

Summary
Over the recent years there has been increased interest in applying NMR spectroscopy for to the characterization of proteins and protein complexes of large molecular weight. The combination of multi-dimensional NMR, novel pulse sequences allowing for the selection of slowly relaxing coherence pathways and the development of a range of labeling techniques have enabled high-resolution NMR analyses of supramolecular systems of even mega-dalton size. Here we describe how NMR can be used to obtain structural information in large systems by using as an example the recent structure determination of the SecA ATPase (204 kDa) in complex with a signal peptide.
1. Introduction
Application of NMR spectroscopy to supramolecular systems (>200 kDa) has been revolutionized by specific labeling of methyl groups (1). The labeling protocol, pioneered by the group of Lewis Kay, is very simple and robust (2, 3). The approach exploits some very favorable properties of methyl groups in proteins: (i) they occur frequently in the hydrophobic cores of proteins, and at the interfaces of biomolecular complexes, and are thus excellent reporters of structure and dynamics; (ii) the three protons of the methyl group all contribute to the intensity of the same signal, and therefore methyl probes are significantly more sensitive than other candidates; (iii) methyl groups are intrinsically optimized for use in TROSY spectroscopy and the simple 1H-13C HMQC experiment can be used to select for pathways with the favorable relaxation properties (4). Currently, the methyl groups of five different amino acids can be labeled in a highly specific and scramble-free manner: Ala, Ile (δ1), Leu, Met, and Val. These five residues are highly abundant, typically accounting for 35–45% of the total number of residues in a protein, they and are distributed throughout the protein and thus provide providing almost complete coverage of the protein space.
The methyl-labeling approach combined with methyl-TROSY currently provides the method of choice for the NMR characterization of large protein systems. Although this approach has proven to be very robust for recording spectra of large proteins with high sensitivity and resolution, a major hurdle in obtaining site-specific information remains the difficulty in obtaining assignments. While the traditional approach of assigning the backbone and subsequently linking the methyl side chains to the backbone has worked efficiently for smaller proteins, it is not applicable to larger systems. The only approach currently is to “disassemble” the supramolecular system. For higher-order oligomeric systems, such as the proteasome (5), this means preparing the subunit in its monomeric form and for large single-chain proteins, such as the SecA (6), preparing isolated domains or fragments.
In principle, determining solution structures of supramolecular protein-ligand complexes by NMR should be feasible, provided that the crystal structures of the free partners are previously known. Because usually only methyl groups can be robustly unambiguously detected for supramolecular systems, in case where complex interactions are mediated by hydrophobic contacts involving methyl-bearing residues, it is likely that intermolecular NOEs can be detected thereby enabling the reliable docking of the complex. Unfavorable motions commonly observed at protein interfaces, however, may result in line broadening and render NOE detection unfeasible. Although the NOE has served as the gold standard for protein structure determination by NMR, the old, but recently resurrected, paramagnetic relaxation enhancement (PRE) (7, 8) technique holds great promise for obtaining both structural and dynamic information in supramolecular protein complexes (6). By combining transferred NOESY, line broadening and PRE experiments the structure of the 204 kDa SecA ATPase in complex with a secretory signal peptide was recently determined (6). Using this system as an example we describe strategies to (i) obtain samples optimally labeled for methyl detection, (ii) assign the methyl resonances of the large protein system and (iii) obtain inter-molecular distance restraints for the structure determination of large protein-ligand complexes.
2. Materials
Frozen, transformed E. coli BL21(DE3) cells to overexpress protein of interest.
M9 medium: 6g/L Na2HPO4, 3g/L KH2PO4, pH 7.0–7.4., 0.5g/L NaCl, 1.0g 15NH4Cl, Autoclave, let the medium cool down and then add 0.1 ml/L of CaCl2 (from 1 M stock ) and 1 mL/L of MgSO4 from 1 M stock) . Stock solutions should be filter-sterilized.
1 M CaCl2 stock: Dissolve 11.0 g of CaCl2 in 100 mL of D2O.
1 M MgSO4 stock: Dissolve 12.04 g of MgSO4 in 100 mL of D2O.
D2O (Cambridge Isotope Laboratories, CIL)
D-[2H,13C]-glucose (CIL)
D-[2H,12C]-glucose (CIL)
15NH4Cl
BIOEXPRESS (CIL)
ISOGRO (Isotec)
13CH3-2H-alanine
α–ketobutyrate (CIL or Isotec)
α–ketoisovalerate (CIL or Isotec)
13CH3-Methionine (CIL)
IPTG
AMICON stir cell (Millipore)
L-broth
(2,2,5,5-tetramethyl-2,5-dihydro-1H-pyrrol-3-yl)methyl methanesulfonothioate (MTSL) (Toronto Research Chemicals Inc.)
3. Methods
3.1 Protein labeling for methyl detection
Pick a freshly transformed colony (see Note 1) of BL21(DE3) cells and inoculate a 1–2 ml culture of L-broth in D2O containing 0.1% of glucose, at 37 °C until cells reach an OD600 of ~0.7–0.8.
Centrifuge the cells at 1,200 ·xg) for 20 min at room temperature and resuspend them in 5–10 ml of sterile M9 medium in D2O (M9/D2O) in a sterile flask to a starting OD600 of ~0.05. Incubate the culture in a shaking incubator (220–250 rpm) at 37 °C until it reaches an OD600 of ~0.6.
Centrifuge, as in step 2, and resuspend the cells in 50–100 mL of M9/D2O (prepared with either D-[2H,13C]glucose (see Note 2) or D-[2H, 12C]glucose (see Note 3) and 15NH4Cl) containing 2% of BIOEXPRESS (CIL) or ISOGRO (Sigma) (see Note 4). The starting OD600 should be ~0.1. Incubate the culture in a shaking incubator (220–250 rpm) at 37 °C until the OD600 is ~0.6.
Centrifuge, as in step 2, and resuspend the cells in 1 L of M9/D2O and grow until the OD600 is ~0.25.
- Add reagents for methyl labeling:
- At this point, amino acid precursors for methyl labeling can be added.
- a. Add precursors (see Note 5) or amino acids 30–60 mins prior to IPTG induction; add the following quantities (final concentration): 100 mg/L of 13CH3-2Hα-alanine (see Note 6) for Ala labeling; 45–50 mg/L of α–ketobutyrate for isoleucine labeling; 85–100 mg /L of α–ketoisovalerate for leucine and valine labeling (see Note 7); 250 mg /L of [13CH3]-methionine for Met labeling. The methyl groups of all five residues can be labeled in one sample in a scramble-free manner (see Note 8) (Figure 1a).
- b. Continue incubating the culture for approximately 1 h. The OD600 should reach a value of ~0.3–0.4
Add IPTG to 0.5 mM to induce protein over-expression.
Continue post-induction growth for 6–8 h (see Note 9).
Harvest the cells by centrifugation at 5,000 ×g for 15 minutes at 4 °C.
Freeze the wet cell pack at −80 °C
Figure 1.
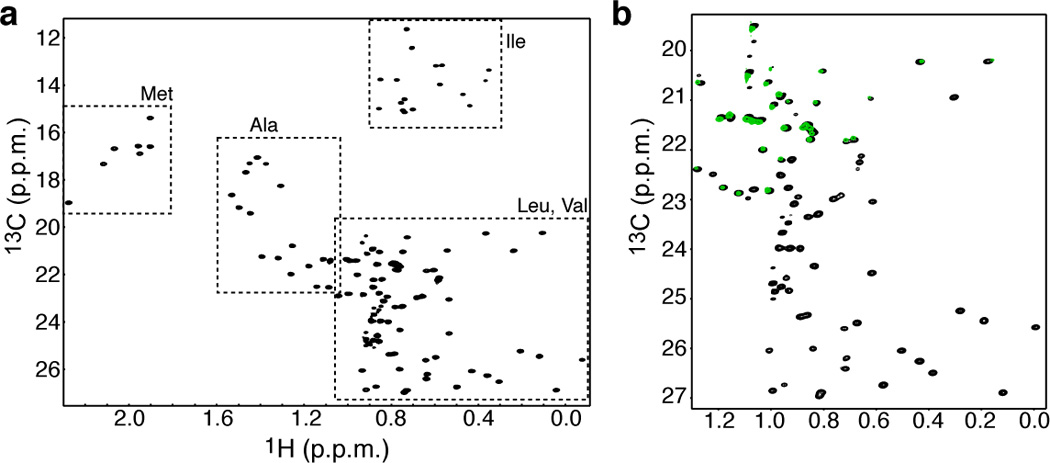
(a) 1H-13C HMQC of [U-2H,12C], Ala-, Leu-, Met-, Val-, Ile-δ1-[13CH3] labeled protein. The methyl groups of all five amino acids can be labeled with no scrambling. (b) 1H-13C HMQC of the same protein as in (a, black) but prepared using 10% BIOEXPRESS (green). The Leu methyl groups are completely suppressed whereas the Val methyl groups are only minimally affected.
3.2. NMR assignment
To assign a large protein such as SecA (204 kDa; 901 residues per subunit) a domain-parsing strategy is followed.
Isolate and characterize virtually all domains of the full-length protein and a number of fragments comprising contiguous domains by NMR (see Note 10). The size of the isolated domains and fragments should be such that backbone and side chain assignment is feasible using standard approaches.
Prepare Ala, Ile, Met, Leu and Val methyl labeled samples for the full length protein and the domains thereof (see Note 11). Record methyl-TROSY for all of the samples, overlay and compare the spectra of the individual domains against the spectra of the longer fragments and full length protein. If good resonance correspondence between domains, fragments and the full length protein can be demonstrated then assignment is in principle transferrable.
Record standard triple resonance NMR experiments for isolated domains and obtain backbone assignments. Record the three-dimensional spectra required for the assignment of the methyl groups (see Note 12).
Transfer the methyl assignments obtained for the isolated domains to the larger fragments and finally to the full length protein by visually inspecting the methyl-TROSY spectra. Only the assignment of the obvious and well dispersed resonances can be safely transferred this way.
Record 13C HMQC-NOESY-HMQC spectra (see Note 13) for the methyl-labeled samples. Use the NOE patterns to confirm and extend the assignment transfer from the domains to the full length protein. If a crystal structure is available, it can be used to determine the distances between the methyl groups and assist with the assignment.
Prepare site-directed mutations to assign ambiguous resonances and further extend and confirm the assignments (see Note 14).
3.3. Paramagnetic Relaxation Enhancement (PRE) Measurements
To prepare nitroxide spin label (MTSL)-derivatized ligand via cysteine-specific modification of engineered ligand derivatives containing single-solvent-accessible cysteine residues at sites of interest (see Note 15),add MTSL from a concentrated stock in acetonitrile to the ligand solution (free from any reducing agent) at a 10-fold molar excess over the ligand and allow the reaction to proceed at 4 °C for ~12 hrs. If available, confirm the completion of the reaction by mass spectrometry.
Remove excess MTSL by extensive dialysis using an Amicon stirred cell ).
Determine PRE-derived distances from 1H-13C HMQC spectra by measuring peak intensities before (paramagnetic) and after (diamagnetic) reduction of the nitroxide spin label with ascorbic acid (see Note 16).
Convert PRE values to distances by using a modified Solomon-Bloembergen equation for transverse relaxation (7).
Incorporate distance (inter-molecular) restraints into the structure calculation protocol of the complex.
Restrain resonances strongly affected by the presence of the spin label in the ligand (Ipara/Idia < 0.15) and whose resonances broaden beyond detection in the paramagnetic spectrum with only an upper bound distance estimated from the noise of the spectrum plus 4 Å.
Restrain resonances that appear in the paramagnetic spectra (Ipara/Idia < 0.85) as the calculated distance with ±4 Å upper/lower bounds.
3.4. Structure determination
Determine the interface between the ligand and the large protein using differential line broadening (9). The residues affected by complex formation can be used as ambiguous restraints.
-
If the ligand is a flexible peptide, use transferred-NOESY (10) to determine the structure of the peptide in the complex.
Determine the structure of the complex by using a CNS-based software such as HADDOCK (11) or Xplor-NIH (12). Use the crystal structure of the large protein to define the starting conformation, and both unambiguous and ambiguous restraints obtained from NOE, PRE, line broadening and chemical shift perturbation experiments.
Footnotes
Freshly transformed colonies give always better protein yield.
[U-2H,13C]-glucose should be used when uniform 13C labeling is desired or when all side chain carbons of the methyl-bearing residues are to be 13C labeled for magnetization transfer from methyls to the backbone. In this case the uniformly 13C-labeled ketoacid precursor must be used.
[U-2H,12C]-glucose should be used when a sample with all carbon positions 12C labeled, except the methyl carbons of interest, is desired. 1H-13C HMQC spectra of such samples are recorded without the use of the constant time version and typically provide the best resolution. Such a sample can also be used for relaxation experiments (1).
Up to ~2.5% of a rich labeling medium can be used to increase the protein yield with no effect on the specific labeling of the methyl groups.
Precursors can be purchased in protonated form and dissolved in D2O for exchange to take place: at pH 12.5 (45 °C), 2–3 h for α-ketoisovalerate, and at pH 10.5 (45 °C), 12–14 h for α-ketobutyrate; the pH values are optimized for exchange and prevent the generation of dimers through condensation of two ketoacid molecules.
13CH3-2Hα-alanine can be prepared by using the tryptophan synthase enzyme to catalyze the proton-to-deuterium exchange of the α hydrogen, as described by Matthews and coworkers (13).
Incorporation of 13CH3/12CD3 isotope labels into the isopropyl moieties of Val and Leu residues should be used for very large proteins as the inter-methyl dipolar relaxation is significantly reduced. The methyl-TROSY spectra show significant gains in resolution with practically no losses in sensitivity despite the 2-fold dilution of the NMR-active methyls in such samples. Precursors have also become available that allow any of the methyl isotopomers (13CHD2, 13CH2D and 13CH3) to be incorporated into the protein (1). The different isotopomers can be used for relaxation experiments.
In this case addition of ~2% of a rich labeling medium (e.g. BIOEXPRESS or ISOGRO) is required to suppress scrambling associated with the addition of the alanine amino acid. Interestingly, further increase of the rich labeling medium (~10%) suppress completely the methyl labeling of Leu while having a minimal effect on the methyl labeling of Val (Figure 1b). Since the methyl groups of these two residues often overlap this labeling scheme can be used to differentiate between the methyl groups of them.
Critical step: Excessively prolonged growth after induction should be avoided to prevent generation of methyl groups with undesired isotopomers.
The design of domains and fragments thereof that would retain their fold and be soluble in isolation can be quite tricky. In this respect, the availability of a crystal structure can be of tremendous help.
When the methyl residues of all five residues are labeled in the same sample of a very large protein signal may be significantly compromised due to enhanced inter-methyl relaxation. The preparation of multiple samples each containing a single amino acid labeled may be desirable in such a case.
An arsenal of pulse sequences are available for methyl assignment (14).
The highly deuterated background suppresses spin diffusion and thus the mixing time for the NOESY experiments can be set as high as 500 ms allowing for NOEs to be observed between methyl groups as far as ~8 Å.
Amino acids should be typically substituted by an isosteric amino acid to prevent significant changes in the local environment and protein packing, which could introduce significant chemical shift effects.
Non-reactive Cys residues can be judged by the Elman's test. Sites for MTSL incorporation should be selected so that they cause no or minimal effect on protein structure. This can be assessed by NMR.
PRE rates should typically be measured using several MTSL-derivatized ligands, each containing a single MTSL at a different site. Because PRE rates provide long-range distance information, in the absence of available NOE data, a large number of PREs are required to properly determine the structure of a protein-ligand complex. The complex between SecA and the signal peptide was determined using 160 PRE-derived inter-molecular restraints.
References
- 1.Ruschak AM, Kay LE. Methyl groups as probes of supra-molecular structure, dynamics and function. J. Biomol. NMR. 2010;46:75–87. doi: 10.1007/s10858-009-9376-1. [DOI] [PubMed] [Google Scholar]
- 2.Goto N, Gardner K, Mueller G, Willis R, Kay L. A robust and cost-effective method for the production of Val, Leu, Ile (delta 1) methyl-protonated 15N-, 13C-, 2H-labeled proteins. J. Biomol. NMR. 1999;13:369–374. doi: 10.1023/a:1008393201236. [DOI] [PubMed] [Google Scholar]
- 3.Tugarinov V, Kanelis V, Kay LE. Isotope labeling strategies for the study of high-molecular-weight proteins by solution NMR spectroscopy. Nat. Protoc. 2006;1:749–754. doi: 10.1038/nprot.2006.101. [DOI] [PubMed] [Google Scholar]
- 4.Tugarinov V, Hwang P, Ollerenshaw J, Kay L. Cross-correlated relaxation enhanced 1H-13C NMR spectroscopy of methyl groups in very high molecular weight proteins and protein complexes. J. Am. Chem. Soc. 2003;125:10420–10428. doi: 10.1021/ja030153x. [DOI] [PubMed] [Google Scholar]
- 5.Sprangers R, Kay LE. Quantitative dynamics and binding studies of the 20S proteasome by NMR. Nature. 2007;445:618–622. doi: 10.1038/nature05512. [DOI] [PubMed] [Google Scholar]
- 6.Gelis I, Bonvin A, Keramisanou D, Koukaki M, Gouridis G, Karamanou S, Economou A, Kalodimos CG. Structural basis for signal-sequence recognition by the translocase motor SecA as determined by NMR. Cell. 2007;131:756–769. doi: 10.1016/j.cell.2007.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Battiste J, Wagner G. Utilization of site-directed spin labeling and high-resolution heteronuclear nuclear magnetic resonance for global fold determination of large proteins with limited nuclear overhauser effect data. Biochemistry. 2000;39:5355–5365. doi: 10.1021/bi000060h. [DOI] [PubMed] [Google Scholar]
- 8.Tang C, Schwieters C, Clore G. Open-to-closed transition in apo maltose-binding protein observed by paramagnetic NMR. Nature. 2007;449:1078–1082. doi: 10.1038/nature06232. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi K, Wagner G. NMR studies of protein interactions. Curr. Opin. Struct. Biol. 2006;16:109–117. doi: 10.1016/j.sbi.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 10.Post C. Exchange-transferred NOE spectroscopy and bound ligand structure determination. Curr. Opin. Struct. Biol. 2003;13:581–588. doi: 10.1016/j.sbi.2003.09.012. [DOI] [PubMed] [Google Scholar]
- 11.de Vries SJ, van Dijk M, Bonvin AM. The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 2010;5:883–897. doi: 10.1038/nprot.2010.32. [DOI] [PubMed] [Google Scholar]
- 12.Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J. Magn. Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- 13.Isaacson R, Simpson P, Liu M, Cota E, Zhang X, Freemont P, Matthews S. A new labeling method for methyl transverse relaxation-optimized spectroscopy NMR spectra of alanine residues. J. Am. Chem. Soc. 2007;129:15428–15429. doi: 10.1021/ja0761784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tugarinov V, Kay L. Ile, Leu, and Val methyl assignments of the 723-residue malate synthase G using a new labeling strategy and novel NMR methods. J. Am. Chem. Soc. 2003;125:13868–13878. doi: 10.1021/ja030345s. [DOI] [PubMed] [Google Scholar]