

Abstract
This article focuses on the underlying mechanisms contributing to sleep-disordered breathing. Obstructive sleep apnea (OSA) is the most common sleep-related breathing disorder and is characterized by repetitive narrowing or collapse of the pharyngeal airway during sleep. Conversely, central sleep apnea (CSA), highly prevalent in congestive heart failure, is distinguished by a lack of drive to breathe during sleep resulting in repetitive periods of insufficient ventilation. Both lead to compromised gas exchange, impaired sleep continuity, catecholamine surges and are associated with major co-morbidities including excessive daytime sleepiness and increased risk of cardiovascular disease. While OSA and CSA exist on a spectrum of sleep-disordered breathing, the two entities have overlap in their underlying pathophysiologies. This brief review summarizes the etiology and current understanding of OSA and CSA pathophysiology, the role of the cardiovascular system may play in contributing to disease pathology and, highlights the likely substantial overlap that exists between the various forms of sleep-disordered breathing.
Keywords: arousal, upper airway, ventilatory control stability, control of breathing, apnea threshold
MECHANISMS OF OBSTRUCTIVE SLEEP APNEA
Repetitive cessation of breathing during sleep has substantial impact on the cardiovascular system, emphasizing the importance of understanding the underlying pathophysiology of these disorders. Obstructive sleep apnea (OSA) is a common disorder, characterized by repetitive upper airway collapse during sleep. The airway obstruction results in either greatly reduced (hypopnea) or absent (apnea) ventilation, despite persisting respiratory efforts, such that ventilatory requirements are not met. As a result, hypoxemia and hypercapnia develop, which further stimulate respiratory effort. However, without spontaneous airway opening the increased drive is ineffective at increasing ventilation. Thus, the apnea/hypopnea typically continues until the patient arouses from sleep and terminates the obstruction. Following airway re-opening, hyperventilation occurs to reverse the blood gas disturbances that developed during the respiratory event. The patient then returns to sleep and another obstruction develops (Figure 1). The repetitive nature of these events result in significant sleep fragmentation, which along with intermittent hypoxia likely result in the excessive daytime sleepiness, fatigue (1), neurocognitive dysfunction (2) and increased risk for car accidents (3). As shall be reviewed in detail in this series, OSA has also been implicated in the development of cardiovascular disease likely via blood gas abnormalities, autonomic changes and oxidative stress among other causes.
Figure 1. Obstructive Sleep Apnea.
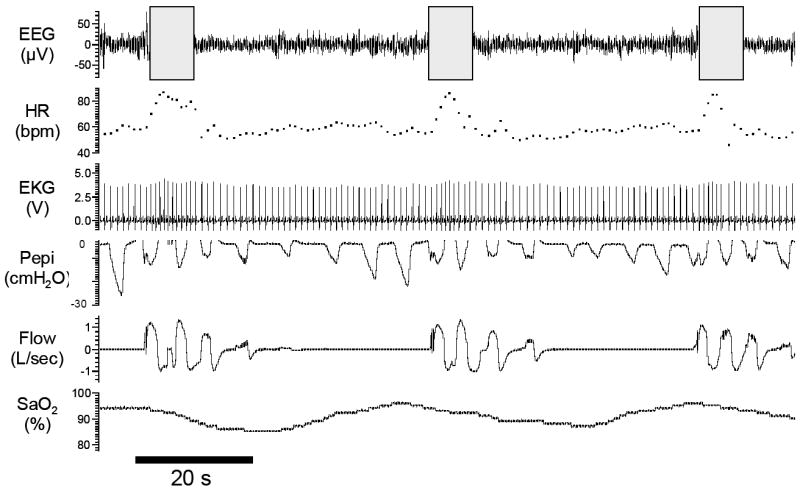
Polysomnographic example of obstructive sleep apnea from an experimental study in a patient with severe disease (apnea/hypopnea index= 75 events per hour). Repeated oxygen desaturations occu due to an absence of airflow (apnea) despite continual breathing efforts (Pepi). Note the associated surges in heart rate that occur with apnea termination and arousal from stage 1 sleep (grey boxes). EEG= electroencephalogram (C3-A2), HR= heart rate, EKG= Electrocardiogram, Pepi= pressure at the level of the epiglottis (as a measure of respiratory effort), Flow measured via nasal mask and pneumotachograph, SaO2= Arterial blood oxygen saturation.
Upper Airway Anatomy
A major abnormality in patients with OSA is a collapsible pharyngeal airway. The degree of collapsibility of a particular airway can be measured by calculation of the pharyngeal critical closing pressure, or Pcrit. Pcrit is the airway pressure below which airway collapse occurs and is therefore usually positive or zero for OSA patients and negative for healthy people, although some overlap between groups exists (4). In most patients the increase airway collapsibility is, at least in part, due to the cross sectional area of the upper airway being smaller in OSA patients than in control subjects (5-7). Fat deposition around the airway is one possible mechanism whereby obesity may predispose to apnea and moreover how some of the cardiovascular manifestations of obesity may be mediated. However, other anatomical factors also likely determine airway collapsibility such as airway length, lateral pharyngeal wall thickness and tongue volume (8, 9).
Upper Airway Dilator Muscle Activity
If impaired airway anatomy were the sole explanation for OSA, then patients would obstruct during both wakefulness and sleep whereas clearly OSA only occurs during sleep. This is largely a result of the high activity of upper airway dilator muscles during wakefulness (10). There are numerous airway dilator muscles which stiffen or dilate various regions of the upper airway and most of these dilator muscles have reduced activity during sleep (11). The largest and most extensively studied airway dilator muscle is the genioglossus, which forms the bulk of the tongue. The genioglossus receives input from sleep/wake centers of the brain, but unlike many other airway dilator muscles it also receives input from the respiratory pattern generating neurons, chemoreceptors and negative pressure receptors in the airway (12-16). Thus at sleep onset, the activity of the genioglossus is reduced (17-19), but as sleep progresses and airway resistance and PETCO2 rise, genioglossus activity returns, often to a level greater than was present during wakefulness (18). Therefore, if genioglossus activity is higher than wakefulness why does the upper airway still collapse during sleep? Collapse likely occurs because the activity of many other dilator muscles still have greatly reduced activity during sleep due to their minimal input from respiratory drive centers and negative pressure receptors. It is also possible that the airway is more prone to collapse because other factors have also changed from wake to sleep such as reduced end-expiratory lung volume (discussed below) or other currently under-recognized factors.
Lung Volume
The airway cross sectional area has long been recognized to increase from residual volume to total lung capacity (20-22). However, more recently, airway collapsibility (23), sleep-disordered breathing severity (24) and continuous positive airway pressure (CPAP) requirements (25) have been shown to be reduced with experimentally induced increases in end-expiratory lung volume during sleep. This information, combined with the observation that healthy normal subjects have between a 200 and 400cc reduction in lung volume from wakefulness to sleep (26, 27), have led to the notion that lung volume changes during sleep likely contribute to OSA pathogenesis. The mechanisms by which increasing lung volume influences the upper airway have been minimally investigated. However, increased lung volume may act by both dilating and stiffening the pharyngeal airway (28). Low lung volume may also contribute to rapid and marked oxygen desaturations for any given respiratory perturbation. Further research is clearly required to investigate the magnitude of sleep related changes in lung volume in OSA patients and the magnitude and effects of such changes.
Ventilatory Control Stability
Early studies of OSA treatment with tracheostomy reported persistent cyclical oscillations in breathing with central apnea-hyperpnea during sleep (29) and led to the suggestion that central breathing control may be abnormal in obstructive sleep apnea. These oscillations have received attention in recent years with OSA patients being found to have more unstable respiratory controllers than healthy controls (30-32). The way in which the central respiratory stability is thought to influence upper airway stability is by promoting periods of hypoventilation. During hypoventilation the activity of both respiratory pump muscles (diaphragm) and upper airway dilator muscles (such as genioglossus) have reduced activity (Figure 2). Therefore, a waxing-waning pattern of central respiratory drive in an individual with an upper airway prone to collapse will predispose to apnea/hypopnea during the hypoventilation due to upper airway hypotonia. The proportion of patients in whom respiratory stability contributes to their OSA is currently unknown but is the focus of ongoing investigation (33).
Figure 2. Reduced Central Respiratory Drive May Lead to Both Central Apnea and Upper Airway Closure.
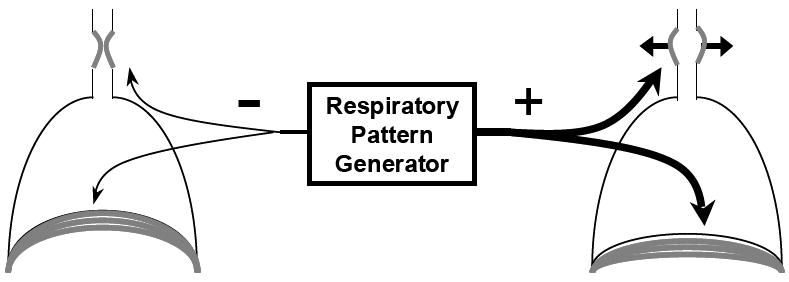
Schematic demonstrating one of the possible links between central sleep apnea and airway obstruction. In the left hand diagram respiratory pattern generator output to both the diaphragm and upper airway dilator muscles are reduced resulting in central apnea and airway closure. In the example on the right, respiratory pattern generator output to both muscles is increased resulting in sufficient ventilation. Note than in obstructive sleep apnea a mismatch between the respiratory pattern generator output to the diaphragm versus the upper airway dilator muscles may contribute to airway collapse.
Arousal from Sleep
Apneas and hypopneas are in many cases terminated with a brief awakening (arousal) from sleep (34). In some cases this likely represents the only way in which the airway can re-open and in this situation arousal is a life saving event. However, when arousal does occur, it enhances the hyperventilation following termination of the respiratory event (34, 35) and thus the arousal can yield hypocapnia and further airway obstruction on return to sleep. However, cortical arousal does not always accompany termination of a respiratory event as at least some of the time patients are able to restore ventilation without arousal. Thus, if arousal occurs very easily (low respiratory effort causes arousal), and is associated with a marked hyperventilatory response, then it may contribute to apnea pathogenesis. Alternatively, if arousal occurs as a last resort to open the airway it may represent a life saving event, at least by preventing severe hypoxemia.
CONSEQUENCES OF OSA ON THE CARDIOVASCULAR SYSTEM
There are several possible mechanisms by which OSA may lead to adverse cardiovascular outcomes. During each obstructive apnea/hypopnea, intrathoracic pressure can become very negative (up to −100 cmH2O), which results in increased cardiac afterload (via increased left ventricular transmural pressure and thus left ventricular wall stress/tension), decreased stroke volume and increased sympathetic activity <cite Malhotra NEJM 2003>. Arterial oxygen saturation also falls during obstructive respiratory events potentially leading to ischemia-reperfusion type injury. Upon arousal from sleep, sympathetic activity, blood pressure, cardiac output and heart rate rapidly increase, resulting in increased oxygen demand by the heart at a time when arterial oxygen saturation is lowest (see Figure 1 for example of increased HR associated with arousal apnea termination). In addition to these changes, OSA patients may have increased platelet activation and aggregation (36, 37), alterations in endothelial function (38) and increased mediators of inflammation (38, 39) that may improve with CPAP treatment. Thus, there are several physiologically plausible pathways by which OSA might increase the risk for cardiovascular disease. The evidence linking OSA with cardiovascular disease and its risk factors will be discussed elsewhere in this series.
MECHANISMS OF CENTRAL SLEEP APNEA
The term central sleep apnea (CSA), defined by cessation of airflow without respiratory effort, is often used to describe several distinct yet interrelated conditions. These include high-altitude periodic breathing, drug or substance-induced CSA, idiopathic CSA, and Cheyne-Stokes breathing (CSB) (Figure 3) (40). Unlike the ongoing respiratory efforts against a compromised upper airway that occur in OSA, the various forms of CSA are characterized by a lack of drive to breathe during sleep. However, as will be discussed, there is likely considerable overlap in the pathophysiology between CSA and OSA, making the distinction between these two disorders somewhat difficult. Typically however, CSA is considered to be the primary diagnosis when ≥50% of apneas are scored as central (i.e. >10 sec of breathing cessation in the absence of respiratory effort) on an overnight polysomnogram. Similar to OSA, CSA can impair sleep continuity due to frequent nighttime awakenings contributing to excessive daytime sleepiness and is associated with increased risk of adverse cardiovascular outcomes (41, 42).
Figure 3. Cheyne-Stokes Breathing.
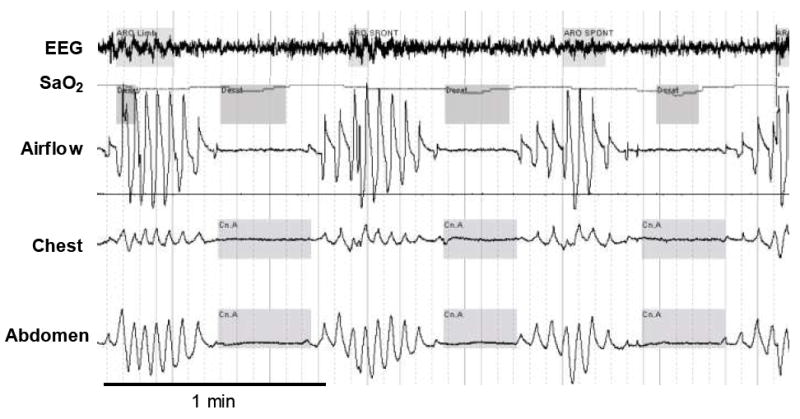
A polysomnographic example of a patient with heart failure experiencing Cheyne-Stokes breathing during sleep. EEG= electroencephalography, SaO2= arterial blood oxygen saturation. Note that arousal occurs at the peak of the crescendo (highlighted in grey on the EEG tracing) and that each oxygen desaturation (highlighted in grey on the SaO2 tracing) corresponds to the previous apnea due to prolonged circulatory delay. Adapted with permission from reference (47).
There is a substantial range in the prevalence between the various forms of CSA. For example, idiopathic CSA is relatively uncommon comprising <5% of patients referred to a sleep clinic (43). Conversely, within certain clinical populations, e.g. heart failure patients with left ventricular ejection fraction <45%, the presence of CSA is reported to be ~40% (44) and even higher (~60%) in patients with more severe heart disease (45). In addition, in some studies OSA is as common as CSA in CHF patients (44). In some cases, OSA is difficult to distinguish from CSA in CHF leading some to combine the entities using the term “sleep disordered breathing”. <cite Mary Maconald JCSM 2/2008>. The primary focus of the following section will be to highlight the possible links between the cardiovascular system and CSA pathophysiology (i.e. idiopathic CSA and CSB) and to describe the etiology and predominant underlying mechanisms for these forms of CSA. For further detail on the pathophysiology of other forms of CSA and sleep-related hypoventilation/hypoxia syndromes (i.e. obesity and congenital central hypoventilation syndromes) the reader is referred to recent review’s on these topics (46, 47).
Regulation of Breathing during Wakefulness and Sleep
During wakefulness, there are several inputs that regulate breathing that are either absent or down-regulated during sleep. For example, non-respiratory behavioral factors (i.e. volitional influences) and an independent increase in respiratory drive known as the “wakefulness drive to breathe” are removed during the transition from wakefulness to sleep (12). Thus, other inputs such as central (i.e. medullary neurons) and peripheral (i.e. carotid body) chemoreceptors along with feedback from respiratory muscle afferents become crucially important in regulating breathing during sleep. However, even in healthy individuals the responsiveness of the chemoreceptors to hypoxia and hypercapnia and the ability for stimulation of respiratory muscle afferents to increase ventilation is reduced, particularly during REM sleep (48-51).
These and other physiological changes that occur at sleep onset such as, decreased upper airway muscle tone, result in reduced ventilation from the waking level (18, 52, 53). While these changes occur in all individuals, if the removal of the wakefulness drive to breathe results in a marked, rapid reduction in ventilation, this in itself may be sufficient to promote hypopnea/apnea as a result of the delay required to elicit an appropriate compensatory response from the chemoreceptors (54). Indeed, dysrhythmic breathing patterns are not uncommon at sleep onset even in healthy individuals. However, over time if stable sleep is achieved the reduction in ventilation at sleep onset is coupled with a gradual rise in PaCO2 (~3-8 mmHg) and a new sleep specific CO2 set point (~45 mmHg) is established (55).
The loss of behavioral influences that strongly oppose breathing cessation during wakefulness and the reliance upon respiratory and chemical control feedback mechanisms place all individuals at risk of developing central apnea/hypopnea during sleep. Specifically, if the PaCO2 falls below a critical threshold known as the “apnea threshold”, central respiratory drive ceases. The apnea threshold is typically ~2-6 mmHg below the sleeping CO2 set point (i.e. similar to the wakefulness eucapnic PaCO2 level or marginally lower) (56-58). The degree of hypocapnia required to induce apnea appears to be crucially mediated by the peripheral chemoreceptors (59). As stated previously, brief arousal from sleep (3-15 sec) typically occurs with each sleep-disordered breathing event and the ease with which an individual wakes from sleep (arousal threshold) likely contributes to apnea pathophysiology. In addition, with arousal the sleeping CO2 set point (~45 mmHg) rapidly shifts to the wakefulness level (~40 mmHg) creating a state of relative hypercapnia and the wakefulness drive to breathe is reintroduced leading to hyperventilation. Upon resumption of sleep the arousal induced-ventilatory response leads to reduced PaCO2 such that central apnea may ensue, if the hypocapnia is sufficient to cross the CO2 apnea threshold (60). Thus, in addition to the difference between the apnea threshold and the CO2 set point being an important determinant of central apnea propensity, the change in ventilation that occurs with arousal is also of key importance.
Cheyne-Stokes Breathing
CSB is characterized by a waxing and waning pattern of breathing (Figure 3) and occurs most frequently in patients with congestive heart failure and left ventricular systolic dysfunction but may also occur in patients with stroke or renal failure (40). Symptoms may include poor sleep continuity, paroxysmal nocturnal dyspnea, orthopnea and daytime fatigue (61). The prolonged circulation time in these patients likely contributes to the longer cycle time associated with CSB (>45 secs) as compared to other forms of CSA (62). During sleep, arousal generally occurs at the peak of the crescendo pattern (63). Following the brief arousal and hyperventilation phase and as the individual falls back to sleep a relative state of hypocapnia ensues. The reduced chemical drive and sudden change of state importantly contribute to the decrescendo/ apnea phase, which most commonly occurs during lighter sleep (stages 1 and 2). However, although far less common than during sleep, in severe cases the CSB pattern can emerge during wakefulness which is associated with poor survival prognosis (45).
Unlike other forms of CSA which may have impaired ventilatory output and hypercapnia during wakefulness (i.e. obesity and congenital central hypoventilation syndromes), patients with heart failure and CSB are typically normocapnic or hypocapnic during wakefulness. Indeed, these patients have high ventilatory drive which likely contributes to CSB pathophysiology (61, 64). Further, there is minimal difference between the wakefulness and sleeping CO2 set points in the absence of a concurrent shift of the apnea threshold making these patients at greater risk of developing central apnea during sleep (65). Other factors such as prolonged circulation time resulting in a mismatch between arterial blood gas concentration with the respiratory controllers (54, 62) and impaired cerebrovascular reactivity to CO2 (66) may also contribute. In support of the role of the cardiovascular system contributing to CSB pathophysiology, ß-blocker therapy appears to dose-dependently decrease CSA in patients with chronic heart failure at least in some studies (67). In animals, pulmonary congestion activates afferent C fibers which can evoke a strong inhibitory reflex causing apnea followed by a period of hyperventilation that is likely to further destabilize breathing (68). In patients with heart failure there is a strong relationship between pulmonary capillary wedge pressure, hypocapnia and CSA severity (69) suggesting that similar mechanisms exist in humans.
Idiopathic Central Sleep Apnea
Patients who are normocapnic or hypocapnic during wakefulness and who have central apneas during sleep without the typical CSB pattern fall into the category of idiopathic CSA (ICSA). In ICSA, central apneas may occur without an obvious pattern or in a repetitive cyclical manner. The duration of the cycle time (~20 to 40 seconds) and the associated blood gas disturbances tend to be less than in CSB. Arousals also characteristically occur at the termination of central apnea in ICSA. Similar to CSB, hypopneas/apneas occur most commonly during lighter sleep (stages 1 and 2). Insomnia and daytime sleepiness are common symptoms. Elevated hypercapnic ventilatory responses (64, 70, 71) leading to increased propensity for hypocapnia and respiratory control instability are believed to be particularly important pathophysiolgical traits in ICSA. While arousal may play an important role in terminating hypopnea/apneas in patients with ICSA, the subsequent hypocapnia that develops appears to play an important role in further destabilization of breathing (60).
CARDIOVASCULAR DISEASE AND APNEA PATHOPHYSIOLOGY
The potential for sleep-disordered breathing to contribute to cardiovascular disease and vice versa and the underlying mechanisms are questions of ongoing investigation. For example, emerging animal data show an increased propensity for apnea and unstable breathing in response to acute elevations in left atrial pressure (72). The presence of intermittent hypoxia may further impair cardiac muscle (73) and therefore increase the risk of developing CSA. Given the link between conditions such as heart and renal failure and sleep-disordered breathing, a series of recent studies have investigated the potential role of fluid shifts contributing to apnea pathophysiology. Chiu and colleagues applied lower body positive pressure to healthy individuals during wakefulness and demonstrated an increase in neck circumference accompanied by increased upper airway resistance (74). A subsequent study employing similar methodology demonstrated reduced upper airway cross sectional area in healthy individuals (75). Furthermore, acute diuretic treatment to reduce airway edema in patients with heart failure and severe OSA reduced the apnea/hypopnea index further supporting the potential role of upper airway fluid accumulation in sleep-disordered breathing (76). To date, similar studies in patients with heart failure and CSA have not been conducted but in theory strategies to reduce fluid accumulation may be beneficial via reduced afferent C fiber stimulation.
Treating sleep-disordered breathing with CPAP improves hemodynamics in heart failure patients (77). However, the largest randomized trial to date in patients with heart failure and CSA did not demonstrate a beneficial survival effect of treatment (78). The efficacy of CPAP in resolving CSA however, varied dramatically between patients and a subsequent analysis of these patients revealed a beneficial effect on survival and other associated outcome measures in “responder” patients whose CSA was reduced below 15 events per hour of sleep (79). Atrial overdrive pacing to improve cardiac function and reduce apnea severity showed some promise in one study (80) but not others (81, 82). Other strategies such as cardiac resynchronization therapy have been shown to be beneficial in reducing the severity of both CSA (83) and OSA (84), presumably by reducing circulatory delay and stabilizing ventilatory control. A 6 month aerobic exercise training program has recently been shown to improve central but not obstructive apnea indices in patients with heart failure and sleep-disordered breathing which may potentially be mediated via exercise-induced changes in chemosensitivity (85).
THE SPECTRUM OF DISEASE: Similarities and differences between OSA and CSA
Although we have initially attempted to separate our description of the underlying mechanisms of OSA and CSA, significant overlap exists and most patients experience varying degrees of both. Indeed, central apneas lead to upper airway closure in the majority of instances (86). This is likely a reflection of the reduced drive to all respiratory muscles (including genioglossus) that occurs with central apnea (Figure 2). Furthermore, patients with OSA have unstable respiratory control (30-32, 87). Thus, the multiple pathophysiological factors that underlie sleep-disordered breathing importantly contribute to where an individual lies on the disease spectrum (i.e. the percentage of central versus obstructive apnea present, Figure 4). In addition, other factors such as alcohol use or body position (supines vs. lateral) for example, may further contribute to night-to-night sifts as to where an individual sits on the disease spectrum. On this note, there has been recent interest regarding the clinical observation that some patients with primarily OSA at baseline develop CSA upon commencement of CPAP treatment. Thus, something about rectifying the collapsible segment of the airway with CPAP shifts an individual from the obstructive to the central side of the disease spectrum (Figure 4). Treatment emergent CSA has been referred to as “complex sleep apnea” and approximately 5-15% of patients diagnosed with OSA develop CSA on the first night of CPAP treatment (88, 89).
Figure 4. The Spectrum of Disease.
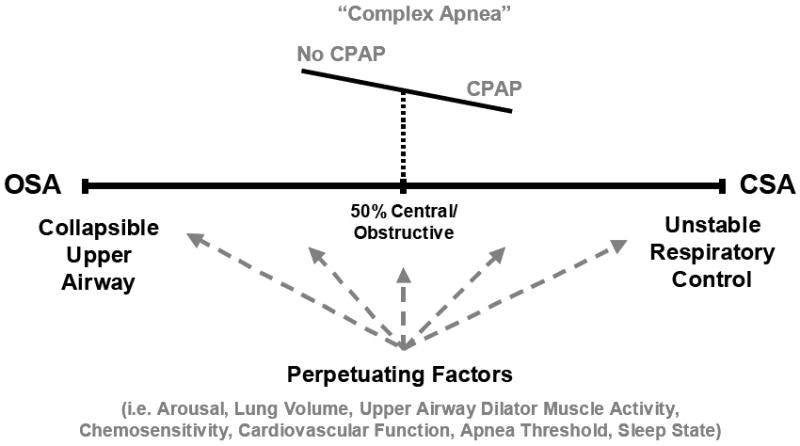
Schematic of the disease spectrum from OSA to CSA and some of the various perpetuating factors that likely influence where a particular patient lies (grey dashed arrows). A proposed example of complex apnea whereby a patient diagnosed with OSA at baseline switches to the CSA side of the disease spectrum upon commencement of CPAP due to currently unknown mechanisms is also depicted. Refer to the text for further detail.
While studies examining the underlying mechanisms for complex sleep apnea are lacking, potential contributing factors may include increased arousals during CPAP titration leading to unstable breathing, stretch reflex-induced apnea due to increased lung volume effects (especially if CPAP is over-titrated), or increased efficiency of CO2 excretion (via reduced upper airway resistance) rendering the patient vulnerable to crossing the apnea threshold. Central apneas typically resolve with ongoing treatment, although their clinical significance remains unclear. Nonetheless, adaptive servo ventilation appears to be a beneficial treatment option for these patients (90). In support of the notion that complex apnea is not a unique pathophysiological condition, but instead, may simply represent a shift on the disease spectrum of a group that lie further towards the midpoint prior to treatment (Figure 4), a recent study suggests that important risk factors are a history of cardiac disease and the presence of some CSA at baseline (89). Clearly, physiological studies to investigate these possibilities are required.
While significant pathophysiological overlap clearly exists between CSA and OSA, and both tend to worsen during lighter stages of sleep (stage 1 and 2) and improve somewhat during deeper sleep stages (stages 3 and 4), they differ considerably during REM sleep. In OSA, hypopneas and apneas worsen during REM (91). Indeed, some patients develop OSA exclusively during REM sleep (92). Decreased upper airway muscle tone (93, 94) and reduced chemosensitivity (49, 50) likely contribute to these REM related effects in OSA. On the other hand, in CSA, apnea severity tends to markedly improve during REM sleep (47), likely due to the increased respiratory neuronal activity that occurs in this sleep state (95).
SUMMARY AND IMPORTANCE OF IMPROVED UNDERSTANDING OF UNDERLYING APNEA MECHANISMS
The stoppages in breathing and the associated events that occur during sleep-disordered breathing clearly have a substantial impact on the cardiovascular system. The underlying mechanisms that contribute to both OSA and CSA are multifactorial. However, both may share common features. Important contributing factors likely include upper airway anatomy, upper airway muscle activity, respiratory control stability, lung volume and arousal from sleep. Cardiovascular disease may also importantly contribute to sleep-disordered breathing pathophysiology particularly in the development of CSA although, recent evidence implicating the role of excess fluid and impaired upper airway function also supports an important role in OSA pathogenesis. Where an individual sits on the disease spectrum and the various pathophysiologal determinants that contribute to the type and severity of sleep-disordered breathing may vary considerably between patients. CPAP treatment for OSA is effective for most patients however, a significant minority may experience a shift towards CSA upon commencement of CPAP. Furthermore, CPAP is only partially effective in treating CSA for most patients although, newer devices such as adaptive servoventilation devices offer promise. The ultimate treatment goal remains to develop approaches that enable determination of the relative importance of the various pathophysiological causes of sleep-disordered breathing on an individual patient basis and to develop novel therapeutic treatment options accordingly (96, 97).
Acknowledgments
SUPPORT: Dr Eckert is a recipient of the Thoracic Society of Australia and New Zealand/Allen and Hanbury’s respiratory research fellowship. Dr. Malhotra is funded by NIH P50 HL060292-09, AG024837-01, RO1-HL73146 and AHA Established Investigator Award. Dr Jordan is supported by the AHA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Danny J. Eckert, Email: deckert@rics.bwh.harvard.edu.
Atul Malhotra, Email: amalhotra1@partners.org.
Amy S. Jordan, Email: ajordan@rics.bwh.harvard.edu.
References
- 1.Punjabi NM, O’Hearn DJ, Neubauer DN, Nieto FJ, Schwartz AR, Smith PL, Bandeen-Roche K. Modeling hypersomnolence in sleep-disordered breathing. A novel approach using survival analysis. Am J Respir Crit Care Med. 1999;159:1703–1709. doi: 10.1164/ajrccm.159.6.9808095. [DOI] [PubMed] [Google Scholar]
- 2.Kim HC, Young T, Matthews CG, Weber SM, Woodward AR, Palta M. Sleep-disordered breathing and neuropsychological deficits. A population-based study. Am J Respir Crit Care Med. 1997;156:1813–1819. doi: 10.1164/ajrccm.156.6.9610026. [DOI] [PubMed] [Google Scholar]
- 3.Young T, Blustein J, Finn L, Palta M. Sleep-disordered breathing and motor vehicle accidents in a population- based sample of employed adults. Sleep. 1997;20:608–613. doi: 10.1093/sleep/20.8.608. [DOI] [PubMed] [Google Scholar]
- 4.Patil SP, Schneider H, Marx JJ, Gladmon E, Schwartz AR, Smith PL. Neuromechanical control of upper airway patency during sleep. J Appl Physiol. 2007;102:547–556. doi: 10.1152/japplphysiol.00282.2006. [DOI] [PubMed] [Google Scholar]
- 5.Schwab RJ, Gupta KB, Gefter WB, Metzger LJ, Hoffman EA, Pack AI. Upper airway and soft tissue anatomy in normal subjects and patients with sleep-disordered breathing. Significance of the lateral pharyngeal walls. Am J Respir Crit Care Med. 1995;152:1673–1689. doi: 10.1164/ajrccm.152.5.7582313. [DOI] [PubMed] [Google Scholar]
- 6.Haponik EF, Smith PL, Bohlman ME, Allen RP, Goldman SM, Bleecker ER. Computerized tomography in obstructive sleep apnea. Correlation of airway size with physiology during sleep and wakefulness. Am Rev Respir Dis. 1983;127:221–226. doi: 10.1164/arrd.1983.127.2.221. [DOI] [PubMed] [Google Scholar]
- 7.Burger CD, Stanson AW, Sheedy PF, 2nd, Daniels BK, Shepard JW., Jr Fast-computed tomography evaluation of age-related changes in upper airway structure and function in normal men. Am Rev Respir Dis. 1992;145:846–852. doi: 10.1164/ajrccm/145.4_Pt_1.846. [DOI] [PubMed] [Google Scholar]
- 8.Malhotra A, Huang Y, Fogel RB, Pillar G, Edwards JK, Kikinis R, Loring SH, White DP. The male predisposition to pharyngeal collapse: Importance of airway length. Am J Respir Crit Care Med. 2002;166:1388–1395. doi: 10.1164/rccm.2112072. [DOI] [PubMed] [Google Scholar]
- 9.Schwab RJ, Pasirstein M, Pierson R, Mackley A, Hachadoorian R, Arens R, Maislin G, Pack AI. Identification of upper airway anatomic risk factors for obstructive sleep apnea with volumetric mri. Am J Respir Crit Care Med. 2003 doi: 10.1164/rccm.200208-866OC. In press. [DOI] [PubMed] [Google Scholar]
- 10.Mezzanotte WS, Tangel DJ, White DP. Waking genioglossal electromyogram in sleep apnea patients versus normal controls (a neuromuscular compensatory mechanism) J Clin Invest. 1992;89:1571–1579. doi: 10.1172/JCI115751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubin L, Davies RO. Mechanisms of airway hypotonia. In: Pack AI, editor. Sleep apnea: Pathogenesis, diagnosis and treatment. New York: Marcel Dekker; 2002. pp. 99–154. [Google Scholar]
- 12.Orem J. The nature of the wakefulness stimulus for breathing. Progress in clinical and biological research. 1990;345:23–30. discussion 31. [PubMed] [Google Scholar]
- 13.Onal E, Lopata M, O’Connor TD. Diaphragmatic and genioglossal electromyogram responses to isocapnic hypoxia in humans. Am Rev Respir Dis. 1981;124:215–217. doi: 10.1164/arrd.1981.124.3.215. [DOI] [PubMed] [Google Scholar]
- 14.Onal E, Lopata M, O’Connor TD. Diaphragmatic and genioglossal electromyogram responses to co2 rebreathing in humans. J Appl Physiol. 1981;50:1052–1055. doi: 10.1152/jappl.1981.50.5.1052. [DOI] [PubMed] [Google Scholar]
- 15.Horner RL, Innes JA, Holden HB, Guz A. Afferent pathway(s) for pharyngeal dilator reflex to negative pressure in man: A study using upper airway anaesthesia. J Physiol (Lond) 1991;436:31–44. doi: 10.1113/jphysiol.1991.sp018537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Horner RL, Innes JA, Murphy K, Guz A. Evidence for reflex upper airway dilator muscle activation by sudden negative airway pressure in man. J Physiol (Lond) 1991;436:15–29. doi: 10.1113/jphysiol.1991.sp018536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mezzanotte WS, Tangel DJ, White DP. Influence of sleep onset on upper-airway muscle activity in apnea patients versus normal controls. Am J Respir Crit Care Med. 1996;153:1880–1887. doi: 10.1164/ajrccm.153.6.8665050. [DOI] [PubMed] [Google Scholar]
- 18.Worsnop C, Kay A, Pierce R, Kim Y, Trinder J. Activity of respiratory pump and upper airway muscles during sleep onset. J Appl Physiol. 1998;85:908–920. doi: 10.1152/jappl.1998.85.3.908. [DOI] [PubMed] [Google Scholar]
- 19.Lo YL, Jordan AS, Malhotra A, Wellman A, Heinzer RC, Eikerman M, Schory K, Dover L, White DP. The influence of wakefulness on pharyngeal airway muscle activity. Thorax. 2007;29:470–477. doi: 10.1136/thx.2006.072488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffstein V, Zamel N, Phillipson EA. Lung volume dependence of pharyngeal cross-sectional area in patients with obstructive sleep apnea. Am Rev Respir Dis. 1984;130:175–178. doi: 10.1164/arrd.1984.130.2.175. [DOI] [PubMed] [Google Scholar]
- 21.Bradley TD, Brown IG, Grossman RF, Zamel N, Martinez D, Phillipson EA, Hoffstein V. Pharyngeal size in snorers, nonsnorers, and patients with obstructive sleep apnea. New England Journal of Medicine. 1986;315:1327–1331. doi: 10.1056/NEJM198611203152105. [DOI] [PubMed] [Google Scholar]
- 22.Brown IG, Zamel N, Hoffstein V. Pharyngeal cross-sectional area in normal men and women. J Appl Physiol. 1986;61:890–895. doi: 10.1152/jappl.1986.61.3.890. [DOI] [PubMed] [Google Scholar]
- 23.Stanchina ML, Malhotra A, Fogel RB, Trinder J, Edwards JK, Schory K, White DP. The influence of lung volume on pharyngeal mechanics, collapsibility, and genioglossus muscle activation during sleep. Sleep. 2003;26:851–856. doi: 10.1093/sleep/26.7.851. [DOI] [PubMed] [Google Scholar]
- 24.Heinzer RC, Stanchina ML, Malhotra A, Jordan AS, Patel SR, Lo Y, Wellman A, Schory K, Dover L, White DP. Effect of increased lung volume on sleep disordered breathing in sleep apnoea patients. Thorax. 2006;61:435–439. doi: 10.1136/thx.2005.052084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heinzer RC, Stanchina ML, Malhotra A, Fogel RB, Patel SR, Jordan AS, Schory K, White DP. Lung volume and continuous positive airway pressure requirements in obstructive sleep apnea. Am J Respir Crit Care Med. 2005;172:114–117. doi: 10.1164/rccm.200404-552OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hudgel DW, Devadatta P. Decrease in functional residual capacity during sleep in normal humans. J Appl Physiol. 1984;57:1319–1322. doi: 10.1152/jappl.1984.57.5.1319. [DOI] [PubMed] [Google Scholar]
- 27.Ballard RD, Irvin CG, Martin RJ, Pak J, Pandey R, White DP. Influence of sleep on lung volume in asthmatic patients and normal subjects. J Appl Physiol. 1990;68:2034–2041. doi: 10.1152/jappl.1990.68.5.2034. [DOI] [PubMed] [Google Scholar]
- 28.Van de Graaff WB. Thoracic influence on upper airway patency. J Appl Physiol. 1988;65:2124–2131. doi: 10.1152/jappl.1988.65.5.2124. [DOI] [PubMed] [Google Scholar]
- 29.Onal E, Lopata M. Periodic breathing and the pathogenesis of occlusive sleep apneas. Am Rev Respir Dis. 1982;126:676–680. doi: 10.1164/arrd.1982.126.4.676. [DOI] [PubMed] [Google Scholar]
- 30.Younes M, Ostrowski M, Thompson W, Leslie C, Shewchuk W. Chemical control stability in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 2001;163:1181–1190. doi: 10.1164/ajrccm.163.5.2007013. [DOI] [PubMed] [Google Scholar]
- 31.Asyali MH, Berry RB, Khoo MC. Assessment of closed-loop ventilatory stability in obstructive sleep apnea. IEEE Trans Biomed Eng. 2002;49:206–216. doi: 10.1109/10.983454. [DOI] [PubMed] [Google Scholar]
- 32.Hudgel DW, Gordon EA, Thanakitcharu S, Bruce EN. Instability of ventilatory control in patients with obstructive sleep apnea. Am J Respir Crit Care Med. 1998;158:1142–1149. doi: 10.1164/ajrccm.158.4.9712105. [DOI] [PubMed] [Google Scholar]
- 33.Eckert DJ, Jordan AS, Wellman A, Smith SA, Malhotra A, White DP. Variability in physiological phenotypic traits in obstructive sleep apnea. Sleep Biol Rhythms. 2007;5:A3. [Google Scholar]
- 34.Younes MK. Role of arousals in the pathogenesis of obstructive sleep apnea. Am J Respir Crit Care Med. 2004;169:623–633. doi: 10.1164/rccm.200307-1023OC. [DOI] [PubMed] [Google Scholar]
- 35.Jordan AS, McEvoy RD, Edwards JK, Schory K, Yang CK, Catcheside PG, Fogel RB, Malhotra A, White DP. The influence of gender and upper airway resistance on the ventilatory response to arousal in obstructive sleep apnoea in humans. J Physiol. 2004;558:993–1004. doi: 10.1113/jphysiol.2004.064238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bokinsky G, Miller M, Ault K, Husband P, Mitchell J. Spontaneous platelet activation and aggregation during obstructive sleep apnea and its response to therapy with nasal continuous positive airway pressure. A preliminary investigation. Chest. 1995;108:625–630. doi: 10.1378/chest.108.3.625. [DOI] [PubMed] [Google Scholar]
- 37.Hui DS, Ko FW, Fok JP, Chan MC, Li TS, Tomlinson B, Cheng G. The effects of nasal continuous positive airway pressure on platelet activation in obstructive sleep apnea syndrome. Chest. 2004;125:1768–1775. doi: 10.1378/chest.125.5.1768. [DOI] [PubMed] [Google Scholar]
- 38.Phillips BG, Narkiewicz K, Pesek CA, Haynes WG, Dyken ME, Somers VK. Effects of obstructive sleep apnea on endothelin-1 and blood pressure. J Hypertens. 1999;17:61–66. doi: 10.1097/00004872-199917010-00010. [DOI] [PubMed] [Google Scholar]
- 39.Yokoe T, Minoguchi K, Matsuo H, Oda N, Minoguchi H, Yoshino G, Hirano T, Adachi M. Elevated levels of c-reactive protein and interleukin-6 in patients with obstructive sleep apnea syndrome are decreased by nasal continuous positive airway pressure. Circulation. 2003;107:1129–1134. doi: 10.1161/01.cir.0000052627.99976.18. [DOI] [PubMed] [Google Scholar]
- 40.American-Academy-of-Sleep-Medicine. International classification of sleep disorders: Diagnostic and coding manual. 2. Westchester, IL: 2005. [Google Scholar]
- 41.Lanfranchi PA, Somers VK, Braghiroli A, Corra U, Eleuteri E, Giannuzzi P. Central sleep apnea in left ventricular dysfunction: Prevalence and implications for arrhythmic risk. Circulation. 2003;107:727–732. doi: 10.1161/01.cir.0000049641.11675.ee. [DOI] [PubMed] [Google Scholar]
- 42.Javaheri S, Parker TJ, Liming JD, Corbett WS, Nishiyama H, Wexler L, Roselle GA. Sleep apnea in 81 ambulatory male patients with stable heart failure. Types and their prevalences, consequences, and presentations. Circulation. 1998;97:2154–2159. doi: 10.1161/01.cir.97.21.2154. [DOI] [PubMed] [Google Scholar]
- 43.Malhotra A, Berry RB, White DP. Central sleep apnea. In: Carney PR, Berry RB, Geyer JD, editors. Clinical sleep disorders. Philadelphia, PA: Lippincott Williams and Wilkins; 2004. pp. 331–346. [Google Scholar]
- 44.Javaheri S. Sleep disorders in systolic heart failure: A prospective study of 100 male patients. The final report. International journal of cardiology. 2006;106:21–28. doi: 10.1016/j.ijcard.2004.12.068. [DOI] [PubMed] [Google Scholar]
- 45.Brack T, Thuer I, Clarenbach CF, Senn O, Noll G, Russi EW, Bloch KE. Daytime cheyne-stokes respiration in ambulatory patients with severe congestive heart failure is associated with increased mortality. Chest. 2007;132:1463–1471. doi: 10.1378/chest.07-0121. [DOI] [PubMed] [Google Scholar]
- 46.Casey KR, Cantillo KO, Brown LK. Sleep-related hypoventilation/hypoxemic syndromes. Chest. 2007;131:1936–1948. doi: 10.1378/chest.06-2334. [DOI] [PubMed] [Google Scholar]
- 47.Eckert DJ, Jordan AS, Merchia P, Malhotra A. Central sleep apnea: Pathophysiology and treatment. Chest. 2007;131:595–607. doi: 10.1378/chest.06.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiegand L, Zwillich CW, White DP. Sleep and the ventilatory response to resistive loading in normal men. J Appl Physiol. 1988;64:1186–1195. doi: 10.1152/jappl.1988.64.3.1186. [DOI] [PubMed] [Google Scholar]
- 49.Douglas NJ, White DP, Weil JV, Pickett CK, Martin RJ, Hudgel DW, Zwillich CW. Hypoxic ventilatory response decreases during sleep in normal men. Am Rev Respir Dis. 1982;125:286–289. doi: 10.1164/arrd.1982.125.3.286. [DOI] [PubMed] [Google Scholar]
- 50.White DP, Douglas NJ, Pickett CK, Weil JV, Zwillich CW. Hypoxic ventilatory response during sleep in normal premenopausal women. Am Rev Respir Dis. 1982;126:530–533. doi: 10.1164/arrd.1982.126.3.530. [DOI] [PubMed] [Google Scholar]
- 51.Douglas NJ, White DP, Weil JV, Pickett CK, Zwillich CW. Hypercapnic ventilatory response in sleeping adults. Am Rev Respir Dis. 1982;126:758–762. doi: 10.1164/arrd.1982.126.5.758. [DOI] [PubMed] [Google Scholar]
- 52.Kay A, Trinder J, Bowes G, Kim Y. Changes in airway resistance during sleep onset. J Appl Physiol. 1994;76:1600–1607. doi: 10.1152/jappl.1994.76.4.1600. [DOI] [PubMed] [Google Scholar]
- 53.Dunai J, Wilkinson M, Trinder J. Interaction of chemical and state effects on ventilation during sleep onset. J Appl Physiol. 1996;81:2235–2243. doi: 10.1152/jappl.1996.81.5.2235. [DOI] [PubMed] [Google Scholar]
- 54.Khoo MC, Gottschalk A, Pack AI. Sleep-induced periodic breathing and apnea: A theoretical study. J Appl Physiol. 1991;70:2014–2024. doi: 10.1152/jappl.1991.70.5.2014. [DOI] [PubMed] [Google Scholar]
- 55.Skatrud JB, Dempsey JA, Badr S, Begle RL. Effect of airway impedance on co2 retention and respiratory muscle activity during nrem sleep. J Appl Physiol. 1988;65:1676–1685. doi: 10.1152/jappl.1988.65.4.1676. [DOI] [PubMed] [Google Scholar]
- 56.Meza S, Mendez M, Ostrowski M, Younes M. Susceptibility to periodic breathing with assisted ventilation during sleep in normal subjects. J Appl Physiol. 1998;85:1929–1940. doi: 10.1152/jappl.1998.85.5.1929. [DOI] [PubMed] [Google Scholar]
- 57.Skatrud JB, Dempsey JA. Interaction of sleep state and chemical stimuli in sustaining rhythmic ventilation. J Appl Physiol. 1983;55:813–822. doi: 10.1152/jappl.1983.55.3.813. [DOI] [PubMed] [Google Scholar]
- 58.Dempsey JA. Crossing the apnoeic threshold: Causes and consequences. Experimental physiology. 2005;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- 59.Smith CA, Chenuel BJ, Henderson KS, Dempsey JA. The apneic threshold during non-rem sleep in dogs: Sensitivity of carotid body vs. Central chemoreceptors. J Appl Physiol. 2007;103:578–586. doi: 10.1152/japplphysiol.00017.2007. [DOI] [PubMed] [Google Scholar]
- 60.Xie A, Wong B, Phillipson EA, Slutsky AS, Bradley TD. Interaction of hyperventilation and arousal in the pathogenesis of idiopathic central sleep apnea. Am J Respir Crit Care Med. 1994;150:489–495. doi: 10.1164/ajrccm.150.2.8049835. [DOI] [PubMed] [Google Scholar]
- 61.Naughton M, Benard D, Tam A, Rutherford R, Bradley TD. Role of hyperventilation in the pathogenesis of central sleep apneas in patients with congestive heart failure. Am Rev Respir Dis. 1993;148:330–338. doi: 10.1164/ajrccm/148.2.330. [DOI] [PubMed] [Google Scholar]
- 62.Hall MJ, Xie A, Rutherford R, Ando S, Floras JS, Bradley TD. Cycle length of periodic breathing in patients with and without heart failure. Am J Respir Crit Care Med. 1996;154:376–381. doi: 10.1164/ajrccm.154.2.8756809. [DOI] [PubMed] [Google Scholar]
- 63.Trinder J, Merson R, Rosenberg JI, Fitzgerald F, Kleiman J, Douglas Bradley T. Pathophysiological interactions of ventilation, arousals, and blood pressure oscillations during cheyne-stokes respiration in patients with heart failure. Am J Respir Crit Care Med. 2000;162:808–813. doi: 10.1164/ajrccm.162.3.9806080. [DOI] [PubMed] [Google Scholar]
- 64.Solin P, Roebuck T, Johns DP, Walters EH, Naughton MT. Peripheral and central ventilatory responses in central sleep apnea with and without congestive heart failure. Am J Respir Crit Care Med. 2000;162:2194–2200. doi: 10.1164/ajrccm.162.6.2002024. [DOI] [PubMed] [Google Scholar]
- 65.Xie A, Skatrud JB, Puleo DS, Rahko PS, Dempsey JA. Apnea-hypopnea threshold for co2 in patients with congestive heart failure. Am J Respir Crit Care Med. 2002;165:1245–1250. doi: 10.1164/rccm.200110-022OC. [DOI] [PubMed] [Google Scholar]
- 66.Xie A, Skatrud JB, Khayat R, Dempsey JA, Morgan B, Russell D. Cerebrovascular response to carbon dioxide in patients with congestive heart failure. Am J Respir Crit Care Med. 2005;172:371–378. doi: 10.1164/rccm.200406-807OC. [DOI] [PubMed] [Google Scholar]
- 67.Tamura A, Kawano Y, Naono S, Kotoku M, Kadota J. Relationship between beta-blocker treatment and the severity of central sleep apnea in chronic heart failure. Chest. 2007;131:130–135. doi: 10.1378/chest.06-0919. [DOI] [PubMed] [Google Scholar]
- 68.Paintal AS. Vagal sensory receptors and their reflex effects. Physiol Rev. 1973;53:159–227. doi: 10.1152/physrev.1973.53.1.159. [DOI] [PubMed] [Google Scholar]
- 69.Solin P, Bergin P, Richardson M, Kaye DM, Walters EH, Naughton MT. Influence of pulmonary capillary wedge pressure on central apnea in heart failure. Circulation. 1999;99:1574–1579. doi: 10.1161/01.cir.99.12.1574. [DOI] [PubMed] [Google Scholar]
- 70.Solin P, Jackson DM, Roebuck T, Naughton MT. Cardiac diastolic function and hypercapnic ventilatory responses in central sleep apnoea. Eur Respir J. 2002;20:717–723. doi: 10.1183/09031936.02.00742002. [DOI] [PubMed] [Google Scholar]
- 71.Xie A, Rutherford R, Rankin F, Wong B, Bradley TD. Hypocapnia and increased ventilatory responsiveness in patients with idiopathic central sleep apnea. Am J Respir Crit Care Med. 1995;152:1950–1955. doi: 10.1164/ajrccm.152.6.8520761. [DOI] [PubMed] [Google Scholar]
- 72.Chenuel BJ, Smith CA, Skatrud JB, Henderson KS, Dempsey JA. Increased propensity for apnea in response to acute elevations in left atrial pressure during sleep in the dog. J Appl Physiol. 2006;101:76–83. doi: 10.1152/japplphysiol.01617.2005. [DOI] [PubMed] [Google Scholar]
- 73.Chen L, Zhang J, Gan TX, Chen-Izu Y, Hasday JD, Karmazyn M, Balke CW, Scharf SM. Left ventricular dysfunction and associated cellular injury in rats exposed to chronic intermittent hypoxia. J Appl Physiol. 2008;104:218–223. doi: 10.1152/japplphysiol.00301.2007. [DOI] [PubMed] [Google Scholar]
- 74.Chiu KL, Ryan CM, Shiota S, Ruttanaumpawan P, Arzt M, Haight JS, Chan CT, Floras JS, Bradley TD. Fluid shift by lower body positive pressure increases pharyngeal resistance in healthy subjects. Am J Respir Crit Care Med. 2006;174:1378–1383. doi: 10.1164/rccm.200607-927OC. [DOI] [PubMed] [Google Scholar]
- 75.Shiota S, Ryan CM, Chiu KL, Ruttanaumpawan P, Haight J, Arzt M, Floras JS, Chan C, Bradley TD. Alterations in upper airway cross-sectional area in response to lower body positive pressure in healthy subjects. Thorax. 2007;62:868–872. doi: 10.1136/thx.2006.071183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bucca CB, Brussino L, Battisti A, Mutani R, Rolla G, Mangiardi L, Cicolin A. Diuretics in obstructive sleep apnea with diastolic heart failure. Chest. 2007;132:440–446. doi: 10.1378/chest.07-0311. [DOI] [PubMed] [Google Scholar]
- 77.Arzt M, Bradley TD. Treatment of sleep apnea in heart failure. Am J Respir Crit Care Med. 2006;173:1300–1308. doi: 10.1164/rccm.200511-1745PP. [DOI] [PubMed] [Google Scholar]
- 78.Bradley TD, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson K, Belenkie I, Pfeifer M, Fleetham J, Hanly P, et al. Continuous positive airway pressure for central sleep apnea and heart failure. N Engl J Med. 2005;353:2025–2033. doi: 10.1056/NEJMoa051001. [DOI] [PubMed] [Google Scholar]
- 79.Arzt M, Floras JS, Logan AG, Kimoff RJ, Series F, Morrison D, Ferguson K, Belenkie I, Pfeifer M, Fleetham J, et al. Suppression of central sleep apnea by continuous positive airway pressure and transplant-free survival in heart failure: A post hoc analysis of the canadian continuous positive airway pressure for patients with central sleep apnea and heart failure trial (canpap) Circulation. 2007;115:3173–3180. doi: 10.1161/CIRCULATIONAHA.106.683482. [DOI] [PubMed] [Google Scholar]
- 80.Garrigue S, Bordier P, Jais P, Shah DC, Hocini M, Raherison C, Tunon De Lara M, Haissaguerre M, Clementy J. Benefit of atrial pacing in sleep apnea syndrome. N Engl J Med. 2002;346:404–412. doi: 10.1056/NEJMoa011919. [DOI] [PubMed] [Google Scholar]
- 81.Simantirakis EN, Schiza SE, Chrysostomakis SI, Chlouverakis GI, Klapsinos NC, Siafakas NM, Vardas PE. Atrial overdrive pacing for the obstructive sleep apnea-hypopnea syndrome. N Engl J Med. 2005;353:2568–2577. doi: 10.1056/NEJMoa050610. [DOI] [PubMed] [Google Scholar]
- 82.Luthje L, Unterberg-Buchwald C, Dajani D, Vollmann D, Hasenfuss G, Andreas S. Atrial overdrive pacing in patients with sleep apnea with implanted pacemaker. Am J Respir Crit Care Med. 2005;172:118–122. doi: 10.1164/rccm.200409-1258OC. [DOI] [PubMed] [Google Scholar]
- 83.Sinha AM, Skobel EC, Breithardt OA, Norra C, Markus KU, Breuer C, Hanrath P, Stellbrink C. Cardiac resynchronization therapy improves central sleep apnea and cheyne-stokes respiration in patients with chronic heart failure. Journal of the American College of Cardiology. 2004;44:68–71. doi: 10.1016/j.jacc.2004.03.040. [DOI] [PubMed] [Google Scholar]
- 84.Stanchina ML, Ellison K, Malhotra A, Anderson M, Kirk M, Benser ME, Tosi C, Carlisle C, Millman RP, Buxton A. The impact of cardiac resynchronization therapy on obstructive sleep apnea in heart failure patients: A pilot study. Chest. 2007;132:433–439. doi: 10.1378/chest.06-2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yamamoto U, Mohri M, Shimada K, Origuchi H, Miyata K, Ito K, Abe K, Yamamoto H. Six-month aerobic exercise training ameliorates central sleep apnea in patients with chronic heart failure. Journal of cardiac failure. 2007;13:825–829. doi: 10.1016/j.cardfail.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 86.Badr MS, Toiber F, Skatrud JB, Dempsey J. Pharyngeal narrowing/occlusion during central sleep apnea. J Appl Physiol. 1995;78:1806–1815. doi: 10.1152/jappl.1995.78.5.1806. [DOI] [PubMed] [Google Scholar]
- 87.Wellman A, Jordan AS, Malhotra A, Fogel RB, Katz ES, Schory K, Edwards JK, White DP. Ventilatory control and airway anatomy in obstructive sleep apnea. Am J Respir Crit Care Med. 2004;170:1225–1232. doi: 10.1164/rccm.200404-510OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Morgenthaler TI, Kagramanov V, Hanak V, Decker PA. Complex sleep apnea syndrome: Is it a unique clinical syndrome? Sleep. 2006;29:1203–1209. doi: 10.1093/sleep/29.9.1203. [DOI] [PubMed] [Google Scholar]
- 89.Lehman S, Antic NA, Thompson C, Catcheside PG, Mercer J, McEvoy RD. Central sleep apnea on commencement of continuous positive airway pressure in patients with a primary diagnosis of obstructive sleep apnea-hypopnea. J Clin Sleep Med. 2007;3:462–466. [PMC free article] [PubMed] [Google Scholar]
- 90.Morgenthaler TI, Gay PC, Gordon N, Brown LK. Adaptive servoventilation versus noninvasive positive pressure ventilation for central, mixed, and complex sleep apnea syndromes. Sleep. 2007;30:468–475. doi: 10.1093/sleep/30.4.468. [DOI] [PubMed] [Google Scholar]
- 91.Findley LJ, Wilhoit SC, Suratt PM. Apnea duration and hypoxemia during rem sleep in patients with obstructive sleep apnea. Chest. 1985;87:432–436. doi: 10.1378/chest.87.4.432. [DOI] [PubMed] [Google Scholar]
- 92.Kass JE, Akers SM, Bartter TC, Pratter MR. Rapid-eye-movement-specific sleep-disordered breathing: A possible cause of excessive daytime sleepiness. Am J Respir Crit Care Med. 1996;154:167–169. doi: 10.1164/ajrccm.154.1.8680674. [DOI] [PubMed] [Google Scholar]
- 93.Sauerland EK, Harper RM. The human tongue during sleep: Electromyographic activity of the genioglossus muscle. Exp Neurol. 1976;51:160–170. doi: 10.1016/0014-4886(76)90061-3. [DOI] [PubMed] [Google Scholar]
- 94.Wiegand L, Zwillich CW, Wiegand D, White DP. Changes in upper airway muscle activation and ventilation during phasic rem sleep in normal men. J Appl Physiol. 1991;71:488–497. doi: 10.1152/jappl.1991.71.2.488. [DOI] [PubMed] [Google Scholar]
- 95.Orem J. Neuronal mechanisms of respiration in rem sleep. Sleep. 1980;3:251–267. doi: 10.1093/sleep/3.3-4.251. [DOI] [PubMed] [Google Scholar]
- 96.Eckert DJ, Malhotra A. Pathophysiology of adult obstructive sleep apnea. Proc Am Thorac Soc. 2008;5:144–153. doi: 10.1513/pats.200707-114MG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.White DP. Pathogenesis of obstructive and central sleep apnea. Am J Respir Crit Care Med. 2005;172:1363–1370. doi: 10.1164/rccm.200412-1631SO. [DOI] [PubMed] [Google Scholar]