70 10.1091/mbc.01-05-0265 (doi:10.1091/mbc.01-05-0265) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang G., Krueger G. R. F. 2010. Computational analysis of mTOR signaling pathway: bifurcation, carcinogenesis, and drug discovery. Anticancer Res. 30, 2683–2688 [PubMed] [Google Scholar]
- 14.Wang G. 2010. Singularity analysis of the AKT signaling pathway reveals connections between cancer and metabolic diseases. Phys. Biol. 7, 046015. 10.1088/1478-3975/7/4/046015 (doi:10.1088/1478-3975/7/4/046015) [DOI] [PubMed] [Google Scholar]
- 15.Giri L., Mutalik V. K., Venkatesh K. V. 2004. A steady state analysis indicates that negative feedback regulation of PTP1B by AKT elicits bistability in insulin-stimulated GLUT4 translocation. Theor. Biol. Med. Model. 1, 2. 10.1186/1742-4682-1-2 (doi:10.1186/1742-4682-1-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gardner T. S., Cantor C. R., Collins J. J. 2000. Construction of a genetic toggle switch in Escherichia coli. Nature 403, 339–342 10.1038/35002131 (doi:10.1038/35002131) [DOI] [PubMed] [Google Scholar]
- 17.Tyson J. J., Albert R., Goldbeter A., Ruoff P., Sible J. 2008. Biological switches and clocks. J. R. Soc. Interface 5(Suppl. 1), S1–S8 10.1098/rsif.2008.0179.focus (doi:10.1098/rsif.2008.0179.focus) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lisman J. E. 1985. A mechanism for memory storage insensitive to molecular turnover: a bistable autophosphorylating kinase. Proc. Natl Acad. Sci. USA 82, 3055–3057 10.1073/pnas.82.9.3055 (doi:10.1073/pnas.82.9.3055) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiong W., Ferrell J. E., Jr 2003. A positive-feedback-based bistable ‘memory module’ that governs a cell fate decision. Nature 426, 460–465 10.1038/nature02089 (doi:10.1038/nature02089) [DOI] [PubMed] [Google Scholar]
- 20.Kapuy O., He E., López-Avilés S., Uhlmann F., Tyson J. J., Novák B. 2009. System-level feedbacks control cell cycle progression. FEBS Lett. 583, 3992–3998 10.1016/j.febslet.2009.08.023 (doi:10.1016/j.febslet.2009.08.023) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Araujo R. P., Liotta L. A., Petricoin E. F. 2007. Proteins, drug targets and the mechanisms they control: the simple truth about complex networks. Nat. Rev. Drug Discov. 6, 871–880 10.1038/nrd2381 (doi:10.1038/nrd2381) [DOI] [PubMed] [Google Scholar]
- 22.Palani S., Sarkar C. A. 2008. Positive receptor feedback during lineage commitment can generate ultrasensitivity to ligand and confer robustness to a bistable switch. Biophys. J. 95, 1575–1589 10.1529/biophysj.107.120600 (doi:10.1529/biophysj.107.120600) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slepchenko B. M., Terasaki M. 2004. Bio-switches: what makes them robust? Curr. Opin. Genet. Dev. 14, 428–434 10.1016/j.gde.2004.05.001 (doi:10.1016/j.gde.2004.05.001) [DOI] [PubMed] [Google Scholar]
- 24.Liu D., Chang X., Liu Z., Chen L., Wang R. 2011. Bistability and oscillations in gene regulation mediated by small noncoding RNAs. PLoS ONE 6, e17029. 10.1371/journal.pone.0017029 (doi:10.1371/journal.pone.0017029) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsai T. Y.-C., Choi Y. S., Ma W., Pomerening J. R., Tang C., Ferrell J. E., Jr 2008. Robust, tunable biological oscillations from interlinked positive and negative feedback loops. Science 321, 126–129 10.1126/science.1156951 (doi:10.1126/science.1156951) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shiraishi T., Matsuyama S., Kitano H. 2010. Large-scale analysis of network bistability for human cancers. PLoS Comput. Biol. 6, e1000851. 10.1371/journal.pcbi.1000851 (doi:10.1371/journal.pcbi.1000851) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutherland W. J. 2005. The best solution. Nature 435, 569. 10.1038/435569a (doi:10.1038/435569a) [DOI] [PubMed] [Google Scholar]
- 28.Cannon W. B. 1929. Organization for physiological homeostasis. Physiol. Rev. 9, 399–431 [Google Scholar]
- 29.Cannon W. B. 1932. Wisdom of the body. New York, NY: W. W. Norton and Company, Inc. [Google Scholar]
- 30.Lazar M. A. 2005. How obesity causes diabetes: not a tall tale. Science 307, 373–375 10.1126/science.1104342 (doi:10.1126/science.1104342) [DOI] [PubMed] [Google Scholar]
- 31.Black P. H. 2003. The inflammatory response is an integral part of the stress response: implications for atherosclerosis, insulin resistance, type II diabetes and metabolic syndrome X. Brain Behav. Immun. 17, 350–364 10.1016/S0889-1591(03)00048-5 (doi:10.1016/S0889-1591(03)00048-5) [DOI] [PubMed] [Google Scholar]
- 32.Neel J. V. 1962. Diabetes mellitus: a ‘thrifty’ genotype rendered detrimental by ‘progress’? Am. J. Hum. Genet. 14, 353–362 [PMC free article] [PubMed] [Google Scholar]
- 33.Nasraway S. A., Jr 2007. Sitting on the horns of a dilemma: avoiding severe hypoglycemia while practicing tight glycemic control. Crit. Care Med. 35, 2435–2437 10.1097/01.CCM.0000286520.50104.51 (doi:10.1097/01.CCM.0000286520.50104.51) [DOI] [PubMed] [Google Scholar]
- 34.Rosenn B., Siddiqi T. A., Miodovnik M. 1995. Normalization of blood glucose in insulin-dependent diabetic pregnancies and the risks of hypoglycemia: a therapeutic dilemma. Obstet. Gynecol. Surv. 50, 56–61 10.1097/00006254-199501000-00027 (doi:10.1097/00006254-199501000-00027) [DOI] [PubMed] [Google Scholar]
- 35.Stannard S. R., Johnson N. A. 2004. Insulin resistance and elevated triglyceride in muscle: more important for survival than ‘thrifty’ genes? J. Physiol. (Lond.) 554, 595–607 10.1113/jphysiol.2003.053926 (doi:10.1113/jphysiol.2003.053926) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cinquin O., Demongeot J. 2002. Positive and negative feedback: striking a balance between necessary antagonists. J. Theor. Biol. 216, 229–241 10.1006/jtbi.2002.2544 (doi:10.1006/jtbi.2002.2544) [DOI] [PubMed] [Google Scholar]
- 37.Lau K.-Y., Ganguli S., Tang C. 2007. Function constrains network architecture and dynamics: a case study on the yeast cell cycle Boolean network. Phys. Rev. E. Stat. Nonlin. Soft Matter Phys. 75, 051907. 10.1103/PhysRevE.75.051907 (doi:10.1103/PhysRevE.75.051907) [DOI] [PubMed] [Google Scholar]
- 38.Airoldi E. M., Huttenhower C., Gresham D., Lu C., Caudy A. A., Dunham M. J., Broach J. R., Botstein D., Troyanskaya O. G. 2009. Predicting cellular growth from gene expression signatures. PLoS Comput. Biol. 5, e1000257. 10.1371/journal.pcbi.1000257 (doi:10.1371/journal.pcbi.1000257) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zaslaver A., Kaplan S., Bren A., Jinich A., Mayo A., Dekel E., Alon U., Itzkovitz S. 2009. Invariant distribution of promoter activities in Escherichia coli. PLoS Comput. Biol. 5, e1000545. 10.1371/journal.pcbi.1000545 (doi:10.1371/journal.pcbi.1000545) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang G., Du C., Chen H., Simha R., Rong Y., Xiao Y., Zeng C. 2010. Process-based network decomposition reveals backbone motif structure. Proc. Natl Acad. Sci. USA 107, 10 478–10 483 10.1073/pnas.0914180107 (doi:10.1073/pnas.0914180107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Scott M., Gunderson C. W., Mateescu E. M., Zhang Z., Hwa T. 2010. Interdependence of cell growth and gene expression: origins and consequences. Science 330, 1099–1102 10.1126/science.1192588 (doi:10.1126/science.1192588) [DOI] [PubMed] [Google Scholar]
- 42.Fox S. 2007. Human physiology, 10th edn Dubuque, IA: McGraw-Hill Science/Engineering/Math [Google Scholar]
- 43.Porte D., Sherwin R. S., Baron A. 2002. Ellenberg and Rifkin's diabetes mellitus, 6th edn. New York, NY: McGraw-Hill Professional [Google Scholar]
- 44.Malaisse W., Malaisse-Lagae F., Wright P. H. 1967. A new method for the measurement in vitro of pancreatic insulin secretion. Endocrinology 80, 99–108 10.1210/endo-80-1-99 (doi:10.1210/endo-80-1-99) [DOI] [PubMed] [Google Scholar]
- 45.Schuit F. C., In't Veld P. A., Pipeleers D. G. 1988. Glucose stimulates proinsulin biosynthesis by a dose-dependent recruitment of pancreatic beta cells. Proc. Natl Acad. Sci. USA 85, 3865–3869 10.1073/pnas.85.11.3865 (doi:10.1073/pnas.85.11.3865) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bosco D., Meda P. 1991. Actively synthesizing beta-cells secrete preferentially after glucose stimulation. Endocrinology 129, 3157–3166 10.1210/endo-129-6-3157 (doi:10.1210/endo-129-6-3157) [DOI] [PubMed] [Google Scholar]
- 47.Topp B., Promislow K., deVries G., Miura R. M., Finegood D. T. 2000. A model of beta-cell mass, insulin, and glucose kinetics: pathways to diabetes. J. Theor. Biol. 206, 605–619 10.1006/jtbi.2000.2150 (doi:10.1006/jtbi.2000.2150) [DOI] [PubMed] [Google Scholar]
- 48.Bergman R. N., Phillips L. S., Cobelli C. 1981. Physiologic evaluation of factors controlling glucose tolerance in man: measurement of insulin sensitivity and beta-cell glucose sensitivity from the response to intravenous glucose. J. Clin. Invest. 68, 1456–1467 10.1172/JCI110398 (doi:10.1172/JCI110398) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sturis J., Polonsky K. S., Mosekilde E., Van Cauter E. 1991. Computer model for mechanisms underlying ultradian oscillations of insulin and glucose. Am. J. Physiol. 260, E801–E809 [DOI] [PubMed] [Google Scholar]
- 50.Tolić I. M., Mosekilde E., Sturis J. 2000. Modeling the insulin–glucose feedback system: the significance of pulsatile insulin secretion. J. Theor. Biol. 207, 361–375 10.1006/jtbi.2000.2180 (doi:10.1006/jtbi.2000.2180) [DOI] [PubMed] [Google Scholar]
- 51.Li J., Kuang Y., Mason C. C. 2006. Modeling the glucose–insulin regulatory system and ultradian insulin secretory oscillations with two explicit time delays. J. Theor. Biol. 242, 722–735 10.1016/j.jtbi.2006.04.002 (doi:10.1016/j.jtbi.2006.04.002) [DOI] [PubMed] [Google Scholar]
- 52.Pontryagin L. S., Boltyanskii V. G., Gamkrelidze R. V., Mishchenko E. F. 1963. The mathematical theory of optimal processes. New York, NY: Wiley-Interscience [Google Scholar]
- 53.Agrachev A. A., Sachkov Y. L. 2004. Control theory from the geometric viewpoint. Berlin, Germany: Springer [Google Scholar]
- 54.Liao Y., Hung M.-C. 2010. Physiological regulation of AKT activity and stability. Am. J. Transl. Res. 2, 19–42 [PMC free article] [PubMed] [Google Scholar]
- 55.Liao Y., et al. 2009. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/AKT stability and activation phosphorylation. Oncogene 28, 2436–2445 10.1038/onc.2009.98 (doi:10.1038/onc.2009.98) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toth M. J., Ward K., van der Velden J., Miller M. S., Vanburen P., Lewinter M. M., Ades P. A. 2011. Chronic heart failure reduces AKT phosphorylation in human skeletal muscle: relationship to muscle size and function. J. Appl. Physiol. 110, 892–900 10.1152/japplphysiol.00545.2010 (doi:10.1152/japplphysiol.00545.2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cannon W. B. 1924. Biographical memoir Henry Pickering Bowditch 1840–1911. Washington, DC: US Government Print Off [Google Scholar]
- 58.Polonsky K. S., Given B. D., Van Cauter E. 1988. Twenty-four-hour profiles and pulsatile patterns of insulin secretion in normal and obese subjects. J. Clin. Invest. 81, 442–448 10.1172/JCI113339 (doi:10.1172/JCI113339) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goldbeter A., Koshland D. E., Jr 1981. An amplified sensitivity arising from covalent modification in biological systems. Proc. Natl Acad. Sci. USA 78, 6840–6844 10.1073/pnas.78.11.6840 (doi:10.1073/pnas.78.11.6840) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Qian H., Cooper J. A. 2008. Temporal cooperativity and sensitivity amplification in biological signal transduction. Biochemistry 47, 2211–2220 10.1021/bi702125s (doi:10.1021/bi702125s) [DOI] [PubMed] [Google Scholar]
- 61.Qian H. 2007. Phosphorylation energy hypothesis: open chemical systems and their biological functions. Annu. Rev. Phys. Chem. 58, 113–142 10.1146/annurev.physchem.58.032806.104550 (doi:10.1146/annurev.physchem.58.032806.104550) [DOI] [PubMed] [Google Scholar]
- 62.O'Reilly K. E., et al. 2006. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates AKT. Cancer Res. 66, 1500–1508 10.1158/0008-5472.CAN-05-2925 (doi:10.1158/0008-5472.CAN-05-2925) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zoncu R., Efeyan A., Sabatini D. M. 2011. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 12, 21–35 10.1038/nrm3025 (doi:10.1038/nrm3025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaelin W. G., Thompson C. B. 2010. Q&A: cancer: clues from cell metabolism. Nature 465, 562–564 10.1038/465562a (doi:10.1038/465562a) [DOI] [PubMed] [Google Scholar]
- 65.Yu C., et al. 2002. Mechanism by which fatty acids inhibit insulin activation of insulin receptor substrate-1 (IRS-1)-associated phosphatidylinositol 3-kinase activity in muscle. J. Biol. Chem. 277, 50 230–50 236 10.1074/jbc.M200958200 (doi:10.1074/jbc.M200958200) [DOI] [PubMed] [Google Scholar]
- 66.Kahn S. E., Hull R. L., Utzschneider K. M. 2006. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 10.1038/nature05482 (doi:10.1038/nature05482) [DOI] [PubMed] [Google Scholar]
- 67.Silveira L. R., Fiamoncini J., Hirabara S. M., Procópio J., Cambiaghi T. D., Pinheiro C. H. J., Lopes L. R., Curi R. 2008. Updating the effects of fatty acids on skeletal muscle. J. Cell. Physiol. 217, 1–12 10.1002/jcp.21514 (doi:10.1002/jcp.21514) [DOI] [PubMed] [Google Scholar]