

Abstract
The aqueous concentrations of heavy metals in soils, sediments, and aquatic environments frequently are controlled by the dissolution and precipitation of discrete mineral phases. Contaminant uptake by organisms as well as contaminant transport in natural systems typically occurs through the solution phase. Thus, the thermodynamic solubility of contaminant-containing minerals in these environments can directly influence the chemical reactivity, transport, and ecotoxicity of their constituent ions. In many cases, Pb-contaminated soils and sediments contain the minerals anglesite (PbSO4), cerussite (PbCO3), and various lead oxides (e.g., litharge, PbO) as well as Pb2+ adsorbed to Fe and Mn (hydr)oxides. Whereas adsorbed Pb can be comparatively inert, the lead oxides, sulfates, and carbonates are all highly soluble in acidic to circumneutral environments, and soil Pb in these forms can pose a significant environmental risk. In contrast, the lead phosphates [e.g., pyromorphite, Pb5(PO4)3Cl] are much less soluble and geochemically stable over a wide pH range. Application of soluble or solid-phase phosphates (i.e., apatites) to contaminated soils and sediments induces the dissolution of the “native” Pb minerals, the desorption of Pb adsorbed by hydrous metal oxides, and the subsequent formation of pyromorphites in situ. This process results in decreases in the chemical lability and bioavailability of the Pb without its removal from the contaminated media. This and analogous approaches may be useful strategies for remediating contaminated soils and sediments.
The Earth’s surface is dominated by the elements O, H, Si, Al, Fe, Ca, Na, K, Mg, Ti, and P. As oxides, these elements account for ≈96% of the total mass of the continental crust (1). Many of the remaining elements in the periodic table (together with C) are essential for life. Many trace elements are toxic to a wide range of organisms when concentrated and some are toxic to most even at very low concentration. These latter contaminants are natural substances (with the exception of the transuranics) and life on Earth evolved in their presence. Human activity has altered the distribution and forms of these elements, locally increasing their relative toxicities and the frequency with which they are encountered by living organisms.
At present, various elements are listed as priority pollutants by the U.S. Environmental Protection Agency. Their concentrations in soils, as well as in surface and ground water, typically are regulated based on total concentration. This system provides a convenient regulatory framework for establishing acceptable levels of contaminant metals and oxoanions in environmental media. However, the environmental science community recognizes that total concentration is not an accurate predictor of the bioavailability or chemical lability of a given contaminant in soils, sediments, or aquatic environments. Rather, the toxicity of a substance, be it an element, an ion, or a molecule typically is controlled by its chemical and physical state, or speciation (2–10). Thus, regulations based on absolute concentration may be convenient, but their scientific validity may be in question.
Knowledge of the link between chemical speciation and bioavailability is not new, nor did it originate with concerns of contaminant toxicity or environmental pollution. The fields of soil chemistry and soil fertility were, in part, created by the recognition that total soil concentration is a poor predictor of the bioavailability of essential nutrients required for plant growth and food production. This recognition has led to extensive research on the identity and form of nutrient elements in soils and fertilizers with emphasis on predicting their bioavailability. An example is the attention paid to the chemistry and mineralogy of P in soils and fertilizers (11–13).
Phosphorous is often a limiting nutrient, and supplementation of soil P through the addition of P-containing amendments has been practiced at least as early as 287 BC (13). The apatite mineral family is the most ubiquitous form of P in the Earth’s crust, as well as the most geochemically stable one in neutral to alkaline environments (14). Although used extensively as fertilizer, its intrinsically low solubilities makes apatite a very poor choice. Instead, more soluble forms of P commonly are used as amendments to P-deficient agricultural soils. Thus, when serving as a nutrient, the total concentration of P in the agricultural amendment is not as important as the form or availability of the P in the amendment material.
In addition to being an essential nutrient, P is an important environmental contaminant. Excessive influx of P into fresh water can lead to increases in primary productivity (photosynthesis) and accelerated sedimentation (15). This process, cultural eutrophication, can result in the growth of deleterious species of algae, depletions of available O2, and production of toxic metabolic products by a number of phytoplankton. Numerous studies of fresh-water systems have shown the inseparable link between the chemical form or species of P entering lakes and the potential for P-induced eutrophication (16). Indeed, bioassay measurements indicated that P present as apatites is much less bioavailable to planktonic algae than dissolved phosphate (16). Presumably, the lower bioavailability of apatites is a result of their low solubility in neutral and alkaline environments (as also in similar agricultural environments). That apatite as well as other solid-phase forms of P are less bioavailable than dissolved P strongly influenced the institutional controls implemented for the protection of the Great Lakes of North America. Initial efforts were focused on the removal of dissolved P from wastewater discharges followed by a later effort to reduce sediment loads from agriculture. The latter is dominated by particulate P (16).
These examples illustrate two important points relevant to contaminant chemistry and ecotoxicology. First, a substance beneficial in one environment (e.g., bioavailable P in an agricultural soil) may be deleterious in another (e.g., bioavailable P in lake waters). Second, the specific form of a substance has a profound impact on its bioavailability with solid-phase forms (including sorbed species) generally controlling its bioavailability in natural environments. It is clear that dissolved substances are generally more labile and bioavailable than solids, but can one generalize about the relative bioavailabilities of two or more different solids, each containing the same element of interest? As discussed below, this question may be assessed by considering the solubility products of the solids.
The relationship between solubility and bioavailability of contaminants has important ramifications for environmental risk assessment and environmental remediation. Adoption of an environmental impact or bioavailability mode of contaminant regulation can on be accomplished if a formal assessment indicates that some parameter other than total concentration (e.g., solubility) controls the bioavailability and ecotoxicology of that substance. Concomitantly, recognition that bioavailability can be tied to solubility rather than total concentration allows one to consider remediation strategies based on in situ reductions solubility of a contaminant, rather than its complete removal.
Solubility and the Solubility Product
The assertion that the bioavailability of a given element in soils or sediments is controlled by its solid-phase form rests on the assumption that uptake of a contaminant by a target organism happens through the solution phase. This is a safe assumption to make when one considers the uptake of an ion by plants. It may be less appropriate for the uptake of contaminants by fauna where ingestion or inhalation of particulate material can represent a significant mechanism of contaminant exposure. Nevertheless, with the possible exception of radiological damage, toxic responses to a contaminant generally require absorption by biological tissue. Even for ingestion, the relative bioavailabilities and toxicities of different mineral forms of a given element are subject to their relative solubility (17, 18). This phenomenon may in part be caused by the linkage between intrinsic solubility and relative dissolution rates, as discussed below. In any event, the equilibrium solubility of a given mineral and its dissolution kinetics profoundly affect the bioavailability and chemical lability of its constituent ions.
For a given solid, MxLy, a general dissolution reaction is:
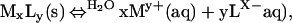 |
1 |
where My+(aq) and Lx−(aq) are the aqueous metal and ligand ions M and L, respectively. An equilibrium constant for this reaction is defined as:
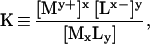 |
2 |
where [ ] denote activities. A solubility product is then defined as:
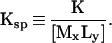 |
3 |
If the solid MxLy is in its standard state then [MxLy] becomes unity and Ksp becomes:
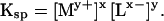 |
4 |
For a fixed activity of Lx−, the solid with the smallest numerical value of Ksp will support the smallest equilibrium activity of My+. Commonly, the toxicity of metal M is directly proportional to the activity of the free metal ion, My+, regardless of whether My+ or some hydrolytic species, or complex ion-pair is the most toxic form of M. This relationship results from the direct relationship between the activity of My+ and all other species of dissolved M (including complex ion-pairs and hydrolytic species). Therefore, the least toxic solid form of M will have the smallest aqueous equilibrium activity of My+. Analogously, the most toxic solid will be that which supports the largest aqueous equilibrium activity of My+.
This so-called solubility product model of contaminant availability has several limitations. First, organisms represent intrinsically dynamic systems, and their reaction with the surrounding environment is typically far from equilibrium. Second, adsorption reactions and mass transfer constraints may lower the aqueous activity of M below that supported by any known phase of M. The formation of solid solutions also may lower the aqueous activity of M below that supported by any known pure phase of M. Finally, the solubility product of MxLy is expressed in terms of its dissolution into its constitutive ions. It does not account for additional side reactions that could lead to consumption of L. Such side reactions could alter the apparent relative solubilities of different M-containing solids. With the exception of the first proviso (local equilibrium) the other limitations of the solubility product model can be addressed by substitution of the appropriate equilibrium constants to account for desorption from particulate surfaces, dissolution of solid solutions, and/or incongruent dissolution reactions. What then, can one do about the assumption of local equilibrium?
The law of detailed balancing (19) indicates that to a first approximation the relative dissolution rates of a series of solids with identical specific surface areas, each containing metal M, will be inversely proportional to their solubility products, (when corrected for side reactions with the surrounding solution). Obviously, in natural systems solids of different chemical composition rarely have identical specific surface areas. Additionally, some solids may undergo surface controlled dissolution under the same conditions that promote diffusion controlled dissolution reactions for other solids containing M. Nevertheless, wide differences in mineral solubilities do produce differential dissolution rates among multiple solids, each containing the metal M. This assertion is true for virtually all aqueous solutions, including surface and ground waters and soil solutions, as well as gastrointestinal tracts. The net outcome is that the solids with greater solubilities generally can be characterized as having greater dissolution rates, resulting in greater bioavailabilities and chemical labilities of their constituent elements.
Recognition that the solubility product serves as a relative constraint on the reactivity and potential toxicity of metals and oxoanions in surficial environments provides a strategy for environmental treatment. Often, it is not possible to remove toxic elements from contaminated soils and sediments. In these cases, inducing changes in the mineralogy of a contaminant (e.g., conversion of a metal-carbonate to a metal-phosphate) may allow one to significantly lower its solubility and its corresponding ecotoxicity. The remainder of this paper will examine the feasibility of this approach with emphasis on the formation of stable Pb precipitates in contaminated soils.
Pb-Phosphates
The orthophosphate ion forms sparingly soluble solids with several toxic metals, including Cd, Zn, Pb, and several of the actinides. Much research has explored the utility of using phosphates to reduce the mobility and bioavailability of these metals in contaminated environments as well as in a number of waste forms. A full discussion of all of these metals and all of these applications is beyond the scope of this paper. For brevity we focus on the formation and bioavailability of Pb-phosphates.
The Pb-phosphates are some of the most insoluble Pb(II)-solids known to form under surficial geochemical conditions (Table 1). At standard state, the Pb-phosphates are at least 44 orders of magnitude less soluble than galena (PbS), anglesite (PbSO4), cerussite (PbCO3), litharge (PbO), and crocoite (PbCrO4), Pb-solids common to soils contaminated by mining and smelting activities and by paint (20, 21). Nriagu (22–24), Santillan-Medrano and Jurinak (25), and Sauvé et al. (26) suggest that Pb-phosphate phases may control the solubility of Pb in noncalcareous soils. Indeed, in oxidized, noncalcareous environments, Pb-phosphates should form at the expense of other Pb solids if sufficient P is present. Natural Pb-phosphate minerals have been identified in soils impacted by the weathering of Pb ores (17, 20, 27, 28) as well as in roadside soils presumably contaminated by automobile emissions, and in urban soils (29). Often these natural Pb-phosphates are present as Pb-Ca solid solutions (17, 27–29) as predicted by Nriagu (24). However, essentially pure Pb-phosphates also have been identified in contaminated soils (20).
Table 1.
Solubility products of selected Pb minerals
| Mineral | Formula | Log Ksp* |
|---|---|---|
| Litharge | PbO | 12.9 |
| Anglesite | PbSO4 | −7.7 |
| Cerussite | PbCO3 | −12.8 |
| Pyromorphite | Pb5(PO4)3Cl | −84.4 |
| Hydroxypyromorphite | Pb5(PO4)3OH | −76.8 |
| Fluoropyromorphite | Pb5(PO4)3F | −71.6 |
| Bromopyromorphite | Pb5(PO4)3Br | −78.1 |
| Corkite | PbFe3(PO4)(SO4)(OH)6 | −112.6 |
| Hindsalite | PbAl3(PO4)(SO4)(OH)6 | −99.1 |
| Plumbogummite | PbAl3(PO4)2(OH)5⋅H2O | −99.3 |
*Data from ref. 20 and references cited therein.
In light of their intrinsically low solubilities and their natural occurrence in some contaminated soils, effort has been given to inducing the formation of Pb-phosphates in Pb-contaminated soils and soil materials through the addition of P. The treatment of Pb-contaminated soils with additions of highly soluble forms of orthophosphate as (Na2HPO4 or KH2PO4) can reduce the bioavailability of Pb as assessed by an in vitro gastrointestinal assay (30, 31) as well as induce the formation of Pb-phosphate particles (32). Unfortunately, treatment with highly soluble P increases the risk of offsite P migration (29, 33) and eutrophication of surrounding surface waters. An alternate approach is to use a lower solubility source of P such as apatite. These Ca-phosphates are prevalent as accessory minerals in igneous rocks and as low-temperature precipitates in soils and sedimentary environments.
Apatite Chemistry
The hexagonal (P63/m) crystals of the apatite group [Ca5(PO4)3X] comprise three dominant end-members, where X = OH in hydroxylapatite, F in fluorapatite, and Cl in chlorapatite. The solubility product constants of the most common hydroxyl and fluoro end members are 10−3.1 and 10−25, respectively. Apatites of geologic origin are dominated by the fluorapatites, exhibiting inhomogeneous solid solution with Cl, OH, and CO3 (11, 34, 35). The Ca in apatites resides in two distinct crystal sites (Fig. 1). The Ca(1) site is coordinated to nine oxygens. The Ca(2) site is comprised of CaO5X octahedron. In fluor- and hydroxylapatites an additional weak bond to O exists (0.15 valence units), resulting in CaO5X(O) polyhedra (34).
Figure 1.

Projection of the hydroxylapatite structure down the c axis, as well as the two cation sites in hydroxylapatite. Structural data from ref. 52. Atom sizes are not to scale.
Natural apatites exhibit extensive substitution with the incorporation of K, Na, Mn, Ni, Cu, Co, Zn, Sr, Ba, Cd, Sn, Y, and rare earth elements in the Ca sites (refs. 11, 34, and 36, and references therein). Substitution occurs at both Ca sites, depending on the ionic radius of the ion in question.
Reaction of Dissolved Pb with Apatites
Apatites have been examined extensively for use in the removal of toxic metal ions from wastewater and aquatic solutions (33, 37–51). Mechanisms of metal uptake vary with identity of the sorbate, the sorbent, and the solution conditions. From changes in solution composition, powder x-ray diffraction (XRD) patterns, and scanning electron micrographs, Ma et al. (33) described the reaction of dissolved Pb2+ with hydroxylapatite by the sequential dissolution and precipitation reactions:
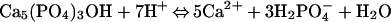 |
5 |
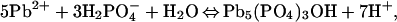 |
6 |
where Pb5(PO4)3OH is the mineral hydroxypyromorphite.
The overall reaction:
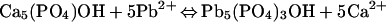 |
7 |
is exergonic with a standard state Gibbs energy change of −137.08 kJ⋅mol−1 (51). The pyromorphite group also includes the mineral pyromorphite [Pb5(PO4)3Cl], fluoropyromorphite [Pb5(PO4)3F], bromopyromorphite [Pb5(PO4)3Br], and various arsenate and vanadate analogs. The solubility of the pyromorphites decreases fluoropyromorphite > hydroxypyromorphite > bromopyromorphite > pyromorphite, with log Ksp of −72, −77, −78, and −84, respectively. The pyromorphites are isostructural with the apatites.
Discrete products of Pb reactions with hydroxylapatite are detectable by powder XRD and by scanning electron microscopy (SEM) in model aqueous systems when the initial aqueous Pb concentrations are >5 mg⋅liter −1 and the initial pH ranges from 3 to 7 (33). Under these conditions, discrete Pb-bearing solids (identified by XRD as hydroxypyromorphite) form in less than 10 min. They have different crystal habits than the original hydroxlyapatites and do not contain Ca (within the detection limits of energy dispersive x-ray analysis). Apparently they form according to the sequential reactions described in Eqs. 5 and 6 and not from ion substitution of Pb for the Ca in the apatite particles (33). Fourier transform IR spectroscopy and x-ray absorption fine structure spectroscopy indicate that the reaction of <1 mg Pb⋅liter−1 with hydroxylapatite still results in the formation of hydroxypyromorphite (52).
Ex situ and in situ atomic force microscopy (AFM) studies of Pb-reacted hydroxylapatite indicate that when initial conditions are far from equilibrium (>1 mg Pb⋅liter−1, pH = 6), pyromorphite can nucleate homogeneously as a result of interactions between dissolved Pb and phosphate (50, 51). In situ, AFM measurements of Pb solutions reacting with hydroxylapatite particles, showed only “clean” apatite surfaces without coatings of pyromorphite crystals (51). Nevertheless, pyromorphite crystals were found in the outflow from the AFM liquid cell, suggesting that homogeneous nucleation had occurred. Additionally, the presence of pyromorphite needles atop the AFM cantilever (Fig. 2) was consistent with precipitation of the Pb-phosphates in solution and not on the surfaces of the hydroxylapatite (51).
Figure 2.

Pyromorphite crystals formed from the reaction of dissolved Pb with hydroxylapatite. (A) An ex situ tapping mode AFM image of effluent from the AFM fluid cell. Scan size = 661 nm on a side. (B) SEM image of pyromorphite crystals deposited atop the AFM cantilever after reaction of dissolved Pb with hydroxylapatite in an AFM liquid cell. [Reproduced with permission from ref. 51 (Copyright 1998, Elsevier Science).]
The initial composition of the solution phase influences the interactions of dissolved Pb with apatites. Ma et al. (42) examined the effects of NO3, Cl, F, SO4, and CO3 on the immobilization of aqueous Pb by hydroxylapatite. Pb concentrations were reduced from an initial 5–100 mg⋅liter−1 to <15 μg⋅liter−1 (the Environmental Protection Agency’s drinking water limit for Pb) except at very high concentrations of CO3. Hydroxylapatite was transformed to hydroxypyromorphite in the presence of NO3, SO4, and CO3, to pyromorphite after reaction with PbCl2, and to fluoropyromorphite after reaction with PbF2. These reaction products were identified by XRD and SEM.
Ma et al. (53) explored the interactions of dissolved Pb with natural fluorapatites and carbonated fluorapatites. These solids varied in their capacity to remove aqueous Pb (from 39% to 100%). The fraction of Pb removed was not related to the initial surface areas of the apatite particles, but rather to their dissolution rates. Fluoropyromorphite and hydrocerussite were the principal Pb phases formed in these experiments.
The exact composition of the products formed by the reaction of aqueous Pb with apatite depends on the solution pH. In the regime of pH 3.1–6.2, Chen et al. (49) found that mixtures of dissolved Pb and carbonate-containing fluorapatite reacted to form fluoropyromorphite by the coupled reactions:
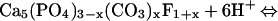 |
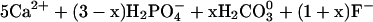 |
8 |
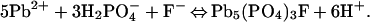 |
9 |
At pH 6.6–6.8, a mixed hydroxylated fluorapatite forms.
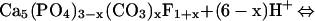 |
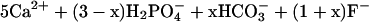 |
10 |
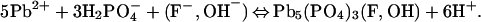 |
11 |
At circumneutral pH, hydrocerussite and carbonated hydroxyl fluoropyromorphite form by the reactions:
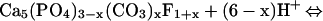 |
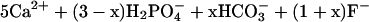 |
12 |
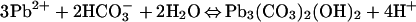 |
13 |
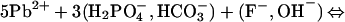 |
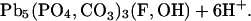 |
14 |
Finally, at pH 10.7–11.9, the reaction products consist of hydrocerrusite, hydroxypyromorphite, and lead oxide fluoride (49). Formation of the lead oxide fluoride is described as:
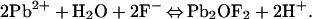 |
15 |
Competition with other metal ions also influences the reactions of aqueous Pb with apatites. At initial solution concentrations of <20 mg⋅liter−1, dissolved Zn, Cd, Ni, Cu, Fe(II), and Al have no discernible effect on the immobilization of 20 mg Pb⋅liter−1, by hydroxylapatite. Additionally, significant quantities of these metals also are removed from solution (43). Cadmium and Zn also have been shown to have no influence on influence the uptake of Pb by carbonated fluorapatite (49). In these latter experiments, the initial concentrations of dissolved Cd, Zn, and Pb all were equimolar.
When the concentration of competing metals far exceeds that of Pb, (M/Pb = 7:1, where M = Zn, Cd, Ni, Cu, Fe(II), or Al and dissolved Pb = 20 mg⋅liter−1) the interactions of Pb with hydroxylapatite can be significantly inhibited (43). Ma et al. (43) reported dissolved Cu was the most effective in inhibiting Pb immobilization by hydroxylapatite, followed by Fe(II), Cd, Zn, Al, and Ni. In all cases, hydroxypyromorphite was the only reaction product detected by XRD besides hydroxylapatite. The intensities of hydroxypyromorphite XRD peaks decreased with increased concentrations of competing metals. Inhibition of hydroxypyromorphite formation was positively correlated with the solubility of known M-phosphates; however, no metal-phosphates other than hydroxylapatite could be detected with XRD or SEM (43). Additionally, geochemical calculations indicated that the solutions were all undersaturated with respect to known Zn-, Cd-, Ni-, Cu-, Fe(II)-, or Al-phosphate phases. The nature of this inhibition is not known, but it possible that these metal ions passivated the surfaces of the apatite, through the formation of sorbed, or surface-precipitated species.
Reaction of Apatites with Sorbed and Solid-Phase Pb
The pyromorphites are much less soluble than the other Pb-solids commonly present in terrestrial and aquatic environments (Table 1). Thus, it is expected that if sufficent soluble P is present, pyromophites will form at the expense of the “native” Pb-solids (including both adsorbed and precipitated Pb). Ma et al. (33) observed XRD-detectable hydroxypyromorphites after the reaction of hydroxylapatite with Pb-saturated cation exchange resins with hydroxylapatite (note, the initial concentration of dissolved Pb was <1 mg⋅liter−1). SEM showed that the spherical particles of the exchange resin were coated with hexagonal hydroxypyromorphite needles after they were reacted with apatite. Precipitates were not detectable on the surfaces of the reacted apatite particles. Zhang et al. (54) observed the formation of pyromorphite when hydroxylapatite particles were reacted with goethite suspensions containing sorbed Pb. In these experiments, the Pb-treated goethites and the apatite particles were separated by dialysis membranes. Hexagonal pyromorphite crystals formed on the inside surfaces of the dialysis membranes, indicating that nucleation on the apatite surface is not required for pyromorphite precipitation as was also suggested by Lower and coworkers (50, 51).
Consistent with thermodynamic predictions hydroxypyromorphite forms at the expense of more soluble Pb-minerals when they are exposed to apatites. Laperche et al. (21) found XRD- and SEM-detectable hydroxypyromorphite when pure systems of PbO and cerussite each were reacted with hydroxylapatite at pH 5, 6, and 7. Decreased pH caused more rapid reaction rates with greater loss of the parent phases and increased formation of hydroxypyromorphite (Fig. 3). Apparently dissolution of the apatite and/or the original Pb-phases was rate limiting.
Figure 3.

SEM images of the reaction products of hydroxylapatite and solid-phase forms of Pb (cerussite) at pH 5 (A) and pH 7 (B). [Reproduced with permission from ref. 21 (Copyright 1996, American Chemical Society).]
In all of these cases, initial precipitation of pyromorphite or hydroxypyromorphite quickly reduced the concentration of dissolved Pb. Growth of the Pb-phosphate crystals required continued dissolution of the apatite particles, desorption of adsorbed Pb from the surfaces of the exchange resin and the goethite, and dissolution of the PbO and cerussite phases. The driving force for these dissolution and desorption reactions was formation of the Pb-phosphates.
Reactions of Apatites with Pb-Contaminated Soils
The interactions of apatites with soil Pb are similar to those observed in model solutions and laboratory mixtures of pure solids. When added to a soil slurry from an automobile battery cracking facility, hydroxylapatite caused a 99% reduction in dissolved Pb (from 3,370 to 36 μg⋅liter−1) over a 5-h period (33). Similarly, amendment of Pb-contaminated soils with natural fluorapatites reduced the leachability of Pb in soil columns (53) and decreased its extractability by neutral salts (MgCl2) and weak acids (Na-acetate) (55, 56).
Laperche et al. (21) used direct physical methods to show apatite-induced formation of pyromorphites in soil slurries. When synthetic hydroxylapatite was added to a soil contaminated by paint residues, these investigators observed decreases in the intensity of the XRD peaks associated with the “native” Pb (cerussite) and appearance of peaks attributed to pyromorphite. The rate and magnitude of changes in the XRD peaks was greater at pH 5 than 7, presumably because of the more rapid dissolution of the cerussite and the hydroxylapatite.
Effects of Apatites on the Bioavailability of Pb in Contaminated Soils
The conversion of soil or sedimentary Pb from highly reactive, chemically labile forms to less reactive solids should result in concomitant decreases in bioavailability. Chlopeka and Adriano (57) evaluated this hypothesis by adding natural apatite from North Carolina to a soil amended with varying amounts of Pb-containing flue dust. These materials then were used as potting mixes in glass-house experiments that produced a single crop of maize (Zea mays, var. Pioneer 3165) followed by single crop of barley (Hordeum vulgare, var. Boone). Apatite amendment resulted in decreases in extractable Pb from the soil as well as decreases in tissue Pb concentrations in both crops.
Laperche et al. (58) investigated the use of apatite minerals to induce in situ formation of Pb-phosphates in contaminated soil and determined the impact of apatites on Pb uptake by plants. Subsamples of a Pb-contaminated soil (containing 37,026 mg Pb⋅kg−1 soil from paint residues) were mixed with sufficient quantities of either synthetic hydroxylapatite or natural fluorapatite to convert 33%, 66%, 100%, and 150% of the native soil Pb to either pyromorphite or hydroxypyromorphite. These materials then were planted with sudax grass (Sorghum bicolor L. Moench) a hybrid of sorgum (Sorghum vulgare L. Moench) and sudan grass (Sorghum vulgare var. sudanese). In all cases, hydroxylapatite amendments decreased the concentrations of Pb in the above-ground biomass (shoots) (by 92–98%) relative to the unamended soil. Fluorapatite-induced reductions in the concentration of Pb in the shoots ranged from 87% to 96%. The effect of apatite amendments on the Pb concentration in the roots was much different. For both apatites, the lowest concentrations of Pb within the root tissue were associated with the smallest levels of apatite amendments. Increased levels of apatite addition corresponded to increases in the quantity of Pb associated with the roots. At the greatest levels of apatite amendment, the root Pb actually exceeded that found in the unamended soil.
Examination of the root surfaces with SEM, energy dispersive x-ray analysis, and XRD analysis of root-associated particles indicated the presence of Ca-substituted pyromorphites on those plants grown in the apatite-treated soils (Fig. 4). Similar particles were not found on the root surfaces of sudax grown in the unamended soils; nor could similar particles be found in the bulk soil after reaction with apatite. Apparently, addition of apatite to the contaminated soil resulted in precipitation of pyromorphite particles on the exterior of the root surfaces. Local acidity within the rhizosphere may have enhanced the local dissolution of apatite grains, facilitating pyromorphite precipitation. Cotter-Howells and Caporn (32) also observed the precipitation of Ca-substituted pyromorphites on plant roots (Agrostis capillaris) grown on Pb-contaminated soils. In this case pyromorphite precipitation may have resulted from root-exudate phosphatase, causing increased local concentrations of phosphate in the rhizosphere. In any event, the formation of pyromorphite likely decreased the bioavailability of Pb.
Figure 4.

SEM micrograph of sudax root grown in Pb-contaminated soil mixed with hydroxylapatite. Note the pyromorphite crystals on the root.
Soils, surface sediments, and surficial aquatic environments are open, dynamic systems best characterized as mixtures of meta-stable solids. It is safe to say that these systems never attain absolute thermodynamic equilibrium, and one can expect to find multiple forms of a given element present. Thus, the bioavailability and chemical lability of toxic elements in these systems are transient properties that are controlled by the reaction dynamics and the total quantity of the most reactive forms of these elements present. Ideally, treatment technologies would facilitate the complete conversion of a toxic element (e.g., Pb) from all pre-existing forms to the most geochemically stable phase; but as we know, kinetic constraints will prevent this from happening. Fortunately, decreases in the chemical reactivity of and bioavailability of a given element (e.g., Pb) can be accomplished by elimination of the most reactive forms (58).
Conclusions
Human activity on this planet has altered the distribution and form of various elements in the periodic table. Often these activities have converted many potentially toxic metals from nonreactive, geochemically stable solids into forms that are more soluble and bioavailable, increasing their effective toxicity. Fortunately, it is often possible to reverse this process, transforming reactive forms of toxic metals to less labile species through appropriate precipitation reactions. Although such an approach does not remove the element in question from the biosphere, it can significantly reduce its bioavailability. In many instances, this approach may be a practical alternative to more invasive methods of environmental restoration (e.g., excavation and removal of contaminated materials). In the event that more extensive treatment is chosen, geochemical stabilization may still be desirable as a rapid response treatment or to increase the chemical stability of excavated materials in landfill environments.
This approach is not limited to Pb, nor is it only possible to induce precipitation of phosphates. Extensive research has been conducted on various treatment technologies designed to remove toxic ions from contaminated waters or to stabilize these elements in soil waste materials or contaminated soils and sediments (5). Many of these efforts involve the formation of geochemically stable solids through precipitation and/or adsorption reactions. In essence, these methods attempt to close the circle, converting labile forms of toxic elements into less reactive solids more consistent with long-term geochemical equilibrium.
ABBREVIATIONS
- XRD
x-ray diffraction
- SEM
scanning electron microscopy
- AFM
atomic force microscopy
References
- 1.Krauskopf K B, Bird D K. Introduction to Geochemistry. New York: McGraw-Hill; 1995. [Google Scholar]
- 2.Hayes K F, Traina S J. In: Soil Chemistry and Ecosystem Health. Huang P M, editor. Madison, WI: Soil Sci. Soc. Am.; 1998. pp. 45–84. [Google Scholar]
- 3.Newman M C, McIntosh A W. Metal Ecotoxicology Concepts and Application. Chelsea, MI: Lewis; 1991. [Google Scholar]
- 4.Adriano D C. Biogeochemistry of Trace Metal. Chelsea, MI: Lewis; 1992. [Google Scholar]
- 5.Iskandar I K, Selim H M. Engineering Aspects of Metal-Waste Management. Chelsea, MI: Lewis; 1992. [Google Scholar]
- 6.Hamelink J L, Landrum P F, Bergman H L, Benson W H. Bioavailability Physical, Chemical, and Biological Interactions. Chelsea, MI: Lewis; 1994. [Google Scholar]
- 7.Ernst W H O. Appl Geochem. 1996;11:163–167. [Google Scholar]
- 8.Davis A, Sellstone C, Clough S, Barrick R, Yare B. Appl Geochem. 1996;11:409–423. [Google Scholar]
- 9.Davies N A, Taylor M G, Simkiss K. Environ Pollut. 1997;96:179–184. doi: 10.1016/s0269-7491(97)00026-2. [DOI] [PubMed] [Google Scholar]
- 10.Peijnenburg W J G M, Posthuma L, Eijsackers H J P, Allen H E. Ecotox Environ Saf. 1997;37:163–172. doi: 10.1006/eesa.1997.1539. [DOI] [PubMed] [Google Scholar]
- 11.Lindsay W L, Vlek P L G, Chien S H. In: Minerals in Soil Environments. 2nd Ed. Dixon J B, Weed S B, editors. Madison, WI: Soil Sci. Soc. Am.; 1989. pp. 1089–1130. [Google Scholar]
- 12.Troeh F R, Thompson L M. Soils and Soil Fertility. New York: Oxford Univ. Press; 1993. [Google Scholar]
- 13.Tisdale S L, Nelson W L, Beaton J D, Havlin J L. Soil Fertility and Fertilizers. New York: Macmillan; 1993. [Google Scholar]
- 14.Lindsay W L. Chemical Equilibria in Soils. New York: Wiley; 1979. pp. 162–209. [Google Scholar]
- 15.Laws E A. Aquatic Pollution: An Introductory Text. New York: Wiley; 1993. [Google Scholar]
- 16.Lee G F, Jones R A, Rast W. In: Phosphorous Management Strategies for Lakes. Loehr R C, Martin C S, Rast W, editors. Ann Arbor, MI: Ann Arbor Science; 1980. pp. 259–308. [Google Scholar]
- 17.Davis A, Drexker J W, Ruby M V, Nicholson A. Environ Sci Technol. 1993;27:1415–1425. [Google Scholar]
- 18.Davis A, Ruby M V, Bloom M, Schoof R, Freeman G, Bergstrom P D. Environ Sci Technol. 1996;30:392–399. [Google Scholar]
- 19.Lasaga A C. Kinetic Theory in the Earth Sciences. Princeton: Princeton Univ. Press; 1998. [Google Scholar]
- 20.Ruby M V, Davis A, Nicholson A. Environ Sci Technol. 1994;28:646–654. doi: 10.1021/es00053a018. [DOI] [PubMed] [Google Scholar]
- 21.Laperche V, Traina S J, Gaddam P G, Logan T J. Environ Sci Technol. 1996;30:3321–3326. [Google Scholar]
- 22.Nriagu J O. Inorg Chem. 1972;11:2499–2503. [Google Scholar]
- 23.Nriagu J O. Geochem Cosmochim Acta. 1973;37:367–377. [Google Scholar]
- 24.Nriagu J O. Geochem Cosmochim Acta. 1974;37:367–377. [Google Scholar]
- 25.Santillan-Medrano J, Jurinak J J. Soil Sci Soc Am Proc. 1975;39:851–858. [Google Scholar]
- 26.Sauvé S, McBride M B, Hendershot W H. Environ Pollut. 1997;2:149–155. doi: 10.1016/s0269-7491(96)00081-4. [DOI] [PubMed] [Google Scholar]
- 27.Cotter-Howells J D, Thornton I. Environ Geochem Health. 1991;12:127–135. doi: 10.1007/BF01734304. [DOI] [PubMed] [Google Scholar]
- 28.Cotter-Howells J D, Champness P E, Charnock J M, Patrrick R A D. J Soil Sci. 1994;45:393–402. [Google Scholar]
- 29.Cotter-Howells J D. Environ Pollut. 1996;1:9–16. doi: 10.1016/0269-7491(96)00020-6. [DOI] [PubMed] [Google Scholar]
- 30.Rabinowitz M B. Bull Environ Contam Toxicol. 1993;51:438–444. doi: 10.1007/BF00201764. [DOI] [PubMed] [Google Scholar]
- 31.Berti W R, Cunningham S C. Environ Sci Technol. 1997;31:1359–1364. [Google Scholar]
- 32.Cotter-Howells J, Caporn S. Appl Geochem. 1996;11:335–342. [Google Scholar]
- 33.Ma Q Y, Traina S J, Logan T J, Ryan J A. Environ Sci Technol. 1993;27:1803–1810. [Google Scholar]
- 34.Hughes J M, Cameron M, Crowley K D. Am Mineral. 1989;74:870–876. [Google Scholar]
- 35.Santos R V, Clayton R N. Am Mineral. 1995;80:336–344. [Google Scholar]
- 36.Rakovan J, Reeder R J. Am Mineral. 1994;79:892–903. [Google Scholar]
- 37.Suzuki T, Hatsushika T, Hayakawa Y. J Chem Faraday Trans 1. 1981;77:1059–1062. [Google Scholar]
- 38.Suzuki T, Hatsushika T, Miyake M. J Chem Soc Faraday Trans 1. 1982;78:3605–3611. [Google Scholar]
- 39.Suzuki T, Ishigaki K, Miyake M. J Chem Soc Faraday Trans 1. 1984;80:3157–3165. [Google Scholar]
- 40.Takeuchi Y, Suzuki T, Arai H J. J Chem Eng Jpn. 1988;21:98–100. [Google Scholar]
- 41.Takeuchi, Arai H J. J Chem Eng Jpn. 1990;23:75–80. [Google Scholar]
- 42.Ma Q Y, Logan T J, Traina S J. Environ Sci Technol. 1994;28:408–419. doi: 10.1021/es00052a011. [DOI] [PubMed] [Google Scholar]
- 43.Ma Q Y, Traina S J, Logan T J. Environ Sci Technol. 1994;28:1219–1228. doi: 10.1021/es00056a007. [DOI] [PubMed] [Google Scholar]
- 44.Xu Y, Schwartz F W. J Contam Hydrol. 1994;15:187–206. [Google Scholar]
- 45.Xu Y, Schwartz F W, Traina S J. Environ Sci Technol. 1994;28:1472–1480. doi: 10.1021/es00057a015. [DOI] [PubMed] [Google Scholar]
- 46.Jeanjean J, Rouchard J C, Tran L, Fedoroff M. J Radioanal Nucl Chem Lett. 1995;201:529–539. [Google Scholar]
- 47.Reichert J, Binner J G P. J Mater Sci. 1996;31:1231–1241. [Google Scholar]
- 48.Chen X, Wright J V, Conca J L, Peurrung L M. Water Air Soil Pollut. 1997;98:57–78. [Google Scholar]
- 49.Chen X, Wright J V, Conca J L, Peurrung L M. Environ Sci Technol. 1997;98:624–631. [Google Scholar]
- 50.Lower S K, Maurice P A, Traina S J, Carlson E H. Am Mineral. 1998;83:147–158. [Google Scholar]
- 51.Lower S K, Maurice P A, Traina S J. Geochim Cosmochim Acta. 1998;62:1773–1780. [Google Scholar]
- 52.Laperche V, Traina S J. In: Adsorption of Metals by Geomedia. Jenne E A, editor. New York: Academic; 1998. pp. 255–276. [Google Scholar]
- 53.Ma Q Y, Logan T J, Traina S J. Environ Sci Technol. 1995;29:1118–1126. doi: 10.1021/es00004a034. [DOI] [PubMed] [Google Scholar]
- 54.Zhang P, Ryan J A, Bryndzia L T. Environ Sci Technol. 1997;31:2673–2678. [Google Scholar]
- 55.Ma L Q, Rao G N. J Environ Qual. 1997;26:788–794. [Google Scholar]
- 56.Ma L Q, Choate A L, Rao G N. J Environ Qual. 1997;26:801–807. [Google Scholar]
- 57.Chlopeka A, Adriano D C. Sci Total Environ. 1997;207:195–206. doi: 10.1016/s0048-9697(97)00268-4. [DOI] [PubMed] [Google Scholar]
- 58.Laperche V, Logan T J, Gaddam P, Traina S J. Environ Sci Technol. 1997;31:2745–2753. [Google Scholar]