

Abstract
Background
Inflammatory bowel disease (IBD) pathogenesis involves an inadequately controlled immune reaction to intestinal microbiota and CD4+ T cells, dependent upon MHC class II (MHC-II) processing and presentation by antigen presenting cells (APC), play important roles The role of professional APC (macrophages and dendritic cells (DC)) and nonprofessional APC (intestinal epithelial cells (IEC)) in microbial driven intestinal inflammation remains controversial.
Methods
We generated transgenic animals on a MHC-II−/− genetic background in which MHC-II is expressed on a) DC via the CD11c promoter (CD11cTg) or b) IEC via the fatty acid binding protein (liver) promoter (EpithTg). These mice were crossed with Rag2−/− mice to eliminate T and B cells (CD11cTg/Rag2−/− and EpithTg/Rag2−/−). Helicobacter bilis (Hb) infection and adoptive transfer (AT) of naïve CD4+ T cells were used to trigger IBD.
Results
CD11cTg/Rag2−/− mice infected with Hb+AT developed severe colitis within three weeks post AT, similar to disease in positive control Rag2−/− mice infected with Hb+AT. CD11cTg/Rag2−/− mice given AT alone or Hb alone had significantly less severe colitis. In contrast, EpithTg/Rag2−/− mice infected with Hb+AT developed mild colitis by three weeks and even after 16 weeks post adoptive transfer, had only mild lesions.
Conclusions
MHC-II expression restricted to DCs is sufficient to induce severe colitis in the presence of T cells and a microorganism such as Hb within three weeks of adoptive transfer. Expression of MHC-II solely on IEC in the presence of a microbial trigger and T cells was insufficient for triggering severe colitis.
Keywords: dendritic cells, intestinal epithelial cells, adoptive transfer induced colitis, MHC II, Helicobacter bilis
Inflammatory bowel diseases (IBD) encompass several related chronic inflammatory disorders of the gastrointestinal tract, notably ulcerative colitis (UC) and Crohn’s disease (CD).1 While CD and UC are distinct from a clinical and histopathological standpoint, important themes have emerged over the past two decades linking their pathogenesis to aberrant T cell immune responses in the intestinal mucosa. These inflammatory T cell responses arise from a complex interaction between host genetics and the diverse microbiota present within the lumen of the intestinal tract.
Data from a variety of animal models highlight the importance of CD4+ T cells in the pathogenesis of 2IBD.2 Perhaps the most commonly used example is the adoptive transfer model in which CD4+ T cells expressing ‘high’ levels of CD45RB induce significant intestinal inflammation in an immunocompromised host.3–6 Importantly, the inflammation observed is strictly dependent on the microbial flora, specifically the presence of commensal organisms such as Helicobacter species that are capable of triggering a pathogenic response in a susceptible host.7 Given the fact that CD4+ T cell responses are dependent on MHC II processing and presentation, these data underscore the importance of MHC II positive antigen presenting cells (APC) in the pathogenesis of IBD.
Dendritic cells (DCs) are the most efficient MHC II expressing APCs with regards to processing and presentation of exogenous antigens capable of stimulating CD4+ T cell responses.8 In the adoptive transfer model of T cell mediated colitis, T cell interactions with activated DCs in the mesenteric lymph nodes (MLN) prior to the onset of disease are important for disease development.9 DCs have also been shown to interact with clusters of transferred CD4+ T cells at the basal crypt epithelium10 prior to development of signs of IBD. Although DCs play a role in priming inflammatory responses, various populations of DCs are found throughout the intestinal mucosa and also play a role in regulating inflammation and immunologic responses that are dysregulated in IBD.11, 12
It is thought that other MHC-II positive populations in the gastrointestinal tract such as epithelial cells that line mucosal surfaces and interact with luminal microbes may also contribute to the inflammatory responses characteristic of IBD depending on the context of epithelial/effector cell interactions. Intestinal epithelial cells (IECs) constitutively express MHC-II molecules, can present protein antigens on both MHC class I and class II molecules, and thus have the capacity to serve as antigen presenting cells (APCs) in the gut.13 They have been shown to serve as APCs to several populations of T cells in vitro and ex vivo13–15 and upregulate CD40 in response to inflammatory cytokines16 as well as other non-classical costimulatory molecules for lymphocyte interations.17 It has not been directly investigated whether interaction of effector cells with MHC-II expressing epithelial cells can drive IBD without the presence of other antigen presenting cells, such as DCs. Indirect evidence involving DC depletion experiments show that DCs are required for naïve T cell expansion,18 which is one of the prerequisites for disease development in the adoptive transfer model of colitis.19, 20
We have developed mouse models that enabled us to determine the importance of MHC-II expression on dendritic cells or on epithelial cells in a bacterial induced adoptive transfer model of colitis. CD45RBhigh T cells were adoptively transferred into rag deficient, MHC-II deficient mice in which MHC-II was only expressed on CD11c+ cells (CD11cTg/Rag2−/−) or only on IEC via the FABPL promoter on a MHC-II−/− genetic background (EpithTg/Rag2−/−). This model system clearly demonstrated that MHC-II expressed solely on DCs was sufficient to induce robust colitis but only in the presence of a bacterial organism, Helicobacter bilis (Hb). Expression of MHC-II only on IECs was insufficient to drive severe colitis even in the presence of Hb infection.
Materials and Methods
Generation of transgenic mice
Transgenic mice in which MHC-II was expressed predominantly on DCs (CD11cTg/MHC-II−/− lines 6175, 6761) or predominantly on IECs (EpithTg/MHC-II−/− lines 5809, 6258) were generated at the University of Washington Transgenic Facility. CD11cTg lines 6175 and 6761 were generated by pronuclear microinjection of a construct where the beta chain of the heterodimeric I-A MHC class II molecule (I-Ab β) was linked to the CD11c promoter.21 The construct was injected into MHC-II-deficient oocytes from B6.129-H2-Ab1tm1Gru N12 mice,22, 23 which lack surface expression of both I-A (because of a deletion of I-Ab β) and I-E MHC-II molecules. EpithTg lines 5809 and 6258 were generated by pronuclear microinjection of two constructs comprised of the rat fatty acid binding protein (liver) [(FABP)L] promoter containing promotor nucleotides −596 to +21.24 The two constructs were co-injected into MHC-II-deficient oocytes.
Transgene expression was confirmed by PCR, FACS analysis and/or immunohistochemistry (see below for methods). The original CD11cTg lines and EpithTg lines were endemically infected with multiple Helicobacter species. Hence, CD11cTg line 6761 and both lines of EpithTg mice were rederived as Helicobacter-free by Caesarian section; subsequent PCR testing confirmed that CD11cTg line 6761 was Helicobacter-free while the EpithTg lines were still infected with a non-Hb Helicobacter spp. Helicobacter-free CD11cTg line 6761 mice, as well as EpithTg line 6258 were later backcrossed C57BL/6-Rag2tm1Cgn/J to generate CD11cTg/Rag2−/− and EpithTg/Rag2−/− mice which are deficient in MHC-II except for that expressed by the transgenes and lack rag. Prior to and during experiments, mice were tested for presence of Hb by fecal PCR using genus- and species-specific primers.25 Animals were housed in a specific pathogen free (SPF) environment in polycarbonate microisolator cages containing Bed-O-Cob (Andersons, Maumee, OH). Mice were fed irradiated Picolab Rodent Diet 20 #5053 (PMI Nutrition International, Brentwood, MO) and autoclaved, acidified water. In order to prevent cross contamination of uninfected mice and Hb infected mice, animals were housed in separate racks within the same room and cages changed in ‘uninfected’ or ‘infected’ changing stations. Sentinel mice were tested quarterly for endo- and ectoparasites, mouse hepatitis virus, mouse parvovirus, and rotavirus and annually for Mycoplasma pulmonis, pneumonia virus of mice, reovirus-3, Sendai virus, and Theiler’s murine encephalomyelitis virus. Also, quarterly sterile colon samples were screened for Citrobacter rodentium, non-lactose fermenting E.coli, Salmonella spp., Klebsiella spp., and Clostridium spp. (Phoenix Laboratories, Seattle, WA). All animal procedures were approved by the University of Washington Institutional Animal Care and Use Committee.
CD4+CD45RBhi adoptive transfer and Helicobacter infection
Adoptive transfer studies were done in EpithTg, CD11cTg, EpithTg/Rag2−/−, CD11cTg/Rag2−/− mice and Rag2−/− mice. Recipient mice were orally given Hb (2×107 CFU) or broth once by oral gavage prior to adoptive transfer (AT) of 2×105 CD4+CD45RBhi cells. Splenic CD4+CD45RBhi cells were prepared by sorting cells enriched for CD4+T cells by negative selection based on CD45RB expression. Hb was a natural isolate and kindly provided by L. Riley (University of Missouri, Columbia, MO) and cultured and prepared as previously described.25 Mice were weighed weekly and monitored for weight loss, dehydration, and diarrhea. Mice were euthanized by CO2 inhalation or cervical dislocation when they developed diarrhea or suffered 20% body weight loss and samples were taken for histopathology and immunohistochemistry.
Colitis pathology
Cecum, colon, and rectum were fixed in 10% neutral-buffered formaldehyde. The colon was prepared in a “Swiss roll” technique26 in order to evaluate the entirety of the proximal, middle, and distal colon. Tissues were routine processed, embedded in paraffin, sectioned at 5 µm, stained with hematoxylin and eosin and coded to the pathologist (HBO) blinded to the experimental status. The cecum, proximal colon, middle and distal colon, and rectum from each mouse were scored on severity of mucosal epithelial changes, degree of inflammation and extent of pathology. The segment score was derived by summing the severity scores [segment score = mucosal score + inflammation score + extent of intestine affected in any manner (Extent 1) + extent of intestine affected at level 3 or 4 (Extent 2).25 The total score for each mouse was derived by summing the scores from the individual segments and the mean derived for each treatment group.
Transgene expression analysis
Expression of transgenes were evaluated using reverse transcriptase-PCR on RNA extracted from tissues indicated in the figures. RNA was extracted from tissues homogenized in TRI REAGENT™ (Sigma, Saint Louis, Missouri). After homogenization, samples were centrifuged at 12,000×g for 10 minutes at 4 °C. Samples were then subjected to extraction with chloroform and precipitation with isopropanol. RNA was washed twice with 75% ethanol and resuspended in RNAse free water. cDNA was generated using MMLV reverse transcriptase and random hexamer primers (Life Technologies). RNAsin (Promega, Madison, WI) was added to the reaction to inhibit RNAses. Primer sequences were P5 (HPRT) 5'-GTTGGATACAGGCCAGACTTTGTTG-3'; P6 (HPRT) 5'-GAGGGTAGGCTGGCCTATGGCT-3'; HGHR 5'-TCTGTTGTGTTTCCTCCCTGTTGG-3'; I-AB(BETA) 5'-AGCAAGATGTTGAGCGGCATC-3'; RBG-R 5'-CTTTGTTCATGGCAGCCAGCAT-3'; I-AB(ALPHA) 5’- GAT CAG GTG GCA CCT CCA GA - 3’
In CD11cTg mice, transgene expression was evaluated using I-AB and the RBG-R primer specific for the rabbit β-Globin Gene contained in the construct used to make the mice. Transgene expression from the I-Abα contruct was evaluated in EpithTg mice using the I-AB(ALPHA) sequence above combined with the HGHR primer specific for human growth hormone contained in the construct. I-Abβ expression was evaluated with I-AB (BETA) and HGHR.
Immunohistochemisty (IHC)
Expression of MHC-II (and CD11c in CD11cTg mice) was assessed in the small and large intestines, thymus, mesenteric lymph nodes, spleen and lungs of CD11cTg/Rag2−/− and MHC-II-deficient mice. IHC was performed using monoclonal antibody clone M5/114 for MHC-II and RM4–5 for CD11c (BD Biosciences, San Diego, CA) on tissues snap frozen in OCT or fixed in formalin using antigen retrieval. Briefly, 5 to 6 µm frozen sections were fixed with acetone/ethanol (volume:75%/25% ) and 5 µm paraffin sections were subjected to heat-induced epitope retrieval in 0.01 M citrate buffer, pH of 6.0 using a microwave oven for 10 minutes as described 27. Endogenous peroxidase activity was inhibited by sodium azide with trace amounts of nascent H2O2 produced by a glucose oxidase/glucose system 28. Sections were incubated for 60 minutes with the primary monoclonal antibodies followed by the secondary biotinylated antibodies for 30 minutes, and detection of antibody-binding with the avidin-biotin immunoperoxidase, Vectastain ABC Kit (Vector Laboratories, Inc., Burlingame, CA). 3,3-diaminobenzadine was used as the chromogen. Negative controls included the exclusion of the primary antibody. All slides were scored by the pathologist ‘blinded’ to experimental group following a scoring system ranging from 0 (no expression) to 5 (intense expression). Microphotographs were taken with a Nikon E400 microscope equipped with a Coolpix5400 camera. Photomicrographs are shown as original after conversion to Tiff-format, but without further manipulations.
Flow cytometric analysis of mesenteric lymph node and spleen cells
Cells were isolated from collagenase digested spleen and mesenteric lymph nodes of Tg and control mice as previously described.29 Cellularity was determined and cells were stained with anti-MHC-II, anti-CD11c, anti-CD40, anti-CD4 and anti-CD11b antibodies (BD Biosciences, San Diego, CA). Cells were analyzed by flow cytometry with an LSR (BD Biosciences, San Diego, CA) and data analyzed utilizing Cellquest software (BD Biosciences, San Diego, CA).
Statistical Analyses
Differences between mean total pathology scores were evaluated by non-parametric ANOVA followed by Dunn's test for differences between treatment groups and the positive control group. FACS data significance was determined by two-way Student's T test. Significance was set at P ≤ 0.05.
Results
Development of mice where MHC-II expression is targeted to intestinal epithelial or dendritic cells
In order to determine the importance of MHC-II expression on subsets of intestinal cells in development of inflammatory bowel disease (IBD), we developed two different types of Tg mice targeting MHC-II expression to either dendritic cells or intestinal epithelial cells. In CD11cTg lines 6175, 6761 expression of the I-Aβ chain was placed under control of the CD11c promoter21 (Figure 1A) on an MHC-II-deficient background22,23 lacking functional expression of I-E and I-Aβ. In transgenic mice in which MHC-II was expressed predominantly on intestinal epithelial cells (EpithTg lines 5809, 6258), I-A expression was driven by the Fatty Acid Binding Protein (liver) FABPL promoter that targeted expression to small intestinal epithelial cells. Because I-Aα expression is not constitutive in epithelial cells, MHC-II oocytes were injected with two constructs, one containing I-Aα and one containing I-Aβ (Figure 1B)
Figure 1. Transgene constructs and expression analysis in transgenic animal tissues.

(A) Construct design for CD11cTg mice. I-Abβ was placed under the control of the CD11c promotor with rabbit β-Globin Gene used as a marker for transgene expression (B) Two constructs, one each for I-Abα and I-Abβ, were co-injected into MHC-II-deficient (I-E/I-Aβ-deficient). Both MHC-II chains were under control of promotor elements of fatty acid binding protein to direct expression to small intestine (FABPL) (C) Representative dot plots showing staining with anti- MHC-II antibody versus anti-CD11c antibody in splenocytes (top row) and MLN cell suspensions (bottom row) from C57BL/6 (WT), Rag2−/−, CD11cTg/Rag2−/− and CD11cNtg/Rag2−/− mice (indicated in figure). Dot plots are gated through a FSC/SSC gate that excluded small (presumably dead) cells. (D) Histograms show MHC-II expression of CD11c+ cells in spleen (left) and MLN (right). (E) RT-PCR showing transgene expression of the I-Abα construct, the I-Abβ construct and HPRT in various tissues of EpithTg line 6258.
Transgene expression for the CD11cTg line was evaluated using flow cytometry of splenocytes to show MHC-II expression only in cells that express CD11c. As this line was crossed onto Rag2−/− mice for use in adoptive transfer studies (see below), data is shown from CD11cTg/Rag2−/− mice. Figure 1C shows FACS plots of MHC-II staining versus CD11c staining in spleen (top panels) and MLN (bottom panels) from WT, Rag2−/−, CD11cTg/Rag2−/− and CD11cNtg/Rag2−/− mice. As expected, CD11cTg/Rag2−/− mice only express Class II on CD11c+ cells compared with Rag2−/− and Wt mice where class II expression is noted on all antigen presenting cells. No MHC-II staining is seen in animals genetically deficient in Class II (CD11cNtg/Rag2−/−). Comparison of MHC-II expression on CD11c+ DCs in spleen and MLN via histograms (Figure 1D) demonstrate that splenic DCs have similar MHC-II expression in Rag2−/− and CD11cTg/Rag2−/− mice and that expression is reduced compared to WT animals, likely due to the absence of T and B cells in Rag2−/− mice. In the MLN, however, Rag2−/− animals express higher levels of MHC-II than CD11cTg/Rag2−/− animals, although still not quite as high as that seen in wild type animals (Figure 1D, right). Transgene expression in the EpithTg mice was evaluated using realtime PCR. In EpithTg mice, despite the use of part of the FABPL promoter targeting expression to small intestinal epithelial cells, transgene expression was detected in cecum and colon as well as small intestine. Expression was also detected in stomach, liver and kidney, although expression was comparatively low in kidney, (Figure 1E and data not shown). Transgene expression was not detected in lung, thymus (Figure 1E) or non-transgenic animals (data not shown).
Immunohistochemical labeling for protein expression of I-A in the colon of CD11cTg mice, demonstrated that CD11c and MHC-II expression in the lamina propria both occur on cells with morphological characteristics compatible with DCs (Figure 2). In contrast, in EpithTg mice MHC-II protein expression (Figure 3) is restricted to the epithelial layer in Tg mice. This expression is in contrast to MHC-II staining in WT mice, where MHC-II staining is primarily in the lamina propria. Treatment of WT mice with IFNγ upregulated epithelial MHC-II staining. Only background staining was detected in MHC-II-deficient controls.
Figure 2. Immunohistochemistry for CD11c and MHC-II in Ntg and Tg mice.
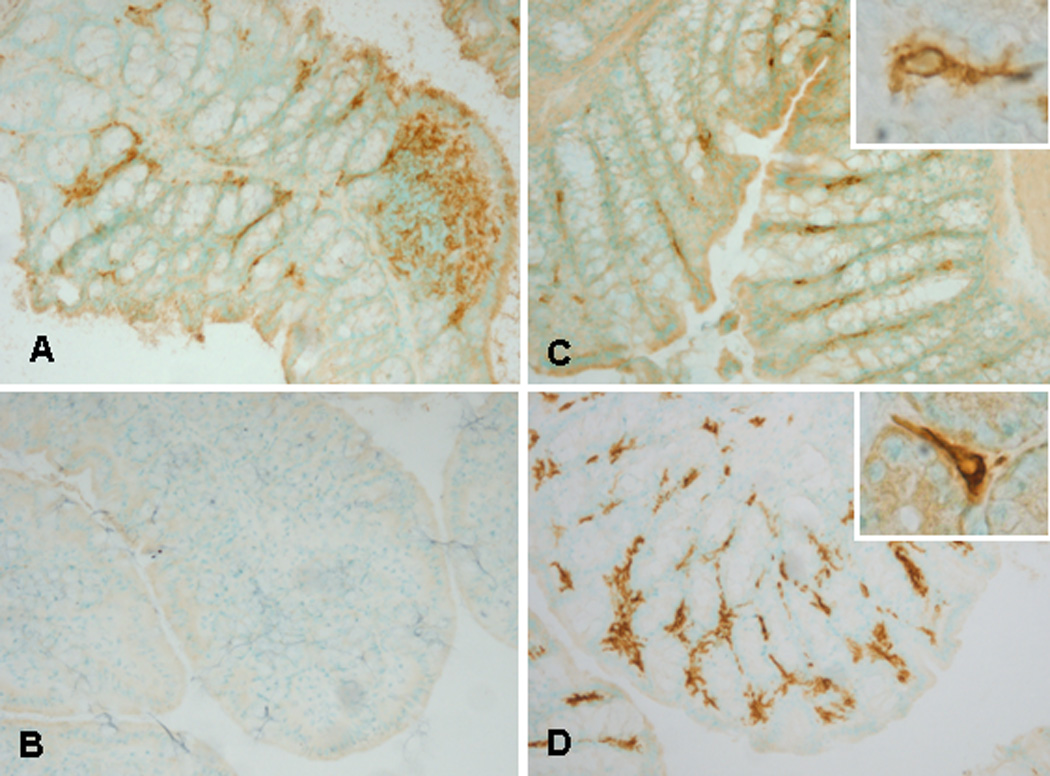
CD11c and MHC-II expressing cells in the colon of Ntg (A, B) and Tg (C, D) mice. CD11c+ cells in lamina propria (A, C) and MHC-II+ cells only noted in Tg animals (D) compared to no MHC-II staining detected in Ntg animals (B).
Figure 3. MHC-II staining in colonic tissue of untreated and IFNγ treated Epith Tg, WT and MHC-II-deficient mice.

MHC-II antibody staining at 20× (left column) and 40× (right column) magnification of indicated animal genotypes/treatment groups. Diffuse staining of epithelial cells in the EpithTg mice contrasts to the staining seen primarily in the colonic lamina propria of WT mice. Treatment of WT mice with IFNγ leads to upregulation of MHC-II staining in epithelial cells as well whereas in EpithTg mice, IFNγ treatment induces more expression of MHC-II (punctate staining within Epith cells but still no expression outside of epithelium). Background staining in MHC-II-deficient mice shows non-specific labeling of mast cells with the secondary reagents.
MHC-II expression on CD11c+ cells is sufficient for development of robust IBD while MHC-II expression on only epithelial cells is insufficient
We used an adoptive transfer model of colitis to investigate whether MHC-II expression only on CD11c+ cells or only on epithelial cells would be sufficient to drive development of IBD. In the adoptive transfer model of colitis, giving Rag2−/− mice naïve (CD4+CD45RBhi) T cells induces colitis depending upon the microflora in the animal facility. In our SPF mouse colony, Rag2−/− mice require prior infection with Helicobacter along with adoptive transfer of naïve T cells for development of a robust colitis (Figure 4A). Hb-infected Rag2−/− mice develop rapid, severe IBD (mean IBD score=60.6, N=5) within 2–3 weeks post adoptive transfer while Rag2−/− mice that receive only (CD4+CD45RBhi cells) or only Hb remain healthy long term (necropsied at 16 weeks post adoptive transfer) and show minimal if any colitis (mean colitis score= 1.6 and 1.75, N= 4 and 5, respectively). In order to compare disease in CD11cTg mice and EpithTg mice in an adoptive transfer model, we crossed CD11cTg line 6761 and EpithTg line 6258 with Rag2−/− mice to generate CD11cTg/Rag2−/− and EpithTg/Rag2−/− mice.
Figure 4. IBD scores in Hb-infected Rag2−/− and CD11cTg/Rag2−/− mice that receive adoptive transfer of naïve T cells.

(A) Rag2−/− mice were infected with Hb 2.8 weeks prior to adoptive transfer of 2×105 CD4+CD45RBhi cells IP and were necropsied 2–3 weeks post adoptive transfer. Other controls, including Rag2−/− infected with Hb (and no adoptive transfer) and Rag2−/− mice receiving adoptive transfer only, remained healthy for 16 weeks post adoptive transfer. (B) CD11cTg/Rag2−/− and CD11cNtg/Rag2−/− mice were infected with Hb one week prior to adoptive transfer with 2×105 CD4+CD45RBhi cells IP. Animals were necropsied at three weeks post adoptive transfer. Rag2−/− mice receiving Hb and adoptive transfer are positive controls for disease development. Three treatment groups in (B) show data from animals in two separate studies (# on x axis labels). Treatment groups were compared using nonparametric one way ANOVA (Kruskal-Wallis) followed by Dunn's post test comparing all treatment groups to positive controls (* P<0.05, ** P<0.01, *** P<0.001).
We found that MHC-II expression only on CD11c-expressing cells was sufficient to drive disease in this adoptive transfer model. Hb-infected-CD11cTg/Rag2−/− mice that received naïve T cells via adoptive transfer developed severe, rapid onset IBD similar to positive control Rag2−/− mice while other control animals only developed mild inflammation. The majority (11/13 mice) of Hb-infected CD11cTg/Rag2−/− mice that received CD4+CD45RBhi cells developed severe IBD (mean IBD score=57.8) within three weeks post adoptive transfer (Figure 4B) and this was highly comparable to the disease induced in positive control Rag2−/− mice at the same time point (mean IBD score = 55.4). In the absence of Hb or adoptive transfer of cells, mice developed minimal disease. CD11cTg/Rag2−/− animals that received adoptive transfer without Hb infection (N=6) or were only infected with Hb (N=5) without adoptive transfer developed mild inflammation at three weeks post adoptive transfer (mean IBD scores = 8.8 and 19.6, respectively). CD11cNtg/Rag2−/− treatment groups had zero to mild inflammation as well. Figure 5 shows the gross and histopathologic comparison of an Hb-infected CD11cTg/Rag2−/− mouse with adoptive transfer with a CD11cNtg/Rag2−/− with the same treatment. Hb-infected CD11cTg/Rag2−/− mice receiving CD4+CD45RBhi cells developed a significant pancolitis (Figure 5A) compared with Hb infected-CD11cNtg/Rag2−/− mice given AT; note formed feces in a normal colon (Figure 5B). Histology of the cecum and colon (Figure 5C and E) of CD11cTg/Rag2−/− mice showed pronounced crypt-proliferation, infiltration of lymphocytes, macrophages and neutrophils in lamina propria and submucosa, and severe submucosal edema, resulting in architectural distortion. Normal mucosal architecture is shown in the cecum and colon (Figure 5D and F) of CD11cNtg/Rag2−/− mice.
Figure 5. IBD in Hb-infected CD11cTg/Rag2−/− mice receiving adoptive transfer.

Hb-infected CD11cTg/Rag2−/− mice that received CD4+CD45RBhi cells (AT) developed colitis (A) compared with Hb infected- CD11cNtg/Rag2−/− mice given AT, showing formed feces in a normal colon (B). Histological sections of the lesions in the cecum (C) and colon (E) of CD11cTg/Rag2−/− mice comprised pronounced crypt-proliferation and infiltration of lymphocytes, macrophages and neutrophils in lamina propria and submucosa, as well as severe submucosal edema, resulting in distortion and obliteration of the normal mucosal architecture, as seen in the cecum (D) and colon (F) of CD11cNtg/Rag2−/− mice. All microphotographs are shown at 10× magnification.
In contrast to the severe disease in CD11cTg/Rag2−/− mice, Hb-infected EpithTg/Rag2−/− mice given adoptive transfer did not develop significant disease in an equivalent period of 3 weeks. As shown in Figure 6A, Hb-infected EpithTg/Rag2−/− mice receiving CD4+CD45RBhi cells (N=5) developed very mild inflammation 3 weeks post adoptive transfer (mean IBD score = 5.6) compared with Hb-infected positive control Rag2−/− mice receiving adoptive transfer (mean IBD score = 59.4, N=5). IBD scores in other control mice, including Hb-infected EpithNtg/Rag2−/− receiving adoptive transfer and EpithTg and Ntg/Rag2−/− mice receiving adoptive transfer only were also very low and comparable to that of Hb-infected EpithTg/Rag2−/− mice post adoptive transfer (Figure 6A). In order to determine if more significant IBD would develop over time in EpithTg/Rag2−/− mice, two additional experiments were performed where mice were followed for 16 weeks post adoptive transfer. Data from these two experiments are combined in the analysis shown in Figure 6B. Even at 16 weeks post adoptive transfer, Hb-infeced EpithTg/Rag2−/− mice receiving CD4+CD45RBhi cells developed only mild inflammation (mean IBD score=4.8) which was significantly different (P<0.001) from positive control Rag2−/− mice receiving the same treatment (mean IBD score= 60.6). At this later time point, it should be noted that five transgenic mice (3/17 EpithTg/Rag2−/− mice receiving only adoptive transfer) and two nontransgenic mice (2/5 Hb-infected EpithNtg/Rag2−/− mice+AT) developed significant IBD by 16 weeks post adoptive transfer. Both EpithTg and Ntg lines were infected with non-Hb-Helicobacter(s) spp. and we suspect that this may have driven IBD development in some animals over the longer 16 week study independent of MHC-II expression. In additional experiments, EpithTg mice that were on Rag2+/+ background developed mild to moderate colitis with Hb infection in the absence of adoptive transfer (data not shown). This disease was independent of transgenic status indicating that disease in these mice was possible even in the absence of MHC-II expression and was likely due to an innate immune response to Hb (data not shown). Other Helicobacter spp. (i.e. H. hepaticus) has been reported to drive colitis in lymphocyte deficient mice.30
Figure 6. IBD in Hb-infected EpithTg/Rag2−/− receiving adoptive transfer.

(A) EpithTg/Rag2−/− and EpithNtg/Rag2−/− mice were infected with Hb one week prior to adoptive transfer with 2×105 CD4+CD45RBhi cells IP. Animals were necropsied at three weeks post adoptive transfer. Rag2−/− mice receiving Hb and adoptive transfer are positive controls for disease development. Other controls received adoptive transfer only. (B) EpithTg (line 6258)/Rag2−/− mice were given Hb and adoptive transfer as in (A) and monitored for 16 weeks post adoptive transfer. Rag2−/− controls developed disease rapidly and were necropsied at 3–4 weeks post adoptive transfer. Three treatment groups in (B) show data from animals in two separate studies (# on x axis labels). Treatment groups were compared using nonparametric one way ANOVA (Kruskal-Wallis) followed by Dunn's post test comparing all treatment groups to positive (Rag2−/−+Hb+AT) control (* P<0.05, ** P<0.01, *** P<0.001).
Immunophenotype of CD11cTg/Rag2−/− mice
Because robust disease development and disease course and severity appeared to be similar between CD11cTg/Rag2−/− and positive control Rag2−/− mice (Hb plus adoptive transfer), we compared subsets of DCs in CD11cTg/Rag2−/− mice with those in Rag2−/− mice. In general, percentages and absolute numbers of DCs or subsets of DCs were very similar between CD11cTg/Rag2−/− and Rag2−/− mice (Table 1). The only notable difference was detected in the MLN, where the percentage of MHC-II+ DCs (P<0.01) as well as CD40+ DCs (P<0.01) was lower in CD11cTg/Rag2−/− animals compared to the control Rag2−/− mice. These observations suggest that DCs in this location are less activated in CD11cTg/Rag2−/− animals (Table I), although this difference did not appear to influence the outcome of disease. Compared to Wt animals, both CD11cTg/Rag2−/− and Rag2−/− animals had fewer absolute numbers of splenic DCs as well as lower percentages of MHC-II and CD40+ DCs in spleen as has been shown previously for mice lacking rag expression.31 CD40+ DC percentages were also lower in CD11cTg/Rag2−/− and Rag2−/− mice compared to WT in MLN, however, DC numbers were similar to WT in the MLN and percent of MHC-II+ cells was only reduced in CD11cTg/Rag2−/− compared to WT. DC subsets separated based on CD11b and CD4 staining were also altered in CD11cTg/Rag2−/− and Rag2−/− mice compared to WT as has been shown previously for mice lacking rag expression.31 Percentages of CD11b− populations were decreased in spleen while CD4−CD11b+ percentages were significantly increased. CD4+ CD11b− DCs were also reduced in MLN while percent of CD4−CD11b− DCs in MLN were similar between all the genotypes.
Table 1.
Flow cytometric analysis of spleens and MLN of CD11cTg/Rag2−/−, CD11cNtg/Rag2−/−, Rag2−/−, and Wt mice.
| Spleen | CD11c+ (%) |
#'s CD11c+ (x 106) |
CD11c+ MHC-II+ (%) |
CD11c+ CD40+ (%) |
CD11c+ CD4+ CD11b− (%) |
CD11c+ CD4− Cd11b− (%) |
CD11c+ CD4− CD11b+ (%) |
CD11c+ CD4+ CD11b+ (%) |
|---|---|---|---|---|---|---|---|---|
|
CD11cTg/ Rag2−/− n=9 |
60.9 ± 5.3** | 6.0 ± 3.0* | 20.0 ± 13.1** | 21.2 ± 8.3* | 2.7 ± 0.5** | 24.8 ± 3.4** | 68.9 ± 3.6** | 1.7 ± 0.8 |
|
Rag2−/− (MHC-II+/+) n=9 |
57.4 ± 7.2** | 6.6 ± 4.9 | 22.3 ± 9.8** | 20.2 ± 9.8** | 1.7 ± 0.7** | 27.3 ± 6.8* | 68.6 ± 6.9** | 0.7 ± 4.1 |
|
CD11cNtg/ Rag2−/− n=4 |
64.4 ± 4.8** | 6.6 ± 3.2* | 0.7 ± 0.9** | 20.3 ± 8.1** | 3.4 ± 1.3** | 21.9 ± 3.6** | 69.9 ± 4.7** | 2.4 ± 1.1 |
|
Wt n=9 |
10.2 ± 6.6 | 11.5 ± 4.3 | 50.6 ± 19.3 | 51.9 ± 10.2 | 11.3 ± 6.7 | 38.8 ± 9.1 | 46.7 ± 15.2 | 1.0 ± 6.7 |
| MLN |
CD11c+ (%) |
#'s CD11c+ (x 105) |
CD11c+ MHC-II+ (%) |
CD11c+ CD40+ (%) |
CD11c+ CD4+ CD11b− (%) |
CD11c+ CD4− Cd11b− (%) |
CD11c+ CD4− CD11b+ (%) |
CD11c+ CD4+ CD11b+ (%) |
|
CD11cTg/ Rag2−/− n=3 |
66.1 ± 9.8** | 4.1 ± 1.8 | 19.4 ± 11.3**£ | 24.1 ± 12.1** | 1.6 ± 0.9** | 35.2 ± 4.3 | 60.8 ± 3.6 | 0.8 ± 0.8** |
|
Rag2−/− (MHC-II+/+) n=7 |
70.9 ± 2.4** | 5.5 ± 1.7 | 66.2 ± 14.7 | 34.7 ± 16.2* | 1.6 ± 0.7 | 42.3 ± 4.7 | 53.6 ± 1.9 | 4.1 ± 3.6 |
|
CD11cNtg/ Rag2−/− n=3 |
63.4 ± 6.8** | 2.4 ± 2.0 | 0.5 ± 0.5** | 31.8 ± 11.9** | 1.4 ± 1.1* | 31.7 ± 5.2 | 64.1 ± 6.9 | 1.1 ± 1.4** |
|
Wt n=5 |
5.9 ± 5.3 | 2.4 ± 2.0 | 68.0 ± 8.4 | 70.4 ± 13.5 | 9.3 ± 6.2 | 46.3 ± 23.4 | 46.5 ± 16.1 | 6.7 ± 1.3 |
P<0.05 compared to Wt
P<0.01 compared to Wt
P<0.01 CD11cTg/Rag2−/− vs Rag2−/− control
MLN-mesenteric lymph nodes
Discussion
The importance of CD4+ T cell responses in inflammatory bowel disease has been shown in many animal models, and is highlighted in studies focused on adoptive transfer of naïve cells into immunodeficient hosts. Dendritic cells are key antigen presenting cells for CD4+ T cells8 and function in many aspects of gut immunobiology, including the maintenance of tolerance to gut antigens as well as initiating appropriate immune responses to inflammatory insults at the gut epithelium.32 Dendritic cells interact with luminal antigens by projecting long processes through interstices of epithelial cells to sample contents.33 Our model where MHC-II expression is restricted to CD11c+ cells provides a tool to study the roll of DCs in disease separate from other cell types expressing MHC-II.
Our studies show that antigen presentation to CD4+ cells solely by DCs is sufficient to drive robust IBD but only in the presence of a bacterial trigger. This indicates that although other cell types expressing MHC-II molecules are known to be present in inflamed gut tissues, their ability to present antigen to CD4+ T cells is neither sufficient nor necessary for disease development in the adoptive transfer model of colitis. Our findings are in agreement with and complementary to several studies detailing the importance of interactions between CD4+ T cells and DCs in development of adoptive transfer colitis. Leitherhauser et al10 showed that adoptively transferred T cells cluster with DC aggregates and proliferate under the basal crypt epithelium prior to disease onset. These T cell/DC clusters, which excluded F4/80+ cells, occurred with highest frequency in the cecum where histopathology of IBD was most severe.10 Additionally, in vitro DCs pulsed with gut bacterial lysates efficiently elicited MHC-II-dependent IFN-γ production by lamina propria T cells isolated from colitic mice.10 The importance of DCs in adoptive transfer colitis is again shown by Malstrom et al in which increases in activated CD134L+ DCs were shown in MLN during adoptive transfer-colitis. Blocking T cell/DC interactions with anti-CD134L inhibited development of colitis which was correlated with reduced T cell proliferation and upregulation of gut homing receptors on MLN T cells.9 Increases in total CD11c+ cells in the GALT was also demonstrated with colitis, although CD134L was not detected on these DCs.9 More recently, a population of E-Cadherin+ DCs has been shown to increase dramatically in GALT and MLN during AT-colitis and appears to be important particularly in T cell mediated colitis but not colitis mediated by innate cells.34 Interestingly, adoptive transfer of BM derived E-Cadherin-positive DCs into mice that had previously received CD45RBhi T cells exacerbated adoptive transfer-colitis compared to transfer of BM derived E-Cadherin-negative− DCs34 once again demonstrating the important role of dendritic cells in the adoptive transfer model of colitis. As our model is one where DCs are the only antigen presenting cells able to present MHC-II restricted antigens to adoptively transferred T cells, we have demonstrated that MHC-II+ DCs alone are sufficient to drive robust colitis.
In addition to being important during the disease development phase of adoptive transfer colitis, DCs are also important in the initial expansion of T cells transferred into lymphopenic hosts. This expansion, termed spontaneous proliferation, is a prerequisite to disease development35 (see Surh and Sprent (2008) for review) and is dependent on MHC-II expression36 where the majority of T cell expansion is in response to certain species of gut flora.19, 20, 37 Using an elegant model of CD11cDTR Tg mice crossed with TCRβ−/− mice, Do and Min18 showed that spontaneous proliferation of transferred cells was dependent upon DCs. Our data complements these findings and suggest that DCs in CD11cTg mice are able to induce sufficient spontaneous proliferation required for robust disease. We did see mild inflammation in CD11cTg/Rag2−/− mice that received only AT-T cells (these were not infected with Hb) perhaps indicating some T cell expansion was occurring without development of robust colitis. Because our focus was on disease development, we did not test the requirement of Hb infection in spontaneous proliferation of adoptively transferred T cells in our model. Interesting data published by Feng et al (2010) showed that although specific antigen presentation in the intestine was required for disease development in an adoptive transfer model using TCR Tg mice, it was not necessarily required for spontaneous proliferation of transferred T cells. Our Tg model where Hb infection is required for robust disease and only DCs present MHC-II antigens may be a useful tool to examine antigen requirements for spontaneous proliferation and disease development in the adoptive transfer model.
The role of MHC-II expression on intestinal epithelial cells in development of colitis is less clear. In vitro studies suggest that epithelial cells can function as nonprofessional APCs. IECs from IBD patients were shown to stimulate CD4+ T cells to proliferate and produce IFN gamma.14 Furthermore, purified mouse IECs from Villin-HA mice have been shown to stimulate proliferation of Foxp3+ as well as Foxp3− HA-antigen-specific TCR Tg CD4+ T cells.15 Additionally, adoptive transfer of (HA)-specific TCR Tg T cells into mice that express HA antigen under control of the Villin promoter demonstrated expansion of FoxP3+ T cells independent of dendritic cells.15 These data indicate that IEC have the potential to trigger both regulatory and inflammatory CD4+ T cell responses and the type of T cell response favored in an immune reaction likely depends on other environmental signals present in the gut at the time of T cell stimulation by IECs. In our model, an essentially ectopic expression of MHC-II on epithelial cells was not sufficient to drive a robust adoptive transfer colitis even in the presence of a bacterial trigger (Hb) but we cannot exclude the possibility that this may have been due to inadequate expansion of adoptively transferred naïve T cells.18, 20
Aberrant CD4+ T cell responses have been strongly implicated in the pathogenesis of IBD in both humans and experimental models. These responses are regulated by a complex interplay between multiple types of MHC-II positive cells within the intestinal mucosa and the high concentration of bacterial antigens within the intestinal lumen. In this manuscript, we have attempted to isolate and independently manipulate various components of this system using transgenic mice that selectively express MHC-II on either CD11c+ cells or IECs using Helicobacter bilis as a trigger for mucosal inflammation. We demonstrate that in DCs, but not IECs, MHC-II expression is sufficient to for development of bacterial-induced IBD.
Acknowledgements
We thank Piper Treuting for help with immunohistochemistry figures, Lela Riley for providing the Hb isolate, Loida Torres, and Betty Yue and Tristan Root for genotyping and care of these mice.
Supported in part by grants NIH R01 DK056204-07 (RMH) and Broad Medical Research Program (RMH).
References
- 1.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. doi: 10.1038/nature06005. [DOI] [PubMed] [Google Scholar]
- 2.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annual review of immunology. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 3.Morrissey PJ, Charrier K. Induction of wasting disease in SCID mice by the transfer of normal CD4+/CD45RBhi T cells and the regulation of this autoreactivity by CD4+/CD45RBlo T cells. Research in immunology. 1994;145:357–362. doi: 10.1016/s0923-2494(94)80200-9. [DOI] [PubMed] [Google Scholar]
- 4.Morrissey PJ, Charrier K, Braddy S, et al. CD4+ T cells that express high levels of CD45RB induce wasting disease when transferred into congenic severe combined immunodeficient mice. Disease development is prevented by cotransfer of purified CD4+ T cells. J Exp Med. 1993;178:237–244. doi: 10.1084/jem.178.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Powrie F, Leach MW, Mauze S, et al. Inhibition of Th1 responses prevents inflammatory bowel disease in scid mice reconstituted with CD45RBhi CD4+ T cells. Immunity. 1994;1:553–562. doi: 10.1016/1074-7613(94)90045-0. [DOI] [PubMed] [Google Scholar]
- 6.Powrie F, Leach MW, Mauze S, et al. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5:1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 7.Cahill RJ, Foltz CJ, Fox JG, et al. Inflammatory bowel disease: an immunity-mediated condition triggered by bacterial infection with Helicobacter hepaticus. Infect Immun. 1997;65:3126–3131. doi: 10.1128/iai.65.8.3126-3131.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shortman K, Liu YJ. Mouse and human dendritic cell subtypes. Nature reviews Immunology. 2002;2:151–161. doi: 10.1038/nri746. [DOI] [PubMed] [Google Scholar]
- 9.Malmstrom V, Shipton D, Singh B, et al. CD134L expression on dendritic cells in the mesenteric lymph nodes drives colitis in T cell-restored SCID mice. J Immunol. 2001;166:6972–6981. doi: 10.4049/jimmunol.166.11.6972. [DOI] [PubMed] [Google Scholar]
- 10.Leithauser F, Trobonjaca Z, Moller P, et al. Clustering of colonic lamina propria CD4(+) T cells to subepithelial dendritic cell aggregates precedes the development of colitis in a murine adoptive transfer model. Laboratory investigations. 2001;81:1339–1349. doi: 10.1038/labinvest.3780348. [DOI] [PubMed] [Google Scholar]
- 11.Niess JH. Role of mucosal dendritic cells in inflammatory bowel disease. World J Gastroenterol. 2008;14:5138–5148. doi: 10.3748/wjg.14.5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coombes JL, Powrie F. Dendritic cells in intestinal immune regulation. Nat Rev Immunol. 2008;8:435–446. doi: 10.1038/nri2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hershberg RM, Mayer LF. Antigen processing and presentation by intestinal epithelial cells – polarity and complexity. Immunology Today. 2000;21:123–128. doi: 10.1016/s0167-5699(99)01575-3. [DOI] [PubMed] [Google Scholar]
- 14.Dotan I, Allez M, Nakazawa A, et al. Intestinal epithelial cells from inflammatory bowel disease patients preferentially stimulate CD4+ T cells to proliferate and secrete interferon-γ. American Journal of Physiology - Gastrointestinal and Liver Physiology. 2007;292:G1630–G1640. doi: 10.1152/ajpgi.00294.2006. [DOI] [PubMed] [Google Scholar]
- 15.Westendorf AM, Fleissner D, Groebe L, et al. CD4+Foxp3+ regulatory T cell expansion induced by antigen-driven interaction with intestinal epithelial cells independent of local dendritic cells. Gut. 2009;58:211–219. doi: 10.1136/gut.2008.151720. [DOI] [PubMed] [Google Scholar]
- 16.Borcherding F, Nitschke M, Hundorfean G, et al. The CD40-CD40L pathway contributes to the proinflammatory function of intestinal epithelial cells in inflammatory bowel disease. The American journal of pathology. 2010;176:1816–1827. doi: 10.2353/ajpath.2010.090461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dahan S, Roth-Walter F, Arnaboldi P, et al. Epithelia: lymphocyte interactions in the gut. Immunological reviews. 2007;215:243–253. doi: 10.1111/j.1600-065X.2006.00484.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Do JS, Min B. Differential requirements of MHC and of DCs for endogenous proliferation of different T-cell subsets in vivo. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:20394–20398. doi: 10.1073/pnas.0909954106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Surh CD, Sprent J. Homeostasis of naive and memory T cells. Immunity. 2008;29:848–862. doi: 10.1016/j.immuni.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 20.Feng T, Wang L, Schoeb TR, et al. Microbiota innate stimulation is a prerequisite for T cell spontaneous proliferation and induction of experimental colitis. J Exp Med. 2010;207:1321–1332. doi: 10.1084/jem.20092253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brocker T, Riedinger M, Karjalainen K. Targeted expression of major histocompatibility complex (MHC) class II molecules demonstrates that dendritic cells can induce negative but not positive selection of thymocytes in vivo. J Exp Med. 1997;185:541–550. doi: 10.1084/jem.185.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grusby MJ, Glimcher LH. Immune responses in MHC class II-deficient mice. Annu Rev Immunol. 1995;13:417–435. doi: 10.1146/annurev.iy.13.040195.002221. [DOI] [PubMed] [Google Scholar]
- 23.Grusby MJ, Johnson RS, Papaioannou VE, et al. Depletion of CD4+ T cells in major histocompatibility complex class II-deficient mice. Science. 1991;253:1417–1420. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- 24.Simon TC, Roth KA, Gordon JI. Use of transgenic mice to map cis-acting elements in the liver fatty acid-binding protein gene (Fabpl) that regulate its cell lineage-specific, differentiation-dependent, and spatial patterns of expression in the gut epithelium and in the liver acinus. Journal of Biological Chemistry. 1993;268:18345–18358. [PubMed] [Google Scholar]
- 25.Burich A, Hershberg R, Waggie K, et al. Helicobacter-induced inflammatory bowel disease in IL-10- and T cell-deficient mice. Am J Physiol Gastrointest Liver Physiol. 2001;281:G764–G778. doi: 10.1152/ajpgi.2001.281.3.G764. [DOI] [PubMed] [Google Scholar]
- 26.Moolenbeek C, Ruitenberg EJ. The "Swiss roll": a simple technique for histological studies of the rodent intestine. Lab Anim. 1981;15:57–59. doi: 10.1258/002367781780958577. [DOI] [PubMed] [Google Scholar]
- 27.Goff BA, Ries JA, Els LP, et al. Immunophenotype of ovarian cancer as predictor of clinical outcome: evaluation at primary surgery and second-look procedure. Gynecol Oncol. 1998;70:378–385. doi: 10.1006/gyno.1998.5094. [DOI] [PubMed] [Google Scholar]
- 28.Andrew SM, Jasani B. An improved method for the inhibition of endogenous peroxidase non-deleterious to lymphocyte surface markers. Application to immunoperoxidase studies on eosinophil-rich tissue preparations. Histochem J. 1987;19:426–430. doi: 10.1007/BF01675753. [DOI] [PubMed] [Google Scholar]
- 29.Seamons A, Perchellet A, Goverman J. Endogenous myelin basic protein is presented in the periphery by both dendritic cells and resting B cells with different functional consequences. Journal of immunology. 2006;177:2097–2106. doi: 10.4049/jimmunol.177.4.2097. [DOI] [PubMed] [Google Scholar]
- 30.Fox JG, Ge Z, Whary MT, et al. Helicobacter hepaticus infection in mice: models for understanding lower bowel inflammation and cancer. Mucosal Immunol. 2011;4:22–30. doi: 10.1038/mi.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shreedhar V, Moodycliffe AM, Ullrich SE, et al. Dendritic cells require T cells for functional maturation in vivo. Immunity. 1999;11:625–636. doi: 10.1016/s1074-7613(00)80137-5. [DOI] [PubMed] [Google Scholar]
- 32.Rescigno M, Di Sabatino A. Dendritic cells in intestinal homeostasis and disease. The Journal of clinical investigation. 2009;119:2441–2450. doi: 10.1172/JCI39134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chieppa M, Rescigno M, Huang AY, et al. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J Exp Med. 2006;203:2841–2852. doi: 10.1084/jem.20061884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siddiqui KR, Laffont S, Powrie F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity. 2010;32:557–567. doi: 10.1016/j.immuni.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng T, Elson CO, Cong Y. Microbiota: dual-faceted player in experimental colitis. Gut Microbes. 2010;1:388–391. doi: 10.4161/gmic.1.6.13727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Min B, Foucras G, Meier-Schellersheim M, et al. Spontaneous proliferation, a response of naive CD4 T cells determined by the diversity of the memory cell repertoire. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:3874–3879. doi: 10.1073/pnas.0400606101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kieper WC, Troy A, Burghardt JT, et al. Recent immune status determines the source of antigens that drive homeostatic T cell expansion. Journal of immunology. 2005;174:3158–3163. doi: 10.4049/jimmunol.174.6.3158. [DOI] [PubMed] [Google Scholar]