

Abstract
Vascular endothelial growth factor/vascular permeability factor (VEGF/VPF or VEGF-A) is a pivotal driver of cancer angiogenesis that is a central therapeutic target in treatment of malignancy. However, little work has been devoted to investigating functions of VEGF that are independent of its pro-angiogenic activity. Here we report that VEGF produced by tumor cells acts in an autocrine manner to promote cell growth through interaction with the VEGF receptor neuropilin-1 (NRP-1). Reducing VEGF expression by tumor cells induced a differentiated phenotype in vitro and inhibited tumor-forming capacity in vivo independent of effects on angiogenesis. Autocrine activation of tumor cell growth was dependent on signaling through NRP-1 and Ras was determined to a critical effector signaling molecule downstream of NRP-1. Our findings define a novel function for VEGF in de-differentiation of tumor cells, expanding its role in cancer beyond its known pro-angiogenic function.
Introduction
Vascular endothelial growth factor/vascular permeability factor (VEGF/VPF, VEGF-A) is a central regulator of physiological and pathological angiogenesis such as tumor angiogenesis (1, 2). The tumor cells secrete a high level of VEGF and other pro-angiogenic factors and promote tumor angiogenesis to maintain the oxygen and nutrition supply in the tumor microenvironment. Treatments involving blocking VEGF to inhibit tumor angiogenesis have gained advances in cancer therapeutics (2). The anti-VEGF monoclonal antibody bevacizumab inhibits tumor angiogenesis by blocking VEGF binding to VEGF receptor 1/2 (VEGFR-1/2) and shows improvement in metastatic colorectal cancer, non-small cell lung cancer (NSCLC), metastatic renal cell carcinoma (RCC), and others (3).
Clear cell RCC is typically highly vascular in nature due to VHL gene mutation. VHL mutation stabilizes HIF1α/2α, which are transcription factors that can induce hypoxia-related gene expression, including constitutive VEGF expression. This formed the rationale for choosing RCC to test the efficacy of anti-VEGF therapy. In fact that despite being approved by FDA for RCC treatment, bevacizumab showed only a marginal improvement in median progression-free survival (PFS) but not a statistically significant advantage in overall survival (4). In addition conflicting reports suggested that bevacizumab could accelerate the tumorigenicity of RCC also compounds the problem (5).
To date, three VEGF receptors have been discovered that include VEGFR-1/2 and Neuropilin-1 (NRP-1). VEGFR-1/2 are both receptor tyrosine kinases. Unlike VEGFR-1/2, NRP-1 lacks kinase domain and is a less defined multi-functional receptor for several ligands, including VEGF (6, 7). Bevacizumab inhibits VEGF interactions with VEGFR-1/2 but not with NRP-1. Contrary to endothelial cells, VEGFR-1/2 expression is not always detectable in certain types of tumor cells (6, 8, 9). In contrast, NRP-1 is the predominantly expressed VEGF receptor expressed in tumor cells, and its expression correlates closely with advanced tumor stages and poor patient prognosis in specific tumor types (7). There are reports showed VEGF controls tumorigenesis properties through an autocrine fashion (9–11). But the mechanisms were still not clear; especially in the case of tumor cells do not express VEGFR-1/2. In the present study we intend to test our hypothesis that VEGF acts through an autocrine signaling pathway mediated by NRP-1 in cancer cells to promote tumorigenesis.
Materials and Methods
Cell Culture
Human RCC cell lines 786-O and A-498 were maintained in DMEM + 10% FBS and MEM + 10% FBS, respectively. Breast cancer cell line MCF7 was maintained in DMEM + 10% FBS. All cell lines were purchased from American Type Culture Collection (ATCC).
Plasmids
Retroviral pZIP-KRas (G12V) and pMMp-NRP-1 plasmid were used in this experiment. Retrovirus was prepared by transfecting the retroviral plasmid together with packaging plasmids into 293T cells using Polyfect (Qiagen).
siRNA and short hairpin RNA (shRNA) transfection
siRNAs for human NRP-1, VEGF, and control were from Qiagen. siRNA transfection was performed with Hiperfect (Qiagen) following the manufacturer's instructions. pLKO.1 and TRIPZ lentiviral shRNA vectors for human VEGF, SOS1/2 and non-targeting control were from Open Biosystems and were prepared as previously described (8). Cells were infected with lentiviral shRNA; and after puromycin selection, stable selected cells were used for the experiments. The sequences of siRNA and shRNA were listed in supplementary table S1.
For the Tet-On inducible pTRIPZ shRNA, 0.2µg/ml of doxycycline was used to induce shRNA expression in the stable infected cells.
Subcutaneous Tumor Model
Subcutaneous tumor models were established in nude mice as described before (8). All animal work was performed under protocols approved by the Mayo Clinic Institutional Animal Care and Use Committee. Briefly, 2×106 cells were injected into the right flank of 6- to 8-week-old female nude mice (purchased from NIH). Tumor size was measured every week. In the case of green fluorescent protein (GFP) or red fluorescent protein (RFP) labeled cells, the GFP and RFP fluorescence signals were monitored and quantified using the IVIS system 200 series (Xenogen Corp).
Ras activity assay
Cells were treated as indicated in each experiment. Cells were lysed and Raf-1 RBD agarose bead pull-down assay was performed. Samples were run on a SDS-PAGE gel, and immunoblotting was performed with the anti-Ras antibody (Millipore).
Statistical Analysis
The independent-samples t-test was used to test the probability of significant differences between two groups. Values of p<0.05 were considered significant.
Additional experimental details are available in the Supplementary Methods.
Results and Discussion
We previously demonstrated that RCC cells express NRP-1 but not VEGFR-1/2 by reverse-transcription PCR (8), and confirmed again by westernblot in this experiment (Supplementary Fig. S1A). Two RCC cell lines A-498 and 786-O, which harbor the VHL mutation and therefore express a high level of VEGF, were chosen for this study. VEGF was knocked down via lentiviral shRNA. Two shRNAs (VEGFsh3 and VEGFsh4) with knockdown efficiencies greater than 90% were selected for the studies (Supplementary Fig. S1B, C). Control and VEGF shRNA transfected A-498 and 786-O cells were injected subcutaneously into nude mice. Eight weeks later the tumors were analyzed. VEGF knockdown significantly decreased tumor development (1298.8 ± 244.6 versus 35.9 ± 34.3; p<0.001 for 786-O-Consh versus 786-O-VEGFsh4, respectively; 625.3 ± 473.3 versus 16.0 ± 9.2; p=0.02 for A-498-Consh versus A-498-VEGFsh3, respectively; mean ± SD, n=5) (Fig. 1A). As expected, the VEGF knockdown tumors showed significantly less vascular density and less proliferation than control tumors (by vWF and PCNA staining; Fig. 1B and supplementary Fig. S1D). Interestingly, the VEGF shRNA expressing tumors showed increased Ksp-Cadherin expression, a marker of mature kidney epithelial cells, suggesting that the tumor cells were undergoing differentiation (Fig. 1B, supplementary Fig. S1E).
Figure 1.
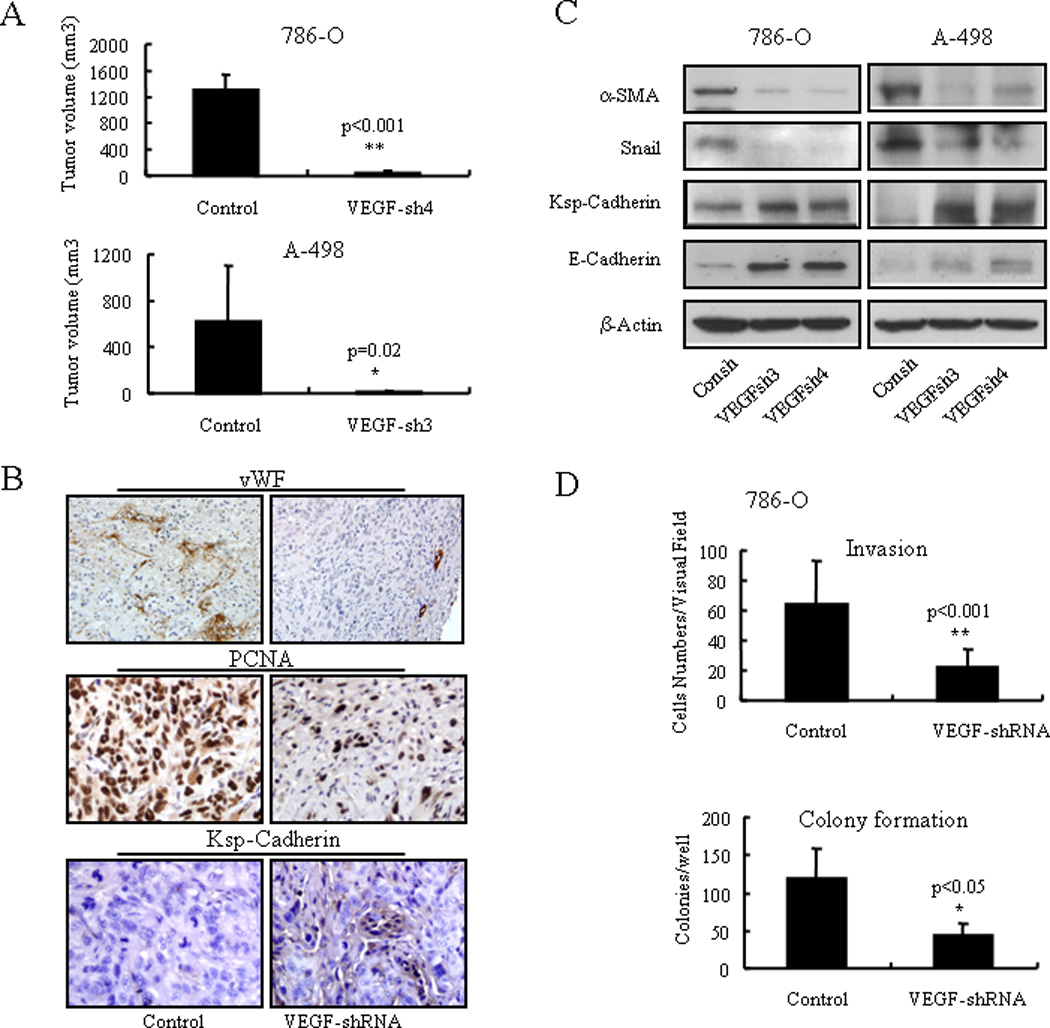
VEGF knockdown restricts tumor growth in vivo and induced differentiated phenotypes in vitro. A) Tumor volume of control and VEGF knockdown 786-O and A-498 tumors eight weeks after injection. B) vWF, PCNA, and Ksp-Cadherin immunohistochemistry staining in control and VEGF knockdown 786-O tumors. C) Western blot analysis of Ksp-cadherin, E-cadherin, α-SMA, and Snail expression in control and VEGF knockdown 786-O and A-498 cells. D) In vitro extracellular matrix invasion ability and anchorage independent clonogenic ability of control and VEGF knockdown 786-O cells.
We further examined the consequences of reduced endogenous VEGF expression in cancer cells with respect to differentiation phenotypes (also was known as mesenchymal to epithelial transition) under in vitro conditions. Upon VEGF knockdown, cancer cells showed increased Ksp-Cadherin and E-Cadherin expression, while Snail and α-SMA levels decreased (Fig. 1C). This profile corresponds to a typical differentiation phenotype of kidney epithelial cells. VEGF knockdown also decreased cancer cell invasion ability (35.4% and 57.9% for 786-O-VEGFsh and A-498-VEGFsh, respectively, as compared to each control; both p<0.01) and in vitro anchorage-independent colony formation ability (36.6% and 35.9% for 786-O-VEGFsh and A-498-VEGFsh, respectively, as compare to each control; both p<0.05) (Fig. 1D, Supplementary Fig. S2). However, loss of VEGF did not significantly affect cancer cell proliferation or survival in vitro (Supplementary Fig. S3).
As described above, the loss of VEGF in cancer cells led to reduced tumor formation in vivo. One intuitive explanation is that inhibition of angiogenesis limited tumor growth and created a “dormant” tumor. However, based on our in vitro data which suggested the cancer cells were less tumorigenic after VEGF knockdown, we sought to investigate whether VEGF had a tumorigenic function in addition to its pro-angiogenesis role in vivo. A major obstacle in this line of inquiry was maintaining normal angiogenesis during tumor growth. To overcome this obstacle and investigate the possibility that VEGF has a tumorigenic role, we designed two independent in vivo experimental approaches.
First, we used a Tet-On inducible VEGF shRNA system (Fig. 2A). Upon Doxycycline (Dox) inducing expression of VEGF shRNA, endogenous VEGF mRNA level was down-regulated significantly within three days. When Dox was removed at day 3, VEGF mRNA level was restored by day 7 (Fig. 2A). In accordance with regular VEGF shRNA results, Dox-induced shRNA expressing cells (referred to as (+)Dox) exhibited less anchorage-independent growth ability (Supplementary Fig. S4) as well as a greater differentiation phenotype compared to non-induced cells (referred to as (−)Dox; Fig. 2B). Despite restoration of VEGF levels by day 7 after removing Dox (referred to as postDox), the cells were still in a differentiated stage (Fig. 2B). Therefore, (−)Dox cells and postDox cells expressed the same level of VEGF, but postDox cells had already started differentiating during the three days of Dox-induced VEGF shRNA expression and could not completely revert back to an undifferentiated state after withdrawal of the Dox. Thus, (−)Dox and postDox cells would have different tumorigenic ability (Fig. 2C; schematic illustration). As expected, tumors formed by postDox A-498 cells were much smaller than those formed by (−)Dox cells (799.6 ± 121 versus 265.3 ± 264.5, p=0.003, for A-498 (−)Dox versus A-498 postDox at eight weeks, respectively; mean ± SD, n=5) (Fig. 2D). This result suggests that the loss of VEGF in tumor cells reduced their tumorigenic ability. As Dox can be used as an anticancer drug, we tested the effect of Dox on wild type A-498 cells. When cells were treated with a relatively low concentration of 0.2µg/ml Dox, which we used for shRNA induction, A-498 cells did not show any significant differences in cell proliferation, apoptosis, and differentiation marker expression in vitro and tumor growth in vivo. (Supplementary Fig. S5). This ruled out the possibility that the less tumor formation by postDox cells was due to Dox treatment.
Figure 2.
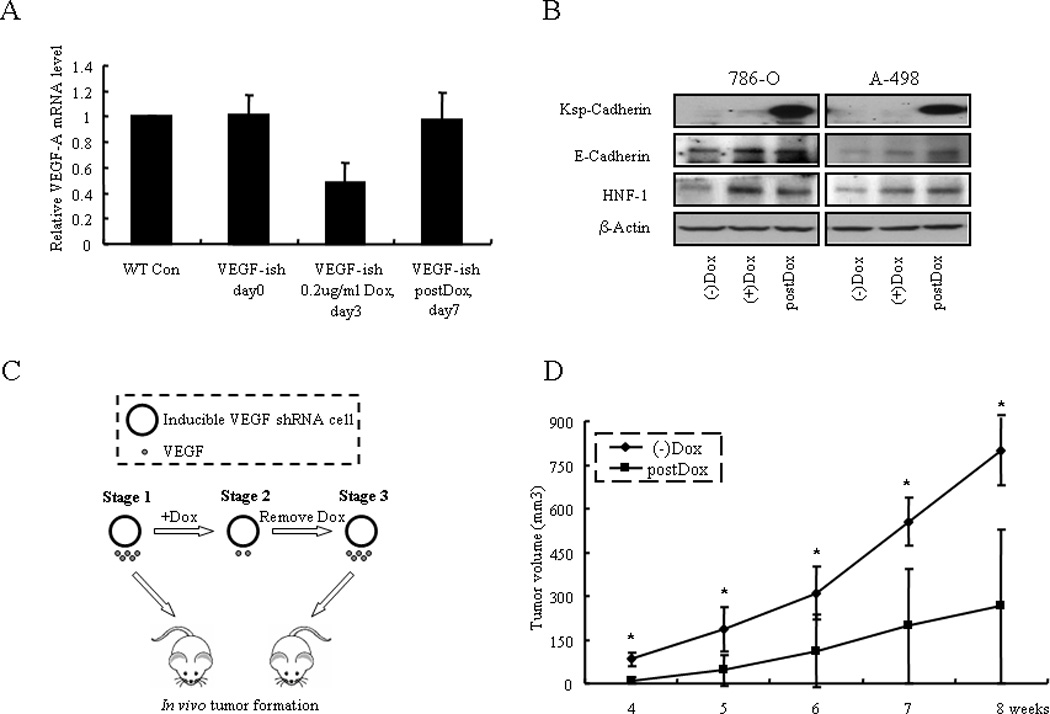
Mouse xenograft model of VEGF in tumorigenesis using a Tet-On inducible system. A) A-498 cells were transfected by the Tet-On inducible VEGF shRNA ((−)Dox), induced by Dox for three days ((+)Dox), and then Dox was removed (postDox). VEGF mRNA levels in the (−)Dox, (+)Dox and postDox RCC cells were examined in A-498 cells. B) Inducible VEGF shRNA expression induced 786-O and A-498 cells differentiation in three days of Dox inducement and then Dox was removed; cells were still in a differentiation state on day 7. C) Schematic illustration of the in vivo experiment. (−)Dox (Stage 1) and postDox (Stage 3) tumor cells, which have the same level of VEGF expression levels, were injected into the nude mice and in vivo tumor formation was examined. D) (−)Dox and postDox induced A-498 cells were analyzed for their tumor forming ability in vivo (n=5 in each group).
In the second in vivo experiment, we labeled A-498 cells with GFP or RFP (referred to as A-498-GFP and A-498-RFP cells) and then knocked down VEGF in the A-498-RFP cells using VEGF shRNA expressing lentivirus. Equal numbers of A-498-GFP and A-498-RFP-VEGFsh cells were mixed and injected subcutaneously into nude mice (n=12). A-498-GFP and A-498-RFP-Consh cells were also mixed and injected as a control group (n=12). Theoretically, in the control group the A-498-GFP and A-498-RFP-Consh cell numbers should be similar as they have the same tumorigenic ability. In the experimental group, if VEGF knockdown does not affect the cancer cell tumorigenic ability in vivo, the cell number of A-498-RFP-VEGFsh should be similar to A-498-GFP; conversely, if the numbers of A-498-GFP and A-498-RFP-VEGFsh cells are not equal, then they would not have the same tumorigenic ability (Fig. 3A, schematic illustration). After five weeks of tumor formation, we used a Xenogen fluorescent imaging system to quantify GFP and RFP signal intensities in the whole tumor. As shown (Fig. 3B and Supplementary Fig. S6), GFP signals in both groups have similar intensities (1144.8 ± 170.6 versus 999.75 ± 255.93; p=0.340 for A-498-GFP/A-498-RFP-Consh versus A-498-GFP/A-498-RFP-VEGFsh, mean ± SD); but RFP signal intensity was significantly less in the A-498-GFP/A-498-RFP-VEGFsh group (7377.5 ± 3681.9) as compared to the A-498-GFP/A-498-RFP-Consh group (19820 ± 4997.2; p=0.004, mean ± SD) (Fig. 3C). By sectioning the tumors, we observed that in the number of the RFP cells was similar to the GFP cells in the A-498-GFP/A-498-RFP-Consh groups; but in the number of RFP cells was much less than the GFP cells in the A-498-GFP/A-498-RFP-VEGFsh group (Supplementary Fig. S7A). Moreover, in both groups the GFP and RFP labeled tumor cells were homogeneously distributed in the later stages of tumor growth and that the vascular density displayed was well distributed in both groups (Supplementary Fig. S7A, B), and this observation ruled out the possibility that the GFP cells were not uniformly providing VEGF to the VEGF-deficient RFP cells within the tumor microenvironment. These results suggest that the A-498-RFP-VEGFsh cells were less tumorigenic than the A-498-RFP-Consh cells and A-498-GFP cells. This evidence supports the hypothesis that VEGF promotes the tumorigenic phenotype within tumor cells through autocrine signaling independently of its pro-angiogenic function.
Figure 3.
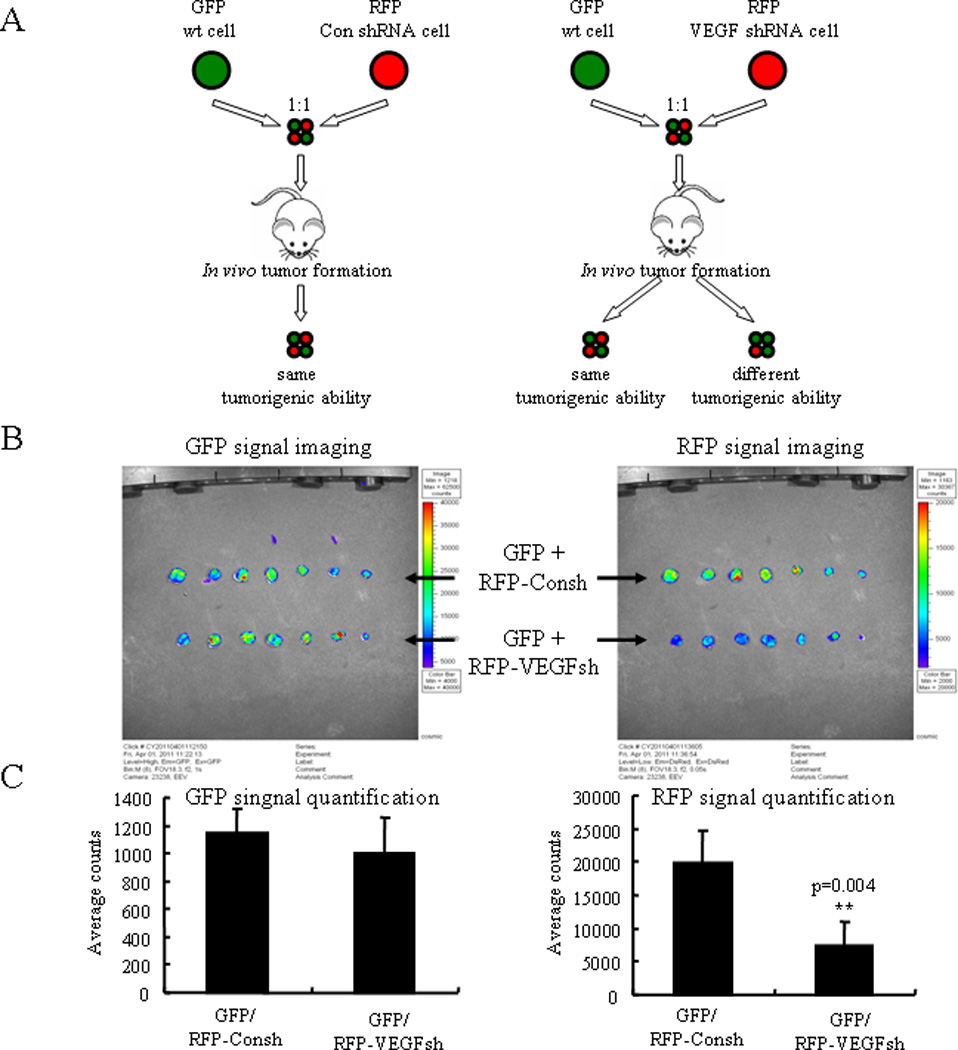
Mouse xenograft model of VEGF in tumorigenesis using a GFP/RFP combination system. A) Schematic illustration of the second in vivo experiment. GFP wild type cells and RFP-VEGFsh cells were mixed equally and injected subcutaneously into nude mice. Two kinds of results were possible. If the numbers of GFP and RFP cells in the tumors were similar, this would indicate that they have the same tumorigenic ability; if the numbers are not equal, this would indicate that they do not have same tumorigenic ability. GFP and RFP-Consh were used as a control group; they should have the same tumorigenic ability, and their cell numbers should be the same in the tumor. B) GFP and RFP signals imaged by Xeongen IVIS Fluorescent Imaging System. Another set of experimental result is shown in Supplementary Fig. S6B. C) Quantification of the imaging data. In the two groups GFP signal strengths were similar, but RFP signals were less in the GFP/RFP-VEGFsh group in comparison to the GFP/RFP-Consh group (p=0.004; n=12).
NRP-1 is a multi-functional receptor for several ligands including SEMA3, VEGF and TGF-β (6, 7, 12). Previous studies by our group proved NRP-1 mediated the migration and survival signaling of VEGF in endothelial cell (13, 14). We have also shown that NRP-1 controls renal cancer cell differentiation and tumorigenesis (8). We propose that NRP-1 mediates VEGF-induced tumorigenesis in cancer cells. NRP-1 doesn’t have a kinase function and several small-GTPases including RhoA and Rac-1 have been reported to be the down stream effector of NRP-1 (8, 13). To elucidate the effector through which VEGF promotes tumorigenesis downstream of NRP-1, we examined whether Ras, the classical small-GTPase and the connector of multiple signaling pathways, is involved in the VEGF/NRP-1 signaling transduction. We observed that VEGF can activate Ras in 786-O cells, which do not express VEGFR-1/2, in a time-dependent manner (Fig. 4A, left). Furthermore, overexpression of NRP-1 or addition of VEGF increased Ras activity in 786-O cells; whereas knockdown of either NRP-1 or VEGF was sufficient to decrease Ras activity (Fig. 4A, right). The phosphorylation of ERK1/2 and AKT proteins which are the down stream of active Ras signaling, were up-regulated by VEGF stimulation and down-regulated by NRP-1/VEGF siRNA knockdown (Supplementary Fig. S8A, B). Next we sought to determine whether introducing the dominant active mutant k-Ras (G12V) to VEGF shRNA expressing cells could override the differentiation phenotype. Indeed, as shown in Fig 4C, k-Ras (G12V) expression was sufficient to reverse the differentiation effect of VEGF knockdown in 786-O cells (Fig. 4B). MEK1 inhibitor PD98059 could also induce the 786-O cells to express the differentiation markers (Supplementary Fig. S8C). To further validate the role of the NRP-1 in the Ras activation by VEGF, we showed in a loss of function experiment that knockdown of NRP-1 in 786-O cells eliminated the Ras activation by VEGF (Fig. 4C left). In comparison, in a gain of function experiment, we utilized the low NRP-1 and VEGF expressing breast cancer cell line MCF-7 and showed that both overexpression of NRP-1 and VEGF were required to activate Ras (Fig. 4C right). Taken together, our data suggest that VEGF functions as a tumor-promoting gene through NRP-1 in a VEGFR-1/2 independent manner and signals through the downstream effector Ras.
Figure 4.
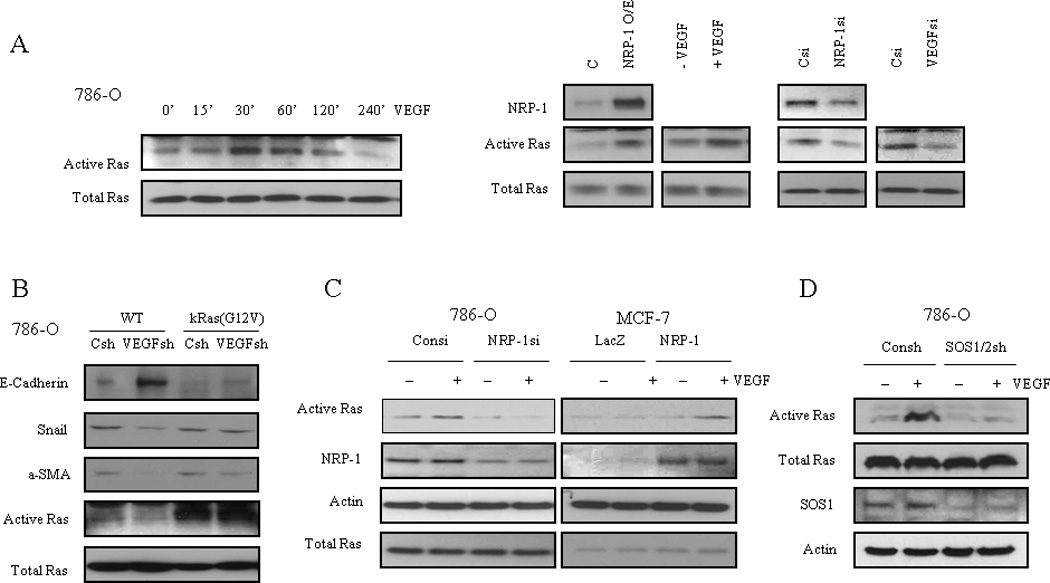
VEGF activation of Ras is NRP-1 dependent. A) Left: VEGF (10ng/ml) activates Ras in a time dependent manner in 786-O cells. Right: Gain of NRP-1 or VEGF increases Ras activityin 786-O cells; loss of NRP-1 or VEGF decreases Ras activity in 786-O cells. B) Introduction of kRas G12V mutation reverses the differentiation effect of VEGF knockdown in 786-O cells. C) Left: Loss of NRP-1 in 786-O cells eliminates the Ras activation by VEGF; right: Gain of NRP-1 plus VEGF in MCF-7 cells activates Ras. D) VEGF/NRP-1 activation of Ras is SOS1/2 dependent. Knockdown of SOS1/2 inhibited Ras activation by VEGF in 786-O cells.
Ras activity is controlled by guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs). We utilized a shRNA capable of knocking down both SOS1 and 2, which are RasGEFs, and demonstrated that VEGF could not activate Ras in the absence of SOS1/2 (Fig. 4D). Additionally, knockdown of SOS1/2 induced cancer cells to express differentiation markers consistent with VEGF knockdown (Supplementary Fig. S9). These results indicate that SOS proteins may be required for the VEGF-induced Ras activation. It is still not clear whether NRP-1 mediates Ras activation directly or not, and the exact mechanism requires further study. Intracellular and extracellular domains are both important for NRP-1 function. It is known that the intracellular domain of NRP-1 does not possess a kinase function, and GIPC is the only known NRP-1 intracellular domain interacting protein so far described (15). Also, evidence exists that the extracellular domain of NRP-1 is responsible for binding with several transmembrane proteins (e.g. Plexins, Integrin-alpha5) and is crucial for NRP-1 function (16, 17). Given that these NRP-1 association proteins have been linked to Ras activation (18, 19), it is presumable that NRP-1 may need adapters or partner proteins to activate Ras.
In this report we demonstrate that VEGF produced by tumor cells can act in an autocrine manner on the tumor cells themselves. This novel in vivo finding extends our understanding regarding the role of VEGF in tumorigenesis, which may translate into modified cancer treatments in the near future. Furthermore, our recent research results showed that the way of autocrine/endogenesis VEGF acting in the cells differed from the paracrine/exogenous VEGF (20). The paracrine VEGF can be more easily blocked by the antibodies than the autocrine VEGF. A future goal is to develop a more efficient method of blocking VEGF and its associated mediators and prevent VEGF from acting in an autocrine fashion on tumor cells to promote tumorigenesis.
Supplementary Material
Acknowledgements
We offer our sincere thanks to Drs. H. Dvorak, K. Alitalo, D. Cheresh, and S. Ananth Karumanchi for their critical comments and helpful suggestions; and Dr. Luke H. Hoeppner for critically reviewing the manuscript.
Grant Support:
This work is partly supported by NIH grants CA78383, CA150190 and a generous gift from Bruce and Martha Atwater Foundation to DM. DM is a visiting Professor of King Saud University.
Footnotes
No potential conflicts of interest were disclosed.
Declarations: While we were preparing this manuscript, another group reported that VEGF could regulate the initiation and stemness of skin tumors (11).
Reference
- 1.Folkman J, Merler E, Abernathy C, Williams G. Isolation of a tumor factor responsible for angiogenesis. J Exp Med. 1971;133:275–288. doi: 10.1084/jem.133.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrara N. VEGF: an update on biological and therapeutic aspects. Curr Opin Biotechnol. 2000;11:617–624. doi: 10.1016/s0958-1669(00)00153-1. [DOI] [PubMed] [Google Scholar]
- 3.Van Meter ME, Kim ES. Bevacizumab: current updates in treatment. Curr Opin Oncol. 2010;22:586–591. doi: 10.1097/CCO.0b013e32833edc0c. [DOI] [PubMed] [Google Scholar]
- 4.Escudier B, Pluzanska A, Koralewski P, Ravaud A, Bracarda S, Szczylik C, et al. Bevacizumab plus interferon alfa-2a for treatment of metastatic renal cell carcinoma: a randomised, double-blind phase III trial. Lancet. 2007;370:2103–2111. doi: 10.1016/S0140-6736(07)61904-7. [DOI] [PubMed] [Google Scholar]
- 5.Grepin R, Guyot M, Jacquin M, Durivault J, Chamorey E, Sudaka A, et al. Acceleration of clear cell renal cell carcinoma growth in mice following bevacizumab/Avastin treatment: the role of CXCL cytokines. Oncogene. 2011 doi: 10.1038/onc.2011.360. [DOI] [PubMed] [Google Scholar]
- 6.Soker S, Fidder H, Neufeld G, Klagsbrun M. Characterization of novel vascular endothelial growth factor (VEGF) receptors on tumor cells that bind VEGF165 via its exon 7-encoded domain. J Biol Chem. 1996;271:5761–5767. doi: 10.1074/jbc.271.10.5761. [DOI] [PubMed] [Google Scholar]
- 7.Bagri A, Tessier-Lavigne M, Watts RJ. Neuropilins in tumor biology. Clin Cancer Res. 2009;15:1860–1864. doi: 10.1158/1078-0432.CCR-08-0563. [DOI] [PubMed] [Google Scholar]
- 8.Cao Y, Wang L, Nandy D, Zhang Y, Basu A, Radisky D, et al. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008;68:8667–8672. doi: 10.1158/0008-5472.CAN-08-2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bachelder RE, Crago A, Chung J, Wendt MA, Shaw LM, Robinson G, et al. Vascular endothelial growth factor is an autocrine survival factor for neuropilin-expressing breast carcinoma cells. Cancer Res. 2001;61:5736–5740. [PubMed] [Google Scholar]
- 10.Lichtenberger BM, Tan PK, Niederleithner H, Ferrara N, Petzelbauer P, Sibilia M. Autocrine VEGF signaling synergizes with EGFR in tumor cells to promote epithelial cancer development. Cell. 2010;140:268–279. doi: 10.1016/j.cell.2009.12.046. [DOI] [PubMed] [Google Scholar]
- 11.Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature. 2011;478:399–403. doi: 10.1038/nature10525. [DOI] [PubMed] [Google Scholar]
- 12.Cao Y, Szabolcs A, Dutta SK, Yaqoob U, Jagavelu K, Wang L, et al. Neuropilin-1 mediates divergent R-Smad signaling and the myofibroblast phenotype. J Biol Chem. 2010;285:31840–31848. doi: 10.1074/jbc.M110.151696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang L, Zeng H, Wang P, Soker S, Mukhopadhyay D. Neuropilin-1-mediated vascular permeability factor/vascular endothelial growth factor-dependent endothelial cell migration. J Biol Chem. 2003;278:48848–48860. doi: 10.1074/jbc.M310047200. [DOI] [PubMed] [Google Scholar]
- 14.Wang L, Dutta SK, Kojima T, Xu X, Khosravi-Far R, Ekker SC, et al. Neuropilin-1 Modulates p53/Caspases Axis to Promote Endothelial Cell Survival. PLoS ONE. 2007;2:e1161. doi: 10.1371/journal.pone.0001161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cai H, Reed RR. Cloning and characterization of neuropilin-1-interacting protein: a PSD-95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J Neurosci. 1999;19:6519–6527. doi: 10.1523/JNEUROSCI.19-15-06519.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakamura F, Tanaka M, Takahashi T, Kalb RG, Strittmatter SM. Neuropilin-1 extracellular domains mediate semaphorin D/III-induced growth cone collapse. Neuron. 1998;21:1093–1100. doi: 10.1016/s0896-6273(00)80626-1. [DOI] [PubMed] [Google Scholar]
- 17.Valdembri D, Caswell PT, Anderson KI, Schwarz JP, Konig I, Astanina E, et al. Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol. 2009;7:e25. doi: 10.1371/journal.pbio.1000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohm B, Rahim B, Kleiber B, Hovatta I, Puschel AW. The semaphorin 3A receptor may directly regulate the activity of small GTPases. FEBS Lett. 2000;486:68–72. doi: 10.1016/s0014-5793(00)02240-7. [DOI] [PubMed] [Google Scholar]
- 19.Schlaepfer DD, Hanks SK, Hunter T, van der Geer P. Integrin-mediated signal transduction linked to Ras pathway by GRB2 binding to focal adhesion kinase. Nature. 1994;372:786–791. doi: 10.1038/372786a0. [DOI] [PubMed] [Google Scholar]
- 20.E G, Cao Y, Bhattacharya S, Dutta S, Wang E, Mukhopadhyay D. Endogenous vascular endothelial growth factor-A (VEGF-A) maintains endothelial cell homeostasis by regulating VEGF receptor-2 transcription. J Biol Chem. 2012;287:3029–3041. doi: 10.1074/jbc.M111.293985. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.