

Abstract
Life history plays a critical role in governing microevolutionary processes such as gene flow and adaptation, as well as macroevolutionary processes such speciation. Here, we use multilocus phylogeographic analyses to examine a speciation event involving spectacular life-history differences between sister species of sea stars. Cryptasterina hystera has evolved a suite of derived life-history traits (including internal self-fertilization and brood protection) that differ from its sister species Cryptasterina pentagona, a gonochoric broadcast spawner. We show that these species have only been reproductively isolated for approximately 6000 years (95% highest posterior density of 905–22 628), and that this life-history change may be responsible for dramatic genetic consequences, including low nucleotide diversity, zero heterozygosity and no gene flow. The rapid divergence of these species rules out some mechanisms of isolation such as adaptation to microhabitats in sympatry, or slow divergence by genetic drift during prolonged isolation. We hypothesize that the large phenotypic differences between species relative to the short divergence time suggests that the life-history differences observed may be direct responses to disruptive selection between populations. We speculate that local environmental or demographic differences at the southern range margin are possible mechanisms of selection driving one of the fastest known marine speciation events.
Keywords: brooding, self-fertilization, isolation-with-migration, ecological speciation
1. Introduction
Disruptive selection that favours the evolution of phenotypic differences among organisms within different habitats can lead to the evolution of reproductive isolation and speciation [1–7], as in adaptive radiations of plant and animal communities of islands or lakes. Such ‘ecological speciation’ events are increasingly acknowledged as an important source of biological diversity at the species level [8–13]. However, in the absence of experimental evidence [14,15] for some specific ecological selective agent(s), there can be uncertainty about the contribution of other factors to divergence and speciation [16,17]. In particular, ecological differences between broadly distributed species that diverged long ago could have arisen in many places at any time after their divergence, and could be a consequence (e.g. of character displacement; [18]) rather than a cause of a speciation process driven by other factors. These include demographic processes of colonization and localized extinction of populations, isolation of populations by geological barriers to dispersal, the strength of sexual selection within populations and the extent of ongoing gene flow between them, plus selection against hybridization between members of diverging populations (regardless of the original cause of their divergence) [5,6,10].
Specific examples of adaptive evolution at spatial and temporal extremes can help us to clarify the role of these various processes in speciation. Life-history traits related to reproduction are a crucial element to understanding speciation this way, because disruptive selection acting specifically on such traits can lead directly to reproductive isolation [15]. Examples of spatial extremes include selection for reduced dispersal of pollen, sperm, seeds or larvae on small geographical scales at range limits [19,20]; temporal extremes include rapid and recent changes in mating system traits such as the evolution of asexuality [21,22]. Population genetic analyses of these spatial and temporal extremes can help us to focus attention on the time and place in which species divergence arose [23], and on the potential selective agents operating in those times and places that favoured divergent phenotypes [18].
Here, we use new population genetic and phylogeographic analyses to investigate an extraordinary example of both geographically localized and geologically instantaneous evolution of some previously described and spectacular life-history differences between sister species of Australian sea stars. We show that a large suite of highly derived life-history traits in Cryptasterina hystera evolved at the southern margin of the geographical range of Cryptasterina about 6000 years ago (and perhaps as recently as 1000 years). We argue that the fast, localized evolution of hermaphroditism, self-fertilization and live-bearing in C. hystera is consistent with paleoecological and comparative evidence for disruptive selection for this highly derived mode of reproduction, and we summarize additional evidence needed to test this ecological speciation hypothesis.
Asterinidae or cushion stars include the broadest range of life-history strategies known among closely related marine animals [24]. Some species retain a suite of ancestral traits shared in common with other sea star families and other classes of echinoderms, including large-bodied gonochoric adults, planktonic spawning of sperm and fertilization of eggs, prolonged planktonic larval development (with high dispersal potential) and dramatic metamorphosis during the ecological transition from planktonic larva to benthic juvenile sea star. The most highly derived life histories include miniaturized adults that are simultaneous hermaphrodites, with internal fertilization, and brood protection of offspring that develop inside the gonad of the parent. This live-bearing life history has evolved at least four times in parallel, including three lineages of Parvulastra and Cryptasterina species in which live-bearing is obligate and some live-borne offspring grow via sibling brood cannibalism, plus a fourth species (Asterina phylactica) with facultative live-bearing [24–30].
Among these four cases, the most striking contrast involves the live-bearer C. hystera and its sister species Cryptasterina pentagona [24,27–29], which is a gonochoric broadcast spawner but so similar in other morphological and ecological respects that the two species were recently recognized as distinct taxa only on the basis of their life-history differences. Eggs of the two species are of similar size and buoyancy, and C. hystera broods fully functional larvae like those of C. pentagona that, if surgically excised from the gonad, can swim, settle, attach, and complete metamorphosis to the juvenile stage outside the maternal environment. Individuals of C. hystera are assumed to self-fertilize (and our genetic data are consistent with the expected effects of selfing on genetic diversity). Lacking specialized anatomical structures for sperm transfer or storage, and possessing fully functional larvae, the transition to live-bearing in C. hystera may represent the minimal level of modification for the evolution of live-bearing [24,25,29], i.e. the evolution of internal self-fertilization and the loss of female spawning behaviour.
These two species have adjacent geographical ranges: C. pentagona is found from Central Queensland, Australia, to further north in the Indo-Pacific; C. hystera is found to the south of this range, on a few beaches near Yeppoon in central Queensland and on One Tree Island off the Great Barrier Reef [27]. This latitudinal difference mirrors the geography of live-bearing in other asterinid lineages, in which viviparous species (especially Cryptasterina pacifica, Parvulastra vivipara, Asterina phylactica) have short geographical ranges at high latitudes on the margin of the distributions of their non-viviparous sister taxa in the same genera [24,25,30,31]. Our phylogeographic analyses were designed to investigate the history of this geographical distribution and its potential role in the phenotypic and reproductive divergence of the two species.
2. Material and methods
(a). Molecular analyses
We collected tissue samples from 197 individuals of C. pentagona and 194 individuals of C. hystera from six populations per species (see electronic supplementary material, table S1 and figure 1). DNA extraction, PCR amplification, sequencing and genotyping all followed the protocols in Keever et al. [32,33]. All individuals were genotyped at five microsatellite loci (including three shared between species): a108, b202, b227, b231 and c219 were used in C. pentagona; loci a102, b202, b227, c219 and d8 were used for C. hystera. For about 20 individuals per population, two parts of the mitochondrial genome (mtDNA) and two nuclear DNA (nDNA) introns were sequenced. The tRNA/COI fragment was 689 bp for C. pentagona and 690 for C. hystera. The putative control region of the mitochondrion was amplified with primers modified from Waters et al. [34] and was 499 and 500 bp in each species, respectively. For most analyses, both mtDNA sequences were concatenated into a single mtDNA locus. The intron in Glucose-6-Phosphate Isomerase (GPI) was 324 bp long in each species, and sequenced in both the forward and reverse direction. The allelic state of heterozygotes were inferred by matching multiple peak chromatograms to alleles recovered from homozygous individuals, and this approach has been confirmed using next-gen sequencing [35]. All singleton alleles were confirmed by a second amplification and sequencing. The intron in the TaTa Box Protein was amplified with primers from Jarman et al. [36], and was 474 bp in C. pentagona and 471 bp in C. hystera. Heterozygotes were inferred identically to GPI. All sequence data were edited and aligned in Geneious v. 5.4 [37]. All sequence data are available in GenBank accession numbers JX217037–JX217643, JX221071–JX221431, JX221441–JX221555, JX227734–JX227847.
Figure 1.

Geographic distribution of mtDNA sequence variation of Cryptasterina pentagona (blue) and Cryptasterina hystera (red). Map units are in longitude and latitude. The haplotype network represents all the unique concatenated mtDNA sequences. Each sequence is represented by a circle with the size proportional to the frequency in all populations and colour-coded to match the sampling locations. Each line represents one mutational change between sequences. Hash marks are used when there is more than one mutational step between sequences. Site codes are as follows: BB, Bingil Bay; TV, Rowes Bay; BO, Bowen Beach; DB, Dingo Beach; AB, Airlie Beach; OTI, One Tree Island; SB, Statue Bay; SY, Yeppon. Full site names, and latitude and longitudes can be found in electronic supplementary material, Table S1.
(b). Molecular diversity indices
Heterozygosity, gene diversity and number of average pairwise differences were calculated in ARLEQUIN 3.5 [38]. Microsatellite rarefied allelic richness was calculated in FSTAT [39].
(c). Phylogenetic analysis
Phylogenetic networks were constructed using the median joining and mp algorithms of NETWORK [40,41] with default parameters. All samples were used.
(d). Population genetic analyses
All global and pairwise measures of population structure were calculated in GENODIVE [42,43]. Isolation by distance was examined via Mantel tests using Spearman's r.
(e). Bayesian clustering
The program TESS [44,45] estimated genetic clusters from microsatellite genotypes and geographical locations of individuals using a 100 000 step burn-in and 1 000 000 sweeps of the CAR model with spatial interaction optimization using mean distance.
(f). Coalescent analyses
Only sequence loci were used for coalescence analyses in IMa2 [46]. The microsatellite data did not conform to a stepwise mutation model, preventing us from accurately coding allele sizes as number of repeats, and the programme requires repeat sizes instead of allele sizes. MCMCMC analyses were run for burn-in until flat trend lines appeared, and afterward 300 000 genealogies per locus were saved. Using multiple pairwise comparisons limits, the number of parameters that are simultaneously estimated, helping to ensure convergence within each analysis, and this method has been shown to recover the same patterns seen in analyses of three or more populations [47]. Therefore, we employed a multiple pairwise population framework to calculate effective population size, migration rates and divergence times between the species pairs [46,47]. Six different coalescent analyses were run, one with each C. pentagona population and the same representative C. hystera population (OTI). Only one C. hystera population was used because of the extremely high levels of similarity of all populations at the sequence level such that all population comparisons give the same result (see table 1 and the electronic supplementary material, S1). We used a conservative mutation rate estimate for the concatenated mtDNA loci of 9.0156 × 10−6 mutations per year. This rate is based on the 4.57 per cent COI divergence seen in ‘geminate’ echinoderm specie pairs separated by the Isthmus of Panama [48], which, assuming 3 million years of isolation, gives a rate of 7.6 × 10−9 mutations per site per year. Because the concatenated mtDNA sequences in our study include the faster evolving control region of the mitochondrion [49], this rate is especially conservative.
Table 1.
Species-level allelic diversity. The number of microsatellite alleles was averaged across loci.
| species | microsatellites | mtDNA | TBP | GPI |
|---|---|---|---|---|
| total number of alleles or haplotypes per locus | ||||
| Cryptasterina pentagona | 7.2 | 41 | 19 | 9 |
| Cryptasterina hystera | 1.4 | 4 | 1 | 1 |
3. Results
(a). Molecular diversity
There is a striking difference in levels of genetic diversity between species (see table 1 and the electronic supplementary material, S1). C. pentagona has relatively modest levels of genetic diversity when compared with other asterinid sea stars with planktonic development [32,50]; however, C. hystera shows a dramatic reduction in nucleotide diversity, allelic diversity and most notably heterozygosity (see electronic supplementary material, table S1). Only one microsatellite locus is polymorphic across the entire known range of this species, and this locus is completely fixed within populations. Every other nuclear locus is completely monomorphic, giving an overall estimate of zero heterozygosity for C. hystera across seven different nuclear loci. Nucleotide diversity and haplotype diversity are also low for the mitochondrial locus, suggesting that C. hystera has experienced a severe population bottleneck. C. hystera may also be self-fertile [29], and while the lack of heterozygosity is consistent with selfing, it also precludes direct detection of selfing by paternity analysis of broodmates. We suspect that some level of selfing does occur; however, selfing alone would not account for the low levels of genetic diversity at the mtDNA locus.
(b). Geographic distribution of genetic diversity
We used median-joining networks to depict the spatial structure of genetic diversity revealed by the sequenced mitochondrial and nuclear loci. C. pentagona and C. hystera show less than 1 per cent divergence at all of these loci, but no haplotypes or alleles are shared between species. At the concatenated mtDNA locus (figure 1), C. pentagona and C. hystera exhibit complete lineage sorting between species; however, this is not the case for either nDNA locus (see figure 2 and the electronic supplementary material, figure S1): C. hystera is fixed for one allele at each nDNA locus, and this C. hystera allele is found in the middle of both of the C. pentagona nDNA networks. Considering that nDNA loci tend to have slower mutation rates and much larger effective populations sizes, this result is consistent with expectations for the effects of genetic drift following a recent reproductive isolation [49].
Figure 2.
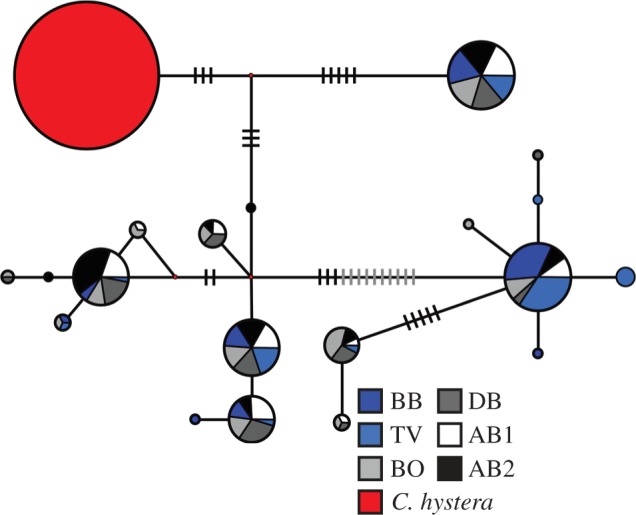
Allelic network for locus TBP. The median-joining allele network represents all the unique TBP sequences. Each sequence is represented by a circle with the size proportional to the frequency in all populations and colour-coded to match the sampling locations or species. Each line represents one mutational change between sequences. Hash marks are used when there is more than one mutational step between sequences.
For C. pentagona, the mtDNA network has a ‘star-shaped’ pattern with most haplotypic diversity produced by singleton haplotypes that differ by only one mutation from a more common haplotype. In the nDNA networks, most alleles are not singletons and tend to be several mutational steps away from each other, indicating a possible population contraction before the expansion shown in the mtDNA network [49]. In both the mtDNA locus and the TBP locus (figures 1 and 2), C. pentagona genetic diversity seems to be slightly partitioned between northern and southern populations, with common haplotypes or alleles often showing a northern or southern polarity. This pattern is largely absent from the GPI locus (see electronic supplementary material, figure S1); however, this may be due to low levels of genetic diversity in that marker.
(c). Dating the origin of Cryptasterina hystera
Using a multiple pairwise population coalescence analysis, we found a clear geographic pattern to the estimates of divergence times between pairs of C. pentagona and C. hystera populations (figure 3). The two most southern populations of C. pentagona are more recently diverged from C. hystera compared with other populations. For the southernmost C. pentagona population (AB1), divergence from C. hystera is estimated at 6159 years before present (ybp) with more narrow confidence limits (95% HPD of 905–22 628) compared with more northern C. pentagona populations. This coalescent pattern strongly suggests that AB1 (or AB2) is the population of origin for C. hystera and that the two species began to diverge at the C. pentagona southern range margin around 6000 ybp. These recent divergence times are consistent with the low molecular diversity within C. hystera populations and the small numbers of mutational steps separating them from some C. pentagona alleles in the coalescent gene trees.
Figure 3.

Estimates of divergence times between C. hystera and each C. pentagona population. Divergence times were estimated from six different IMa2 coalescent analyses, one with each C. pentagona population and the same representative C. hystera population (OTI1).
Other demographic parameters estimated for the same population pairs were consistent with a scenario of recent divergence at the southern end of the C. pentagona range. All migration rates, in either coalescent direction, are estimated at zero, consistent with complete reproductive isolation of these two species. In general, all C. pentagona and C. hystera population sizes were smaller than the estimated ancestral population size, but the effect was stronger in C. hystera with effective population size of the order of tens to hundreds of individuals, one to two orders of magnitude smaller than C. pentagona (see electronic supplementary material, figure S2). Populations AB1 and AB2 also have smaller estimated effective population size compared with other C. pentagona populations (see electronic supplementary material, figure S3), as would be expected for peripheral populations at the range margin [49].
(d). Spatial Bayesian clustering and population genetic structure
Our analyses of differentiation within each of these two species were all consistent with our coalescent isolation-with-migration analyses of divergence between them, and showed (i) spatial clustering of C. pentagona populations with strong genetic differentiation among them, plus (ii) striking loss of genetic diversity and fixation of differences among C. hystera populations (following the speciation event).
Geographical locations of individuals and microsatellite genotypes were used to infer significant clusters of individuals over genetic and physical space in C. pentagona [44,45]. Four significant clusters were found (figure 4) and they largely correlate with geographical distance, suggesting that contemporary gene flow in this species is restricted to only local levels. Global and pairwise F′ST values were calculated for both species using all polymorphic microsatellite loci, and the concatenated mtDNA locus and the TBP locus were also used for C. pentagona [42,43]. Locus GPI had very little within species polymorphism, and was subsequently omitted from within species genetic structure analyses for both species. Both species showed high levels of global genetic structure: For C. hystera, global F′ST = 1.0 (p < 0.001) and for C. pentagona, F′ST values range from 0.22 to 0.40 across loci (table 2).
Figure 4.

Four significant clusters of C. pentagona populations. The posterior probability of individuals being in any one cluster is spatially interpolated using the geographic locations of individuals and their genotypes. (a) Cluster 1, (b) cluster 2, (c) cluster 3, and (d) cluster 4.
Table 2.
Global genetic structure of Cryptasterina pentagona.
| loci | F′ST | p-value |
|---|---|---|
| mtDNA | 0.37 | <0.001 |
| TBP | 0.22 | <0.001 |
| microsatellite | 0.40 | <0.001 |
In C. hystera, this global F′ST is driven by the seemingly haphazard fixation of populations for a single microsatellite allele, with values alternating between 0 and 1 (figure 5). Sites OTI1 and OTI4 are separated by less than 250 m from each other yet are fixed for alternate allelic states. On the other hand, populations OTI2, OTI4 and SY are all fixed for the same allele, despite SY being almost 200 km away from OTI. This lack of correlation between geographical and genetic distance at any scale suggests that gene flow is extremely limited among all populations.
Figure 5.
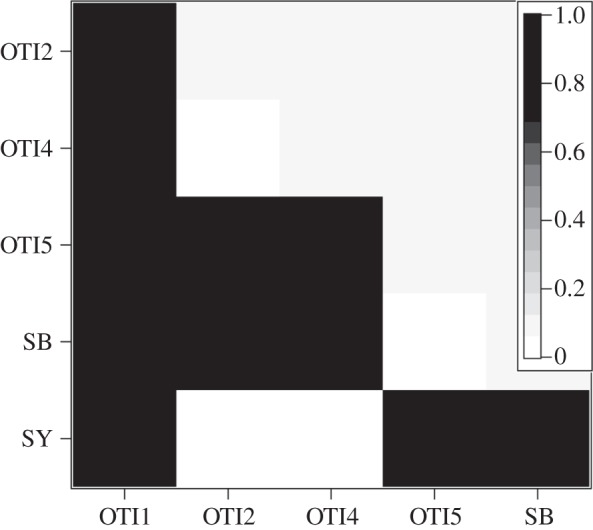
Cryptasterina hystera pairwise F′ST values using microsatellite loci. For all non-zero values, p < 0.001.
The global genetic structure of C. pentagona, in contrast, appears to be largely driven by an isolation-by-distance pattern. In both the mtDNA and TBP loci, the highest pairwise F′ST values are associated with the two most geographically isolated populations, BB and TV (figures 1 and 6). Mantel tests confirm that there is a significant relationship between genetic isolation and geographical distance (mtDNA: R2 = 0.5025, p < 0.0459; TBP: R2 = 0.5435, p < 0.0495). However, there is no correlation between geographical distance and estimates of genetic structure using the microsatellite loci (R2 = 0.02, p = 0.25). Instead, almost all population pairs have significant pairwise F′ST values with the two populations at the end of the sampling range having the highest levels of genetic isolation from all other populations (see electronic supplementary material, figure S4). The microsatellite pairwise F′ST values are more consistent with a pattern of local/regional genetic isolation in C. pentagona that matches well with the genetic and spatial clustering results. On the other hand, these populations may also not fully be in migration-drift equilibrium, and the larger effective population size of microsatellite loci, compared with the mtDNA marker, and the higher chance of homoplasy compared with the nDNA sequence marker, may reduce the power to detect an emergent isolation-by-distance population structure. Considering all three datasets, the genetic connectivity of C. pentagona most likely follows an isolation-by-distance pattern, but not all loci, especially those with larger effective population sizes, are in migration-drift equilibrium.
Figure 6.

Cryptasterina pentagona pairwise F′ST values using (a) TBO and (b) mtDNA loci. For all non-zero values p < 0.05 except for BO/AB2 and DB/AB2, which are non-significant.
4. Discussion
Our coalescent analyses of Australian Cryptasterina sea stars date their speciation at approximately 6000 ybp and suggest that C. hystera originated from the southernmost population(s) of C. pentagona. The origin, spread, and fixation of the derived C. hystera life history was completed in at most about 22 000 years (the upper confidence limit on the divergence time from C. pentagona), and perhaps as little as about 1000 years (the lower confidence limit). Even the oldest of these times is long after other significant biogeographic events in the history of the Australian fauna (such as the colonization of the continent by one primate species), and represents exceedingly rapid life-history evolution on ecological time scales in comparison with similar traits in other organisms (e.g. the single ancient origin and evolutionary conservatism of live-bearing in eutherian mammals).
The timing of these evolutionary changes is critically important for testing and rejecting some alternative hypotheses for speciation in Crypasterina. In particular, our results are not consistent with the slow, gradual loss of shared alleles (and of reproductive compatibility) via genetic drift in allopatry (classic geographical speciation), or with divergent adaptation to broadly sympatric microhabitat differences such as different intertidal heights [13,51]. Instead, the geographically localized evolution of simultaneous hermaphrodites with internal self-fertilization, brood protection and live birth was extraordinarily rapid and implies selection on ancestral C. hystera populations that favoured this life history as an adaptive response to some environmental variable. Other alternative hypotheses, such as the evolution of these life-history traits as adaptations after divergence, or as correlated responses to selection on other traits, are not entirely consistent with our data. For example, there is no evidence that these species were ever sympatric, and all available evidence points to peripatric divergence. Given the low levels of genetic connectivity observed in both species, a range disjunction of 375 km represents a significant barrier to dispersal; therefore, the more parsimonious inference is that this speciation is the result of an extreme dispersal event, followed by geographical and reproductive isolation in conjunction with rapid adaptation to a new environment. However, additional genetic and experimental work would be needed to rule out alternatives and confirm this speciation hypothesis.
Our results suggest that disruptive selection for different life-history phenotypes between sea stars in different habitats may be a short pathway to rapid evolutionary divergence and speciation. The location of larval development (benthic versus planktonic) has been argued to determine the tempo and mode of evolution in marine species: such life-history differences may facilitate or limit opportunities for geographical speciation in allopatry [52,53] by isolating populations with self-recruitment (in species with benthic development of embryos) or by promoting gene flow and limiting genetic drift (in species with planktonic development). However, such arguments do not address the selective causes of the origin of live-bearing and other forms of benthic reproduction without larval dispersal in specific lineages. Viviparity, despite its association with limited dispersal ability, has evolved several times across many different types of marine invertebrate taxa, including multiple lineages of Asterinidae [24,29,30,52,54,55], and it is consistently associated with species in colder water temperatures and higher latitudes [53,56,57]. For example, in cowries, the loss of planktonic development seems to be directly tied to environmental factors specifically associated with temperature and productivity [45], suggesting that environmental selection may be playing a key role in the evolutionary changes in life-history traits and associated speciation.
Unfortunately, the available evidence does not identify a specific ecological factor that selected for live-bearing at high-latitude range margins in cushion stars. Here, we speculate about the nature of two such factors. The first is environmental changes in water temperature associated with post-Pleistocene climate variation. The geographical range disjunction between the two Cryptasterina species in Australia is about 375 km and approximately coincides with the location where colder eastern Pacific waters mix with warmer tropical waters transported by the East Australian Current (EAC) [58]. Our estimate of 6000 ypb for the speciation event is much more recent than the last glacial maximum (LGM), but it coincides with other significant Holocene oceanographic events. During the LGM, the current cold water boundary between the Coral Sea and the Tasman Sea (the Tasman Front), located around 34° S, was shifted northward to about 23–26° S [59], and this change would have most likely extirpated populations of warm-adapted C. pentagona from the majority of its present-day range in Australia. Around 11 000 ybp, the geographical distribution of warmer and cooler sea surface temperatures returned to patterns similar to present-day temperatures [59], which may have allowed C. pentagona to recolonize the southern portion of its contemporary range at that time. This contraction and expansion of C. pentagona populations is consistent with both the coalescent estimates of effective and ancestral population sizes and patterns seen in the phylogenetic networks. Finally, around 5000 ybp, the EAC became reestablished [59] and may have provided a potential warm water pathway for C. pentagona larvae to colonize habitats further south. This speculative scenario is consistent with the geographical and temporal origins of live-bearing in Australian Cryptasterina, and with the evolution of live-bearing at high-latitude range margins in other asterinids [24,29,30,52,54,55] and other organisms [28–29], and again highlights the potential role of disruptive selection in facilitating rapid evolutionary change and speciation.
A second potential factor is small effective population size. Our population genetic analyses all indicate a significant population bottleneck associated with the speciation event, especially in southern C. pentagona populations and most pronounced in C. hystera. An associated Allee effect—scarcity of mates—may have strongly selected for self-fertilization as a form of reproductive assurance [60,61]. The associated inbreeding depression may be slight if populations are already highly inbred, e.g. owing to a population bottleneck before the evolution of selfing [30]. Under some models, selfing and small population size can facilitate adaptive molecular and phenotypic divergence [61,62].
We hypothesize that a small number of C. pentagona individuals or larvae were able to colonize the present-day range of C. hystera, and that this habitat [58] provided a divergent selective regime that led to the loss of planktonic development and other associated life-history changes, with reproductive isolation and speciation as a consequence. The speed of this speciation event suggests that it occurred via selection upon preexisting genetic variation on an ecological time scale, because it is unlikely that a complex suite of derived life-history characters including hermaphroditism, selfing and live-bearing could so quickly evolve via drift- and mutation-driven processes [9,63,64]. The specific genetic targets of this selection are usually poorly known [65], but the evolution of the same suite of life-history traits in a northern temperate Cryptasterina species (and in other asterinid genera) suggests that the genetic variation needed for this adaptation may be widespread among cushion stars [25,31]. It is not known whether these similarities represent convergences (based on responses to selection at the same selected loci) or parallelisms (at different loci).
Testing this hypothesis will require several types of new evidence, especially reciprocal transplants or common garden experiments that reveal fitness differences between Cryptasterina species associated with specific environmental differences between their habitats [15], and hybridization experiments that reveal whether pre- or post-zygotic reproductive incompatibility is associated with the recent localized evolution of life-history differences. Evidence for differential fitness in common gardens or in the field, and of gamete compatibility and hybrid viability, would bolster our argument for life-history traits as the targets of selection (and ecological speciation) in the origin of Cryptasterina and other asterinid species.
Acknowledgments
The authors thank Sergio Barbosa, Alan Dartnall, Tom Prowse and the entire staff of the One Tree Island Research Station of the University of Sydney. This work was funded by a US National Science Foundation grant OCE-0623678 to R.J.T. and R.K.G. This is contribution 1505 from the Hawai‘i Institute of Marine Biology and 8694 from the School of Ocean and Earth Sciences and Technology (SOEST). Lastly, this manuscript benefited the comments of two anonymous reviewers.
References
- 1.Schluter D. 2001. Ecology and the origin of species. Trends Ecol. Evol. 16, 372–380 10.1016/S0169-5347(01)02198-X (doi:10.1016/S0169-5347(01)02198-X) [DOI] [PubMed] [Google Scholar]
- 2.Dobzhansky T. 1937. Genetics and the origin of species. New York, NY: Columbia University Press [Google Scholar]
- 3.Mayr E. 1942. Systematics and the origin of species. New York, NY: Columbia University Press [Google Scholar]
- 4.Mayr E. 1954. Geographic speciation in tropical echinoids. Evolution 8, 1–18 10.2307/2405661 (doi:10.2307/2405661) [DOI] [Google Scholar]
- 5.Rundle H. D., Nosil P. 2005. Ecological speciation. Ecol. Lett. 8, 336–352 10.1111/j.1461-0248.2004.00715.x (doi:10.1111/j.1461-0248.2004.00715.x) [DOI] [Google Scholar]
- 6.Via S. 2001. Sympatric speciation in animals: the ugly duckling grows up. Trends Ecol. Evol. 16, 381–390 10.1016/S0169-5347(01)02188-7 (doi:10.1016/S0169-5347(01)02188-7) [DOI] [PubMed] [Google Scholar]
- 7.Palumbi S. R. 1994. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 25, 547–572 10.1146/annurev.es.25.110194.002555 (doi:10.1146/annurev.es.25.110194.002555) [DOI] [Google Scholar]
- 8.Rocha L. A., Robertson D. R., Roman J., Bowen B. W. 2005. Ecological speciation in tropical reef fishes. Proc. R. Soc. B 272, 573–579 10.1098/2004.3005 (doi:10.1098/2004.3005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schluter D., Conte G. L. 2009. Genetics and ecological speciation. Proc. Natl Acad. Sci. USA 106, 9955–9962 10.1073/pnas.0901264106 (doi:10.1073/pnas.0901264106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schluter D. 2009. Evidence for ecological speciation and its alternative. Science 323, 737–741 10.1126/science.1160006 (doi:10.1126/science.1160006) [DOI] [PubMed] [Google Scholar]
- 11.Pereyra R. T., Bergström L., Kautsky L., Johannesson K. 2009. Rapid speciation in a newly opened postglacial marine environment, the Baltic Sea. BMC Evol. Biol. 9, 70. 10.1186/1471-2148-9-70 (doi:10.1186/1471-2148-9-70) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Conde-Padin P., Carvajal-Rodriguez A., Carballo M., Caballero A., Rolán-Alvarez E. 2007. Genetic variation for shell traits in a direct-developing marine snail involved in a putative sympatric ecological speciation process. Evol. Ecol. 21, 635–650 10.1007/s10682-006-9142-8 (doi:10.1007/s10682-006-9142-8) [DOI] [Google Scholar]
- 13.Bird C. E., Holland B. S., Bowen B. W., Toonen R. J. 2011. Diversification of sympatric broadcast-spawning limpets (Cellana spp.) within the Hawaiian archipelago. Mol. Ecol. 20, 2128–2141 10.1111/j.1365-294X.2011.05081.x (doi:10.1111/j.1365-294X.2011.05081.x) [DOI] [PubMed] [Google Scholar]
- 14.Greenberg A. J., Moran J. R., Coyne J. A., Wu C.-I. 2003. Ecological adaptation during incipient speciation revealed by precise gene replacement. Science 302, 1754–1757 10.1126/science.1090432 (doi:10.1126/science.1090432) [DOI] [PubMed] [Google Scholar]
- 15.Hall M. C., Willis J. H. 2006. Divergent selection on flowering time contributes to local adaptation in Mimulus guttatus populations. Evolution 60, 2466–2477 [PubMed] [Google Scholar]
- 16.Noor M. F., Feder J. L. 2006. Speciation genetics: evolving approaches. Nat. Rev. Genet 7, 851–861 10.1038/nrg1968 (doi:10.1038/nrg1968) [DOI] [PubMed] [Google Scholar]
- 17.Sobel J. M., Chen G. F., Watt L. R., Schemske D. W. 2010. The biology of speciation. Evolution 64, 295–315 10.1111/j.1558-5646.2009.00877.x (doi:10.1111/j.1558-5646.2009.00877.x) [DOI] [PubMed] [Google Scholar]
- 18.Marko P. B. 2005. An intraspecific comparative analysis of character divergence between sympatric species. Evolution 59, 554–564 [PubMed] [Google Scholar]
- 19.Dytham C. 2009. Evolved dispersal strategies at range margins. Proc. R. Soc. B 276, 1407–1413 10.1098/rspb.2008.1535 (doi:10.1098/rspb.2008.1535) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaston K. J. 2009. Geographic range limits: achieving synthesis. Proc. R. Soc. B 276, 1395–1406 10.1098/rspb.2008.1480 (doi:10.1098/rspb.2008.1480) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Law J. H., Crespi B. J. 2002. Recent and ancient asexuality in Timema walkingsticks. Evolution 56, 1711–1717 [DOI] [PubMed] [Google Scholar]
- 22.Schwander T., Crespi B. J. 2009. Multiple direct transitions from sexual reproduction to apomictic parthenogenesis in Timema stick insects. Evolution 63, 84–103 10.1111/j.1558-5646.2008.00524.x (doi:10.1111/j.1558-5646.2008.00524.x) [DOI] [PubMed] [Google Scholar]
- 23.Marko P. B. 1998. Historical allopatry and the biogeography of speciation in the prosobranch snail genus nucella. Evolution 52, 757–774 10.2307/2411270 (doi:10.2307/2411270) [DOI] [PubMed] [Google Scholar]
- 24.Byrne M. 2006. Life history diversity and evolution in the Asterinidae. Integr. Comp. Biol. 46, 243–254 10.1093/icb/icj033 (doi:10.1093/icb/icj033) [DOI] [PubMed] [Google Scholar]
- 25.Byrne M., Hart M. W., Cerra A., Cisternas P. 2003. Reproduction and larval morphology of broadcasting and viviparous species in the Cryptasterina species complex. Biol. Bull. 205, 285–294 10.2307/1543292 (doi:10.2307/1543292) [DOI] [PubMed] [Google Scholar]
- 26.Hart M. W., Byrne M., Smith M. J. 1997. Molecular phylogenetic analysis of life-history evolution in asterinid starfish. Evolution 51, 1848–1861 10.2307/2411007 (doi:10.2307/2411007) [DOI] [PubMed] [Google Scholar]
- 27.Dartnall A. J., Byrne M., Collins J., Hart M. W. 2003. A new viviparous species of asterinid (Echinodermata, Asteroidea, Asterinidae) and a new genus to accommodate the species of pan-tropical exiguoid sea stars. Zootaxa 359, 1–14 [Google Scholar]
- 28.Hart M. W., Byrne M., Johnson S. L. 2003. Patiriella pseudoexigua (Asteroidea: Asterinidae), a cryptic species complex revealed by molecular and embryological analyses. J. Mar. Biol. Assoc. UK 83, 1109–1116 10.1017/S002531540300835Xh (doi:10.1017/S002531540300835Xh) [DOI] [Google Scholar]
- 29.Byrne M. 2005. Viviparity in the sea star Cryptasterina hystera (Asterinidae)—conserved and modified features in reproduction and development. Biol. Bull. 208, 81–91 10.2307/3593116 (doi:10.2307/3593116) [DOI] [PubMed] [Google Scholar]
- 30.Strathmann R. R., Strathmann M. F., Emson R. H. 1984. Does limited brood capacity link adult size, brooding, and simultaneous hermaphroditism? A test with the starfish Asterina phylactica. Am. Nat. 123, 796–818 10.1086/284240 (doi:10.1086/284240) [DOI] [Google Scholar]
- 31.Komatsu M., Kano Y. T., Oguro C. 1990. Development of a true ovoviviparous sea star, Asterina pseudoexigua pacifica. Biological Bulletin 179, 254. 10.2307/1542316 (doi:10.2307/1542316) [DOI] [PubMed] [Google Scholar]
- 32.Keever C. C., et al. 2009. Discordant distribution of populations and genetic variation in a sea star with high dispersal potential. Evolution 63, 3214–3227 10.1111/j.1558-5646.2009.00801.x (doi:10.1111/j.1558-5646.2009.00801.x) [DOI] [PubMed] [Google Scholar]
- 33.Keever C. C., Sunday J., Wood C., Byrne M., Hart M. W. 2008. Discovery and cross-amplification of microsatellite polymorphisms in asterinid sea stars. Biol. Bull. 215, 164–172 10.2307/25470697 (doi:10.2307/25470697) [DOI] [PubMed] [Google Scholar]
- 34.Waters J. M., O'Loughlin P. M., Roy M. S. 2004. Cladogenesis in a starfish species complex from souethern Australia: evidence for vicariant speciation? Mol. Phylogenet. Evol. 32, 236–245 10.1016/j.ympev.2003.11.014 (doi:10.1016/j.ympev.2003.11.014) [DOI] [PubMed] [Google Scholar]
- 35.Puritz J. B., Addison J. A., Toonen R. J. 2012. Next-generation phylogeography: a targeted approach for multilocus sequencing of non-model organisms. PLoS ONE 7, e34241. 10.1371/journal.pone.0034241 (doi:10.1371/journal.pone.0034241) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jarman S. N., Ward R. D., Elliott N. G. 2002. Oligonucleotide primers for PCR amplification of coelomate introns. Mar. Biotechnol. 4, 347–355 10.1007/s10126-002-0029-6 (doi:10.1007/s10126-002-0029-6) [DOI] [PubMed] [Google Scholar]
- 37.Drummond A. J., et al. 2011. Geneious v. 5.4.
- 38.Excoffier L., Lischer H. E. L. 2010. Arlequin suite v. 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Res. 10, 564–567 10.1111/j.1755-0998.2010.02847.x (doi:10.1111/j.1755-0998.2010.02847.x) [DOI] [PubMed] [Google Scholar]
- 39.Goudet J. 1995. FSTAT (v. 1.2): a computer program to calculate F-statistics. J. Hered. 86, 485–486 [Google Scholar]
- 40.Bandelt H. J., Forster P., Röhl A. 1999. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 10.1093/oxfordjournals.molbev.a026036 (doi:10.1093/oxfordjournals.molbev.a026036) [DOI] [PubMed] [Google Scholar]
- 41.Polzin T., Daneshmand S. V. 2003. On Steiner trees and minimum spanning trees in hypergraphs. Oper. Res. Lett. 31, 12–20 10.1016/S0167-6377(02)00185-2 (doi:10.1016/S0167-6377(02)00185-2) [DOI] [Google Scholar]
- 42.Meirmans P. G. 2006. Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution 60, 2399–2402 [PubMed] [Google Scholar]
- 43.Meirmans P. G., Van Tienderen P. H. 2004. Genotype and genodive: two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 4, 792–794 10.1111/j.1471-8286.2004.00770.x (doi:10.1111/j.1471-8286.2004.00770.x) [DOI] [Google Scholar]
- 44.Durand E., Jay F., Gaggiotti O. E., François O. 2009. Spatial inference of admixture proportions and secondary contact zones. Mol. Biol. Evol. 26, 1963–1973 10.1093/molbev/msp106 (doi:10.1093/molbev/msp106) [DOI] [PubMed] [Google Scholar]
- 45.Chen C., Durand E., Forbes F., François O. 2007. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol. Ecol. Notes 7, 747–756 10.1111/j.1471-8286.2007.01769.x (doi:10.1111/j.1471-8286.2007.01769.x) [DOI] [Google Scholar]
- 46.Hey J. 2010. Isolation with migration models for more than two populations. Mol. Biol. Evol. 27, 905–920 10.1093/molbev/msp296 (doi:10.1093/molbev/msp296) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hey J. 2010. The divergence of chimpanzee species and subspecies as revealed in multipopulation isolation-with-migration analyses. Mol. Biol. Evol. 27, 921–933 10.1093/molbev/msp298 (doi:10.1093/molbev/msp298) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lessios H. A., Kessing B. D., Pearse J. S. 2001. Population structure and speciation in tropical seas: global phylogeography of the sea urchin Diadema. Evolution 55, 955–975 10.1554/0014-3820(2001)055[0955:PSASIT]2.0.CO;2 (doi:10.1554/0014-3820(2001)055[0955:PSASIT]2.0.CO;2) [DOI] [PubMed] [Google Scholar]
- 49.Avise J. C. 2004. Molecular markers, natural history, and evolution, 2nd edn. Sunderland, MA: Sinauer Associates [Google Scholar]
- 50.Puritz J. B., Toonen R. J. 2011. Coastal pollution limits pelagic larval dispersal. Nat. Commun. 2, 226. 10.1038/ncomms1238 (doi:10.1038/ncomms1238) [DOI] [PubMed] [Google Scholar]
- 51.Butlin R. K., Galindo J., Grahame J. W. 2008. Sympatric, parapatric or allopatric: the most important way to classify speciation? Phil. Trans. R. Soc. B 363, 2997–3007 10.1098/rstb.2008.0076 (doi:10.1098/rstb.2008.0076) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jablonski D. 1986. Larval ecology and macroevolution in marine invertebrates. Bull. Mar. Sci. 39, 565–587 [Google Scholar]
- 53.Meyer C. P. 2003. Molecular systematics of cowries (Gastropoda: Cypraeidae) and diversification patterns in the tropics. Biol. J. Linnean Soc. 79, 401–459 10.1046/j.1095-8312.2003.00197.x (doi:10.1046/j.1095-8312.2003.00197.x) [DOI] [Google Scholar]
- 54.Strathmann R. R. 1993. Hypotheses on the origins of marine larvae. Annu. Rev. Ecol. Syst. 24, 89–117 10.1146/annurev.es.24.110193.000513 (doi:10.1146/annurev.es.24.110193.000513) [DOI] [Google Scholar]
- 55.Strathmann R. R. 1990. Why life histories evolve differently in the sea. Integr. Comp. Biol. 30, 197–207 10.1093/icb/30.1.197 (doi:10.1093/icb/30.1.197) [DOI] [Google Scholar]
- 56.Thorson G. 1950. Reproductive and larval ecology of marine bottom invertebrates. Biol. Rev. 25, 1–45 10.1111/j.1469-185X.1950.tb00585.x (doi:10.1111/j.1469-185X.1950.tb00585.x) [DOI] [PubMed] [Google Scholar]
- 57.McLay C. L., Lim S. S. L., Ng P. K. L. 2001. On the first zoea of Lauridromia indica (Gray, 1831), with an appraisal of the generic classification of the Dromiidae (Decapoda: Brachyura) using larval characters. J. Crustacean Biol. 21, 733–747 10.1651/0278-0372(2001)021[0733:OTFZOL]2.0.CO;2 (doi:10.1651/0278-0372(2001)021[0733:OTFZOL]2.0.CO;2) [DOI] [Google Scholar]
- 58.Burrage D. M., Steinberg C. R., Skirving W. J., Kleypast J. A. 1996. Mesoscale circulation features of the Great Barrier Reef region inferred from NOAA satellite imagery. Remote Sensing Environ. 56, 21–41 10.1016/0034-4257(95)00226-X (doi:10.1016/0034-4257(95)00226-X) [DOI] [Google Scholar]
- 59.Bostock H. C., Opdyke B. N., Gagan M. K., Kiss A. E., Fifield L. K. 2006. Glacial/interglacial changes in the East Australian Current. Clim. Dyn. 26, 645–659 10.1007/s00382-005-0103-7 (doi:10.1007/s00382-005-0103-7) [DOI] [Google Scholar]
- 60.Gascoigne J., Berec L., Gregory S., Courchamp F. 2009. Dangerously few liaisons: a review of mate-finding Allee effects. Popul. Ecol. 51, 355–372 10.1007/s10144-009-0146-4 (doi:10.1007/s10144-009-0146-4) [DOI] [Google Scholar]
- 61.Levin D. A. 2010. Environment-enhanced self-fertilization: implications for niche shifts in adjacent populations. J. Ecol. 98, 1276–1283 10.1111/j.1365-2745.2010.01715.x (doi:10.1111/j.1365-2745.2010.01715.x) [DOI] [Google Scholar]
- 62.Rajon E., Masel J. 2010. Evolution of molecular error rates and the consequences for evolvability. Proc. Natl Acad. Sci. USA 108, 1082–1087 10.1073/pnas.1012918108 (doi:10.1073/pnas.1012918108) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Barrett R. D. H., Schluter D. 2008. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 10.1016/j.tree.2007.09.008 (doi:10.1016/j.tree.2007.09.008) [DOI] [PubMed] [Google Scholar]
- 64.Hermisson J., Pennings P. S. 2005. Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169, 2335–2352 10.1534/genetics.104.036947 (doi:10.1534/genetics.104.036947) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nosil P., Schluter D. 2011. The genes underlying the process of speciation. Trends Ecol. Evol. 26, 160–167 10.1016/j.tree.2011.01.001 (doi:10.1016/j.tree.2011.01.001) [DOI] [PubMed] [Google Scholar]