

Abstract
Oncogenic mutations in critical nodes of cellular signaling pathways have been associated with tumorigenesis and progression. The B-Raf protein kinase, a key hub in the canonical MAPK signaling cascade, is mutated in a broad range of human cancers and especially in malignant melanoma. The most prevalent B-RafV600E mutant exhibits elevated kinase activity and results in constitutive activation of the MAPK pathway, thus making it a promising drug target for cancer therapy. Herein, we described the development of novel B-RafV600E selective inhibitors via multi-step virtual screening and hierarchical hit optimization. Nine hit compounds with low micromolar IC50 values were identified as B-RafV600E inhibitors through virtual screening. Subsequent scaffold-based analogue searching and medicinal chemistry efforts significantly improved both the inhibitor potency and oncogene selectivity. In particular, compounds 22f and 22q possess nanomolar IC50 values with selectivity for B-RafV600E in vitro and exclusive cytotoxicity against B-RafV600E harboring cancer cells.
Introduction
The evolutionarily conserved Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) signaling pathway, relaying the proliferative signals generated by cell surface receptors and cytoplasmic signaling components into the nucleus, is a critical regulator of many cellular processes involved in cell proliferation, differentiation, and survival.1–3 Aberrant activation of the MAPK pathway represents a common indicator for a wide variety of proliferative diseases and especially cancer.4, 5 Moreover, accumulating data demonstrates that high-frequency activating mutation6–8 or amplification9–11 at several levels in this pathway greatly prompt multiple tumor oncogenic processes, such as cell immortalization, angiogenesis, invasive growth, metastases and drug resistance.12–14 Therefore, the Ras/Raf/MEK/ERK signaling cascade has become one of the primary candidates for novel molecular targeted cancer therapies.15–17
The Raf serine/threonine kinase family was initially identified as a cellular homolog of the retroviral oncogene v-Raf and consists of three isoforms named Raf-1 (C-Raf), A-Raf and B-Raf.18–20 Distinct from other Raf paralogs, B-Raf requires fewer post-translational modifications for optimal activation and possesses substantially greater basal activity.21, 22 In addition, B-Raf has also been shown to be the major Raf effector in the MAPK signaling cascade. 23–26 All of these unique properties in cellular physiology provide a molecular shortcut for B-Raf activation and greatly substantiate the potential of B-Raf to be a bona fide oncogene.27 The importance of B-Raf in oncogenesis is highlighted by the finding that it is mutated in approximately 7% of human cancers, with the highest incidence in malignant melanoma (60%–70%), thyroid (30%–50%), ovarian (~30%) and colorectal (5%–20%) carcinomas, and with a relatively lower frequency (1%–3%) in a wide variety of other cancers. 7, 28 Among more than 75 identified B-Raf mutations in human cancers, a Glu for Val substitution at residue 600 in the kinase domain accounts for over 90% of B-Raf mutations, which harbors 500-fold higher intrinsic kinase activity. 29 The B-RafV600E could effectively circumvent the requirement of phosphorylation-dependent regulation by Ras, and result in constitutive activation of the MAPK signaling pathway, even in the absence of extracellular mitogenic stimuli, facilitating the malignant transformation.30, 31 The presence of B-RafV600E in certain cancer cohorts also correlates with the increased risk of deterioration and poor prognosis.32, 33 Taken together, these findings strongly suggested that B-Raf is a promising therapeutic target in melanoma and other carcinomas that depend upon B-Raf signaling.
Given the significance of B-Raf in tumorigenesis and progression, the inhibitors targeting B-Raf and especially the oncogenic forms of B-Raf, have increasingly come into the limelight of the drug discovery arena. To date, a number of oncogenic B-Raf inhibitors have been reported in various developmental stages.34–39 Some of them have entered clinical trials in recent years and exhibit encouraging clinical efficiency.40–42 Among them, Vemurafenib, which was approved by the FDA and EMA (European Medicines Agency) for the treatment of unresectable or metastatic melanoma, is characterized as a selective B-RafV600E inhibitor.43 It demonstrated complete or partial tumor regression in the majority of patients harboring the B-RafV600E mutation and with an overall increased median survival time after the treatment, which validates the effectiveness of targeting the oncogenic B-Raf in cancer therapy.40, 44 However, the emerging data also indicates that some mutant B-Raf tumors have intrinsic resistances to B-Raf inhibitors accessed clinically to date, and the acquired resistance occurred through a variety of mechanisms.45–47 Furthermore, a series of side effects have also been observed, in particular the induction of keratoacanthoma was frequently observed in Vemurafenib treated patients.44 Therefore, there is clearly an unmet therapeutic need to develop novel, potent and specific B-Raf inhibitors possessing different properties to benefits the patients with B-RafV600E-driven cancers.
Structure based virtual screening (SBVS) has become a powerful tool in the medicinal chemists’ toolkit, and the incorporation of the characteristic information from the known active ligands into the virtual screening campaign could further improve the hit enrichment as well as the potential for scaffold hopping.48, 49 In the present study, a combined ligand- and structure-based virtual screening protocol was employed to discover novel B-RafV600E inhibitors. Nine structurally novel hit compounds with high ligand efficiency were successfully identified. Subsequent scaffold-based analogue searching and structure optimization significantly increased the inhibitory potency and B-RafV600E selectivity in both enzymatic and cellular assays. In particular, compound 22q possesses excellent in vitro and ex vivo B-RafV600E inhibitory potency and exhibits cellular selectivity towards B-RafV600E harboring cancer cell lines.
Results and Discussion
Virtual screening
To strengthen the efficiency of scaffold hopping and hit discovery rate, we designed an integrated ligand- and structure-based virtual screening procedure aimed to counterbalance their fundamental limitations.49 The SHAFTS algorithm, which is a hybrid 3D molecular similarity calculation approach designed to combine the strength of pharmacophore matching and volumetric overlay, exhibiting satisfactory “scaffold hopping” capability against several representative kinases,50–52 was utilized for ligand-based virtual screening (LBVS). Since the shape and volume of the ATP binding pockets are altered upon different ligand binding, two crystal structures of B-Raf in the active conformation (PDB entry: 3og7,40 2fb835) varied in binding pocket architecture were used for conducting the structure-based virtual screening with the GLIDE program,53 which achieved the best performance in our pilot study (see Supporting Information for detailed results). A schematic representation of the overall virtual screening procedure in this study is presented in Figure 1. Taking the respective bioactive conformations of Vemurafenib (Template 1) and SB-590885 (Template 2) in complex with B-Raf as queries,35, 40 SHAFTS was utilized to search the SPECS vendor databases which contains more than 200,000 compounds. The top 1000 molecules with similarity scores >1.0 were reserved for each template,which resulted in 581 hits for template 1 and 1000 hits for template 2. These compounds were then docked into their corresponding structures with GLIDE using the standard precision (SP) mode, and the top 500 compounds for each structure were submitted for further docking validation using the GLIDE extra precision (XP) mode and visual inspection. The candidate molecules were selected based on the following criteria: (1) The molecular weight of the ligand was lower than 500 Da, rendering it suitable for further optimization. (2) Both geometric and chemical features matching existed between the ligand and the ATP binding site of B-Raf. (3) The binding poses and chemical structures were reasonable. Some highly ranked molecules that displayed either high strain energy or excessive rotatable bonds were rejected from further evaluation. (4) At least one H-bond was formed by the ligand with the backbone atoms of the hinge region residues (such as Q530 and C532), which are critical for anchoring the inhibitors in the active site. 54(5) The hydrophobic pocket surrounding the gatekeeper residue (back pocket) was occupied by the corresponding lipophilic groups in the ligand, which plays important roles in determining both the kinase selectivity and inhibitor binding affinity.55, 56(6) The ligands were prioritized for selection if at least one H-bond interaction was formed with the “DFG-motif” of B-Raf, based on the presumption that it may stabilize the active conformation and improve oncogenic B-Raf selectivity. 38, 40 Consequently, 327 compounds that met above criteria were selected for further scaffold diversity analysis with the Cluster Molecules component in Pipeline Pilot 7.5. One or two candidate molecules with relatively simple chemical structure and higher GLIDE score (G-score) within each structural cluster were retained. Finally, 113 compounds were designated for purchase for B-Raf inhibition activityassessment.
Figure 1.

Schematic representation of the virtual screening strategy adopted in the present study.
In vitro inhibition assay against B-RafWT and B-RafV600E kinases
The 113 candidate molecules selected by virtual screening were tested for B-RafWT and B-RafV600E inhibition as previously described.36, 39 Nine compounds with diverse chemotypes demonstrated significant inhibition for B-RafV600E activity at 5μM in the preliminary test, and the IC50 values towards both B-RafWT and B-RafV600E were then determined for the two most potent hit molecules (Table 1). Among the two hits possessing low-micromolar IC50 values against both wide-type and B-RafV600E, compound 1 was characterized as a structurally novel B-Raf inhibitor with higher ligand efficiency (0.42 kcal mol−1 per heavy atom versus 0.24 kcal mol−1 per heavy atom of hit 5)57 in inhibiting B-RafWT and B-RafV600E activity. Moreover, compound 1 also exhibited encouraging anti-proliferation activity. It could efficiently inhibit the proliferation of both A375 (B-RafV600E mutant) metastatic melanoma and HCT-116 (Ras mutant) colon carcinoma cells at 10μM in which the hyperactivated MAPK pathway was the main molecular driver for tumorigenesis (Table S4), implicating that the cytotoxicity of hit 1 may be attributed to its in vivo MAPK pathway inhibition. It’s also noteworthy that the B-RafV600E harboring A375 melanoma cells were more sensitive to hit 1 than that of HCT-116 cancer cells that contain B-RafWT. Taken together, these data suggested that hit 1 represented a novel B-RafV600E selective chemotype with high ligand efficiency and promising anti-proliferation activity, offering a good starting point for further optimization.
Table 1.
Inhibition rates (%) or IC50 values against B-RafV600E of the hits derived from virtual screening.
| Compound | IC50 (μM) or Inhibition rate (%) a |
|---|---|
| 1 | 3.189 (3.238) b |
| 2 | 79% |
| 3 | 52% |
| 4 | 60% |
| 5 | 3.984 (3.699) b |
| 6 | 53% |
| 7 | 86% |
| 8 | 50% |
| 9 | 67% |
Inhibition rate at 5μM of compound.
The IC50 values for both B-RafV600E and wild-type B-Raf (in the parentheses) were determined for the 2 most potent compounds against B-RafV600E.
Scaffold-based analogue searching
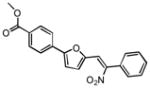
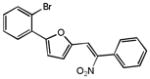
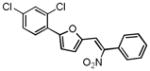
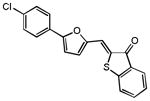
Upon close examination of the binding mode of compound 1 (Figure S2), we noted that the nitro group attached to the exocyclic methylene could form a hydrogen bond with the amide nitrogen of Cys532 in the hinge area, and the 5-phenylfuran moiety exhibited significant shape complementation with the ATP-binding site to make extensive hydrophobic interactions with the back pocket around the gatekeeper residue. On account of the fact that the nitro group generally functioned as a relatively weak H-bond acceptor in molecular interactions and has a relatively small VDW volume,58 we then assumed that a novel hinge region binder with more favorable H-bond and VDW interacting capability may improve both the inhibitory potency and oncogenic B-Raf selectivity. Therefore, a substructure-based analogue searching with 2-phenyl-5-vinylfuran (highlighted by red in Figure 2) as a query template was performed to hop the potential hinge region binders, and a focused library comprising of all of the hit compounds was extracted from the SPECS database. Then the focused library was docked into the active site of B-Raf with GLIDE program and ranked by G-score. After visual inspection, nineteen compounds with the same scaffold but with diverse substitutions on the exocyclic methylene and phenyl were purchased from the vendor database and their inhibitory activities against both B-RafWT and B-RafV600E were assessed. As shown in Table 2, 12 out of 19 compounds exhibited more than 50% inhibition of B-RafV600E activity at 5μM. Apparently, independent of the distinct substitutes on the exocyclic methylene, smaller hydrophobic substitutes (e.g. halogen in compound 17–19, 24) could be well tolerated in the para position of the phenyl group, and kinase inhibitory activity was completely abolished in the case of ortho- & para- double substitutes (e.g. 15, 20, 21). However, the broad activity spectrum for both the ortho- and meta- substituted compounds indicated that the potency and selectivity against B-RafV600E was remarkably affected by the different furan-2-methylene substitutes (e.g. compound 11 and 26, or compound 12, 23 and 27), which was consistent with our assumption and underlined the importance to select proper hinge binders for further optimization. It is also notable that most of the inhibitors showed varying degrees of selectivity against B-RafV600E over B-RafWT, especially for compound 11, 19 and 22, which showed about 2~3 fold selectivity, respectively. In summary, although no significant improvement in inhibitory potency was observed for these analogues over the original hit, they did provide novel chemotypes with enhanced oncogene selectivity for further evaluation. In view of the biological potency, oncogene selectivity and chemical feasibility, compounds 16, 17, 19 and 22, which contained the 2,3-dihydro-1H-inden-1-one, indolin-2-one, thiazolidine-2,4-dione and 2-thiobarbituric acid moiety respectively, were selected for hit optimization and structure-activity relationship (SAR) study.
Figure 2.

Chemical structures of the query templates employed in SHAFTS searching and hit compounds identified from virtual screening. The structural moiety of hit 1 used in the substructure search is highlighted in red.
Table 2.
Structure and potency of the compounds identified from the substructure-search
| ID | Structure | IC50a (μM) or Inhibition rate(%) b
|
|
|---|---|---|---|
| B-Raf | B-RafV600E | ||
| 1 |
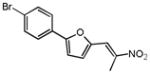
|
3.24c | 3.19c |
| 10 |
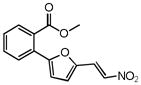
|
1.38 | 1.96 |
| 11 |
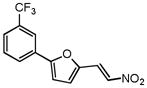
|
4.04 | 2.78 |
| 12 |
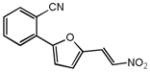
|
43% 17% |
28% 18% |
| 13 |
|
20% n.a. |
56% 27% |
| 14 |
|
50% 21% |
40% 30% |
| 15 |
|
n.a. | n.a. |
| 16 |
|
62% 50% |
78% 61% |
| 17 |
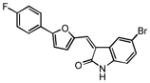
|
2.98 | 3.85 |
| 18 |
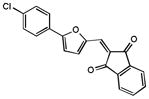
|
40% 14% |
67% 38% |
| 19 |
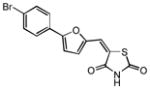
|
4.99 | 2.26 |
| 20 |
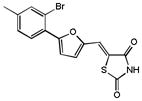
|
n.a. | n.a. |
| 21 |
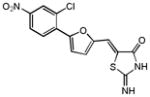
|
n.a. | n.a. |
| 22 |
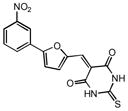
|
6.60 | 2.09 |
| 23 |
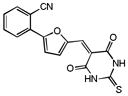
|
62% 36% |
75% 42% |
| 24 |
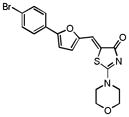
|
46% n.a. |
53% 30% |
| 25 |
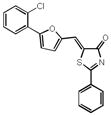
|
43% 24% |
72% 58% |
| 26 |
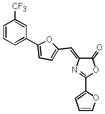
|
n.a. | n.a. |
| 27 |
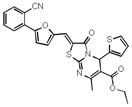
|
30% 14% |
68% 49% |
| 28 |
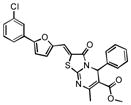
|
32% 17% |
47% 40% |
IC50 values were determined if the inhibition rate at 2μM was larger than 65%.
For each compound, the values listed on the upper and bottom panel of the cell refer to the inhibition rate (%) at 5μM and 2μM, respectively.
Values listed for comparison.
n.a.=not active
Hit optimization and SAR analysis
To confirm the effectiveness of the hits identified through the scaffold searching procedure and explore their preliminary structure-activity relationship (SAR), we initiated the hit optimization campaign. According to isosteric replacement theory and synthetic feasibility, the bivalent sulfur atom in the 2,3-dihydro-1H-inden-1-one moiety of hit 16 was replaced by a methylene group, for which the activity was confirmed by subsequent experimental evaluation (16e). In total, 35 compounds were designed and synthesized, and their chemical structures are shown in Table 3–5.
Table 3.
Chemical structures and activities for the analogues of compound 16
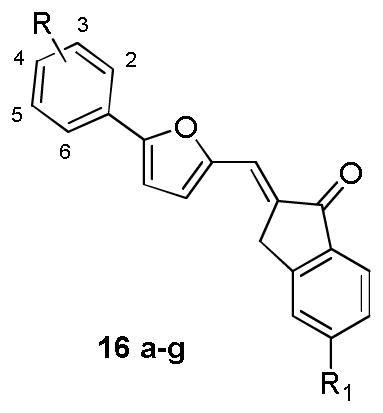
| ||||
|---|---|---|---|---|
| Compound | Substituent | Inhibition rate (%) a | ||
|
| ||||
| R | R1 | B-Raf | B-RafV600E | |
| 16a | 2-NO2 | H | 11% n.a. |
64% 47% |
| 16b | 3-CF3 | H | n.a. n.d. |
22% n.d. |
| 16c | 3-CF3 | Cl | n.a. n.a. |
59% 42% |
| 16d | 4-NO2 | H | n.a. n.d. |
34% n.d. |
| 16e | 4-Cl | H | n.a. n.a. |
52% 41% |
| 16f | 4-Br | H | n.a. n.d. |
42% n.d. |
| 16g | 2-Cl, 4-CF3 | H | n.a. n.d. |
44% n.d. |
For each compound, the values listed on the upper and bottom panel of the cell refer to the inhibition rate (%) at 2μM and 0.5μM, respectively.
n.a.= not active. n.d.=not determined.
Table 5.
Chemical structures and activities for the analogues of compound 19 and 21
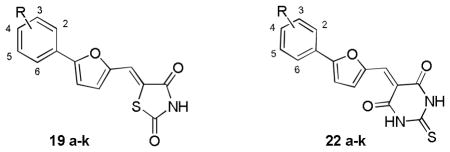
| |||
|---|---|---|---|
| Cmpd. | Substituent | IC50a or Inhibition rate (%)b
|
|
| B-Raf | B-RafV600E | ||
| 19a | 2-NO2 | n.a. n.d. |
7% n.d. |
| - | - | - | - |
| 19b | 3-OCH3 | n.a. n.d. |
34% n.d. |
| - | - | - | - |
| 19c | 4-NO2 | n.a. n.d. |
48% n.d. |
| 19d | 4-CO2Et | n.a. n.d. |
n.a. n.d. |
| 19e | 4-Cl | n.a. n.d. |
n.a. n.d. |
| 19 | 4-Br | 4.99 c | 2.26 c |
| 19g | 4-Me | n.a. n.d. |
41% n.d. |
| 19h | 4-OCH3 | 3% n.d. |
41% n.d. |
| 19i | 4-tBu | 17% n.d. |
34% n.d. |
| 19j | Naphthyl | n.a. n.d. |
51% n.d. |
| 19k | 2-Cl,5-CF3 | 18% n.d. |
37% n.d. |
| 22a | 2-NO2 | 38% n.a. |
61% 48% |
| 23 | 2-CN | 36% 13% |
42% 19% |
| 22b | 3-OCH3 | 6% n.a. |
63% 34% |
| 22 | 3-NO2 | 6.60 c | 2.09 c |
| 22c | 4-NO2 | 55% n.a. |
59% n.a. |
| 22d | 4-CO2Et | n.a. n.d. |
38% n.d. |
| 22e | 4-Cl | 15% n.a. |
69% 33% |
| 22f | 4-Br | 0.60 | 0.47 |
| 22g | 4-Me | 17% n.a. |
59% 50% |
| 22h | 4-OCH3 | 3.69 | 2.69 |
| 22i | 4-tBu | 29% n.a. |
57% 36% |
| 22j | Naphthyl | 1.93 | 1.07 |
| 22k | 2-Cl,5-CF3 | 1.49 | 1.09 |
IC50 values were determined if the inhibition rate at 0.5μM was larger than 50%.
For each compound, the values listed on the upper and bottom panel of the cell refer to the inhibition ratio (%) at 2μM and 0.5μM, respectively.
IC50 values were listed for comparison.
n.a.= not active. n.d.= not determined.
The general synthetic route of the designed compounds 16a-g, 17a-f, 19a-k and 22a-q is depicted in Scheme 1. 5-formyl-2-furanboronic acid was treated with various substituted bromobenzene, Pd(PPh3)4 and K2CO3 to yield compound 30. Then the final products (16a-g, 17a-f, 19a-k and 22a-q) were obtained using the Knoevenagel condensation between compound 30 and various substituted 1-indanone, 2,3-dihydroindol-2-one, 2,4-thiazolidinedione and 2-thiobarbituric acid, respectively. The crude products were purified by washing with MeOH or re-crystallizating in EtOH to yield pure products.
Scheme 1.

a
a Reagents and conditions: (a) Pd(PPh3)4, K2CO3, PhMe, 100°C, 4h; (b) for the compound 16 series: substituted 1-indanone, NaOH, EtOH, room temperature, 3–4h; for the compound 17 series: substituted 2,3-dihydroindol-2-one, NaOH, EtOH, room temperature, 3–4h; for the compound 19 series: 2,4-thiazolidinedione, β-alanine, acetic acid, 90°C, 2h; for the compound 22 series: 2-thiobarbituric acid, acetic acid, 90°C, 2h.
The in vitro kinase inhibition assay clearly indicated that distinct substitutes on the exocyclic methylene had a significant effect on both the inhibitory potency and B-RafV600E selectivity. Of the synthetic derivatives tested, 13 compounds (e.g. 16a, 16c, 16e; 22a-c, 22e-k) exhibited more than 50% inhibition against B-RafV600E at 2μM, wherein most of the compounds were close analogues of hit 22, which contained a 2-thiobarbituric acid moiety as the potential hinge binder (Table 3–5). However, on the whole, the majority of the derivatives of hit 16, 17 and 19 displayed relatively weak inhibition in comparison with that of hit 22, which was more visible when the same substitutes were presented on the benzene ring (e.g. 22a vs 16a and 19a, 22f vs 16f and 19, 22k vs 17d and 19k). Moreover, while the three analogues of hit 16 (16a, 16c, 16e) exhibited encouraging inhibitory activity at 2μM, their activities declined at lower concentration. In contrast, significant inhibitory potency could still be observed for five derivatives of hit 22 at the same concentration (22f-h, 22j, 22k) and was exemplified by the 2~5 fold improvement in their IC50 values compared with the original hit (Table 5). Collectively, the data from the preliminary optimization highlighted the optimal performance of the 2-thiobarbituric acid analogues with respect to B-RafV600E inhibition.
Analyzing the data in Table 5, we found that either small or bulky hydrophobic substitutes, such as methyl, methoxyl, halogens and tertiary butyl retained or improved the inhibition activity to varying degrees (e.g. 22e-i), with the inhibition rate ranging from 30%~60% at 0.5μM, and it was more evident for compound 22f which produced a potency of nearly 5-fold over compound 22. However, the activities substantially decreased when introducing the more polar groups (22c and 22d). Accordingly, the activity is highly sensitive to physiochemical properties rather than the steric size of the substitutes at the para position, indicating the existence of a relatively bulky hydrophobic cavity. Similarly, a hydrophobic substituent was desirable at the meta-position when comparing the activity differences between 22b and 22. Moreover, the presence of a cyano group at the ortho position of the benzene ring showed reduced activity relative to the nitro substitution, which may be attributed to the insufficient complementarity with the ATP binding site due to its linear configuration. Nevertheless, the B-RafV600E selectivity for these analogues did not show concomitant improvement as their biological potency. For example, compound 22f possessed nanomolar activity but exhibited rare discrimination between B-RafV600E and B-RafWT. Consequently, we undertook a second round of hit optimization, mainly focusing on increasing B-RafV600E selectivity, which is deemed to be critical for maximum therapeutic benefit owing to low cytotoxicity toward normal cells. Meanwhile, with the SAR data on hand, most of the efforts were concentrated on exploring the synergistic effects of the substitutions on the benzene ring. As presented in Table 6, the double substitution at ortho- & para- positions, as well as the meta- & para- positions did not show the expected “additive” effect, albeit the corresponding mono-substitute could be well accommodated on the benzene ring (e.g. 22n vs 22a and 22e, 22p vs 22l and 22e). Considering the observation that the presence of a H-bond acceptor at the meta-position of the benzene improved the B-RafV600E selectivity, for example with compound 22, and that the configurational freedom of the methoxyl group may hinder the optimal H-bond formation, we speculated that the introduction of a conformationally constrained H-bond partner with enhanced H-bond forming potential, as well as the relatively hydrophobic nature at both meta- & para- positions may potentiate the B-RafV600E selectivity. Encouragingly, compound 22q, with the 3,4-methylendioxy substituent on the benzene ring, exhibited the expected improvement in both potency and selectivity. It possessed nanomolar inhibitory activity against B-RafV600E and about 4-fold selectivity over B-RafWT, which validated the effectiveness of our speculations and underlined the indispensable contributions of a suitable H-bond acceptor substituent at the meta-position for B-RafV600E selectivity.
Table 6.
Chemical structures and activities for the analogues of 22 in the second round optimization
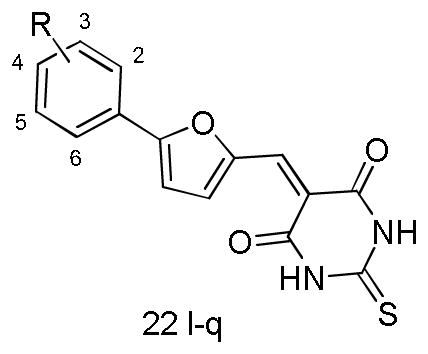
| |||
|---|---|---|---|
| Compound | Substituent | IC50a or Inhibition rate ([I]=1μM) | |
| B-Raf | B-RafV600E | ||
| 22l | 3-CF3 | 30% | 45% |
| 22m | 4-COOH | n.a. | n.a. |
| 22n | 2-NO2, 4-Cl | 20% | 22% |
| 22o | 2-CF3, 4-Cl | 24% | 49% |
| 22p | 3-CF3, 4-Cl | 37% | 48% |
| 22q | 3,4-methylendioxy | 1.15 | 0.30 |
IC50 values were determined if the inhibition rate at 1μM was larger than 70%.
n.a.= not active.
To evaluate the effects of B-Raf kinase inhibition on cellular characteristics, the anti-proliferation activity of selected B-Raf inhibitors was investigated. As shown in Table 7, compound 22e, 22f, 22g, 22h, 22i and 22q demonstrated potent growth inhibition activity against the B-RafV600E harboring A375 malignant melanoma cells at 10μM. Notably, virtually complete growth inhibition for A375 cell lines was achieved by compound 22g, 22h and 22i. On the contrary, anti-proliferation activity toward B-RafWT containing SW620 human colonic carcinoma cells was observed to modest levels for most of the tested inhibitors, indicating cellular selectivity for B-RafV600E over B-RafWT by these compounds. Moreover, the negligible anti-growth activity that was observed for the selected analogues of compound 16 and 19 was consistent with their relatively weak B-RafV600E inhibitory activity in vitro (Table 2), suggesting that the selective targeting of B-RafV600E was responsible for their cytotoxicity of the analogs of compound 22.
Table 7.
In vitro anti-proliferation activity of the selected B-RafV600E inhibitor at 10μM
| Compound | Tumor growth inhibition rate ([I]=10μM)
|
|
|---|---|---|
| A375(B-RafV600E) | SW620 (B-Raf) | |
| 22a | n.a. | n.a. |
| 22b | 26.2 | n.a. |
| 22c | 46.3 | n.a. |
| 22e | 82.9 | 9.0 |
| 22f | 65.3 | n.a. |
| 22g | 84.0 | 3.6 |
| 22h | 94.4 | 13.0 |
| 22i | 95.4 | 7.6 |
| 22j | 6.9 | n.a. |
| 22k | 18.3 | n.a. |
| 22q | 77.7 | 47.7 |
| 19 | n.a. | n.a. |
| 16a | 16.5 | n.a. |
| 16c | 3.2 | n.a. |
| 16e | 5.2 | n.a. |
n.a.= not active.
To further ascertain this selectivity profile, the anti-proliferation effects of compound 22f and 22q possessing the most potent B-RafV600E inhibition potency and selectivity at the enzymatic level were explored in detail against four other human-derived cells. The dose-dependent inhibition profile exemplified in Figure 3 clearly indicates that B-RafV600E bearing tumor cell lines (1205Lu and WM983B) were significantly inhibited following incubation with compounds 22f or 22q, whereas normal human primary fibroblasts (FF2508) and malignant cells not expressing mutant B-Raf (WM3918) were significantly more resistant to compound treatment. Notably, the inhibition of tumor growth at 10μM was more pronounced upon treatment with compound 22q than compound 22f (Table 6 and Figure 3). These results in cells were consistent with the biochemical assays demonstrating that compound 22q was more potent and selective than compound 22f against B-RafV600E (Tables 5 and 6). This correlation between the enzymatic and cellular inhibitory potency again suggested that the anti-growth activity of the compounds can be well related to their capacity to selectively inhibit oncogenic B-RafV600E over B-RafWT. Intriguingly, all of the compounds possessing potent and selective anti-proliferation activity contained the hydrophobic substituent at the para-position on the benzene ring, whereas the corresponding polar substitution essentially abolished compound potency. Since the ability of the compounds to penetrate the cell membrane is a prerequisite to cytotoxicity, the para-position of the benzene ring may represent a functional site to orchestrate the permeability and cellular activity for 2-thiobarbituric acid B-RafV600E inhibitors. Taken together, the ex vivo anti-proliferation activity data presented here indicates that the biochemical selectivity of the inhibitors (e.g. 22q) against B-RafV600E translate to the cellular selectivity, reminiscent of the Vemurafenib and SB-590885 compounds that were utilized as the query template during our ligand-based virtual screening and specially targeted the active conformation of B-Raf.
Figure 3.

Effects of the compounds 22f (A) and 22q (B) on the proliferation of human-derived cells. Cellular proliferation of melanoma cell lines (WM3918, 1205Lu, WM983B), and human fibroblasts (FF2508) were assessed with the MTS assay. Results are shown as the mean value of four data sets, including standard errors, and are representative of replicated independent experiments. Proliferation is shown as the percent change compared to the vehicle control values.
Kinase inhibition profile
To investigate the specificity and selectivity of the identified inhibitors against B-Raf over other kinases, we performed a selectivity profile against a panel of 16 protein kinases at both 5μM and 1μM for the two most potent inhibitors 22f and 22q. As shown in Table 8, compound 22f showed only modest levels of inhibition for most of the tested tyrosine- and serine/threonine-kinases except for FGFR1 and IKKβ kinase, which was significantly inhibited at 5μM. It is noteworthy that the most potent and selective inhibitor 22q, which was derived from the second round optimization campaign, demonstrated an improved selectivity profile. 22q significantly inhibited B-RafV600E at both concentrations but showed only modest levels of inhibition for these other 16 distinct kinases. Taken together, this data demonstrates that compound 22q is a B-RafV600E inhibitor with more than 10-fold selectivity over other tested protein kinases. Collectively, the promising results from this small kinase profiling suggests that compounds 22f and 22q, and 22q in particular, may possess specific inhibitory activity for B-RafV600E. In addition, these studies also suggest that synergy between in silico screening and hierarchical hit optimization can lead to novel B-Raf inhibitors with significant kinase selectivity.
Table 8.
Kinase inhibition profile of compound 22f and 22q against a panel of 16 protein kinases
| Kinase | Inhibition rate (%) for compounds at 5μM and 1μM
|
|||
|---|---|---|---|---|
| 22f (5μM) | 22f (1μM) | 22q (5μM) | 22q (1μM) | |
| SRC | 28.3 | 8.6 | 15.2 | 6.2 |
| LCK | 10.5 | 10.5 | 0.8 | n.a. |
| KDR | 6.1 | 3.9 | 1.2 | n.a. |
| ABL | n.a. | n.a. | n.a. | n.a. |
| P38α | 7.7 | n.a. | n.a. | n.a. |
| CDK2 | 39.7 | 11.6 | 12.6 | 3.3 |
| ERK1 | 30.7 | 14.5 | 13.7 | 10.0 |
| IGF1R | 15.6 | 9.5 | 6.8 | 5.6 |
| IKKβ | 52.1 | 27.8 | 35.7 | 15.0 |
| PKCα | 26.4 | 16.2 | 5.2 | n.a. |
| PDGFRα | 20.4 | 7.2 | 7.7 | 6.0 |
| EGFR | 46.1 | 12.6 | 19.4 | n.a. |
| FGFR1 | 72.2 | 15.0 | 18.6 | 4.9 |
| AKT1 | 20.3 | 10.8 | 14.5 | 4.6 |
| MET | 42.2 | 34.2 | 19.4 | 18.6 |
| cKIT | 40.3 | 18.6 | 20.0 | 18.3 |
n.a.= not active.
Binding mode analysis
To understand the molecular basis for 22q inhibition by B-RafV600E and to explore the potential structural origin for the oncogene selectivity, the molecular docking strategy was utilized to scrutinize the binding poses of 22q. Figure 4 illustrates the predicted binding poses of 22q in the ATP-binding site of B-RafV600E (PDB entry: 3og740). In this model, the 2-thiobarbituric acid moiety is tightly confined within the adenine-binding site of the ATP binding pocket, intercalating between residue W531 and F583 by making π-π stacking interactions. The amide portion of the 2-thiobarbituric acid group makes bidentate hydrogen bonds with the NH group and the carbonyl oxygen of C532 in the hinge region, forming a “handshake” interaction, which effectively anchors the compound into the ATP binding pocket. The 2-phenyl-5-vinylfuran portion of 22q extends towards the activation loop (A-loop) and makes extensive hydrophobic interactions with the surrounding residues. In particular, the 3,4-methylendioxy substituted benzene ring is predicted to occupy the hydrophobic back cavity created by the relatively small gatekeeper residue, mainly comprised of A481, K483, L505, L514, I527, T539 and F595. Gaining access to the kinase back pocket results from the small gatekeeper residue may have the dual effect of not only increasing the biological potency but also improving the selectivity over other kinases that have a larger gatekeeper residue and hence a smaller back pocket, 56, 59, 60 which may account for the selectivity profile of 22q. Notably, the 3,4-methylendioxy substituent on the benzene ring mediates a hydrogen bond interaction with the backbone NH of D594 in the conserved DFG triad.
Figure 4.

Binding mode analysis of compound 22q with B-Raf kinase. (A) B-RafV600E kinase in the active conformation is shown in cartoon diagram. The N-lobe and C-lobe of B-Raf kinase are shown in pink and blue, respectively. Compound 22q occupies the ATP binding site between the two lobes represented as yellow sticks. (B) A close-up view of the key interactions between compound 22q and surrounding residues. Hydrogen bonds are depicted as dotted red lines. (C) Structural comparison of compound 22q with Vemurafenib (in cyan sticks) in the ATP pocket of B-RafV600E. The Raf-selective pocket is represented in surface contour.
The SAR data derived from the two-round structural optimization could be rationalized by the predicted binding mode. The ortho-nitro group on the benzene ring may form a weak H-bond interaction with the sidechain of the gatekeeper T529, thus the lower activity that is observed for the linear cyano substitution may partly be attributed to the non-optimal steric complementarity for H-bond formation. Considering the hydrophobic nature of the back cavity, a lipophilic substitution may be preferred over a polar substitution, which was validated by the aforementioned SAR data. In particular, hydrophobic substitutes such as methyl, methoxyl and halogens on the meta- or para- position of the benzene ring (e.g. 22f, 22h) that would be predicted to better complement the back pocket, achieving elevated shape and chemical feature complementarity with the ATP binding site, greatly contributed to the improved biological potency of the corresponding compounds. Consistent with this notion, the more polar substitutions on the meta- or para- position of the benzene ring, which would be predicted to be disfavorable due to a high desolvation penalty, showed markedly less potency. For example, nitro, carbethoxy and carboxyl substitutions in these positions showed relatively poor potency (22c, 22d and 22m). Furthermore, the relatively snug but length-limited back pocket could well accommodate rigid and bulky groups (22i and 22j), however, the more extended substitutions such as carbethoxy group would be predicted to cause some steric clashes when presented at the para position of the benzene ring, likely contributing to the weak inhibitory potency of 22d. It is noteworthy that the presence of a conformationally constrained H-bond acceptor in the meta-position of the benzene ring significantly improved B-RafV600E selectivity (22, 22q). Based on the predicted binding mode, the H-bond acceptor at the meta-position of the benzene ring could form a hydrogen bond interaction with the mainchain NH group of D594, reminiscent of a similar interaction observed by Vemurafenib when in complex with B-RafV600E. The sulfamide moiety in Vemurafenib was deprotonated to facilitate the same hydrogen bond interaction with D564 which was deemed to stabilize the DFG-in conformation and thus contributed to the B-RafV600E selectivity.38, 40 Moreover, on account of the fact that the conformational equilibrium of B-Raf was strongly biased toward the active state characterized by the DFG-in conformation by the phosphorylation mimic V600E mutation, inhibitors specially targeting the active conformation of B-Raf likley possess higher selectivity against the oncogenic mutant owing to the relatively small configurational entropy cost upon binding.38–40, 43, 54, 61 In addition, the structural comparison of the active and inactive B-Raf conformation highlights the central role of the mainchain conformation of D594 in directing the conformational switching of the A-loop between the corresponding DFG-in and DFG-out states. Therefore, resembling the important role of the sulfamide in Vemurafenib, the improved selectivity of compound 22 and 22q for B-RafV600E over B-RafWT could be rationalized by the H-bond interaction between the meta-substituted hydrogen bond acceptor and the mainchain NH of D594, which potentially stabilizes the DFG-in conformation and the active conformation of B-Raf.
Conclusions and Future Prospects
Targeted intervention of the B-RafV600E gene product represents a promising approach for cancer therapy. In the present study, we identified a novel class of 2-thiobarbituric acid analogues as selective B-RafV600E inhibitors by using an integrated ligand- and structure-based virtual screening strategy in conjunction with hierarchical hit optimization. First, the combined ligand- and structural-based virtual screening was utilized to the discovery of structurally novel B-RafV600E inhibitors. Nine hit compounds with low micromolar activity against B-RafV600E were successfully identified. Among them, hit 1 was characterized with high ligand efficiency and encouraging anti-proliferation activity, providing an attractive starting point for a hit-to-lead optimization campaign. The molecular modeling study of hit 1 into the ATP-binding site of B-Raf indicated that the biological potency as well as oncogene mutant selectivity could be further optimized by varying the potential hinge region binder and substituent on the benzene ring. The follow-up scaffold-based searching from hit 1 led to a series of B-RafV600E inhibitors bearing a wide variety of potency and B-RafV600E oncogene selectivity, four of the analogues of compound 1 displayed potent activities and synthetic feasibility (16, 17, 19 and 22), so were then selected for hit optimization. Subsequently, the medicinal chemistry effort and SAR analysis was performed to explore the activity and specificity profile of the derivatives for these selected inhibitors, which led to the discovery of 2-thiobarbituric acid analogues with improved inhibitory potency and B-RafV600E selectivity. The most potent inhibitor, compound 22q, inhibited the kinase activity of B-RafV600E with an IC50 value in the nanomolar range and with 4-fold selectivity for B-RafV600E over B-RafWT and selective cytotoxicity against B-RafV600E harboring cancer cell lines. In addition, a small kinase profiling study was consistent with the conclusion that the cytotoxicity of the inhibitor was mainly related to the suppression of B-RafV600E activity.
In addition to providing a novel chemotype with significant biological potency and oncogene selectivity for B-Raf inhibition, the SAR studies and B-RafV600E selectivity analysis for the 2-thiobarbituric acid analogues offers valuable pharmacophore insights for future optimizations aimed to increase the inhibitory potency as well as B-RafV600E mutant selectivity. The structural comparison of 22q with Vemurafenib in complex with B-RafV600E reveals that the most significant difference lies on the propyl group in Vemurafenib, which penetrates into the Raf-selective pocket and was proposed to be the critical binding determinant for Raf kinase specificity.38, 40 In the case of compound 22q, the substitution on the methylene presented in the methylendioxy group with a short alkyl chain was predicted to occupy this Raf-specific pocket, which potentially augmented the potency and specificity of the inhibitor towards the oncogenic B-RafV600E. In addition, the structural comparison also reveals that the lipophilic substituent at the ortho-position of the benzene ring may further potentiate the hydrophobic interactions with the pocket around the gatekeeper residue. However, it is noteworthy that the ortho-substitution may affect the relative orientation between the benzene ring and the central furan group, which in turn may impede the pivotal hydrogen bond interaction with the beginning of the DFG region. We therefore hypothesize that a relatively small substitution, such as fluorine, chlorine or methyl group may be more desirable at the ortho-position. In addition, the NH group in the 2-thiobarbituric acid moiety that does not participate in bidentate hydrogen bonds with the hinge area would be predicted to point towards the solvent. Based on this, we propose that the derivatization at this site such as introducing the solubilizing morpholine fragment may contribute to enhanced solubility and cellular activity.
The study presented here details the optimization process from a fragment-like hit compound derived from virtual screening to a more drug like candidate. This underscores the importance of selecting a proper starting point with low molecular weight and high ligand efficiency to facilitate the subsequent hit-to-lead optimization process. Thus, our study may provide a few useful clues regarding the rational selection of hits for further evaluation. Moreover, the encouraging findings obtained in our study will lay the foundation for future development of more potent and selective oncogenic B-Raf inhibitors for cancer therapy.
Experimental
Computational procedure
Protein preparation
Two representative B-Raf/ligand complexes (PDB entry: 3og7,40 2fb835) in the active conformation with apparently distinct shape and volume in the ATP binding site were used during virtual screening. The protein structures were prepared using the Protein Preparation Wizard Workflow provided in the Maestro graphical user interface of the Schrodinger program suite and the default settings were used. Residues around the co-crystal ligands at a radius of 10 A were defined as the binding region in which the docking grids were created. This radius was large enough to include all of the residues that may be involved in inhibitor binding. The crystallographic coordinates for each ligand (Vemurafenib and SB-590885) in the complex structure were extracted and designated as query templates in the ligand-based virtual screening.
Validation of the docking protocol
To evaluate the performance of various available docking methods (GLIDE 5.5,53 GOLD 5.0,62–64 AutoDock 4.265, 66) against B-Raf, two conventional criteria were employed during the pilot study. One of the measurements is the root mean-square deviation (RMSD) value between the best ranked pose predicted by various docking programs and the corresponding experimental pose. The other refers to the enrichment factors, which is defined as the fraction of active compounds found a certain percentage of ranking list divided bythe fraction of the screened library.
GLIDE 5.5
GLIDE (grid-based ligand docking with energetics) approximates a complete systematic search of chemical space available to the docked ligand.53 In this search, an initial rough positioning and scoring phase that dramatically narrows down the search space is used to locate promising ligand poses. The conformational energy for a few hundred surviving candidate poses is then torsionally flexible minimized at the receptor environment with the OPLS-AA force field.67 Monte Carlo simulation is finally conducted between the three to six lowest energy poses of the previous step to explore nearby torsional minima, which is critical for obtaining an accurate docked pose.
GOLD 5.0
GOLD performs automated molecular docking which employs the hydrogen bond-encoded genetic algorithm (GA) to direct the identification of proper binding modes. 62–64 A maximum of 10000 genetic operations were performed on a set of five islands for each of the 20 independent GA runs. The default weights for three genetic operators (crossover, mutation and migration) and a population size of 100 chromosomes were applied in each docking simulation. The kinase scoring function68 implemented in GOLD were utilized to rank the docked compounds, respectively. Early termination was allowed when the RMSD values for the top three scoring poses was less than 1.5Å.
AutoDock 4.2
AutoDock Tools (ADT) was used to prepare the grid parameter files (gpf) and the docking parameter files (dpf) for each of the docked compound. A grid spacing of 0.375Å was applied for the energetic map calculations, which centered on the co-crystal ligands and with the grid number of 60, 60, 60 in the x, y, and z directions. The Lamarckian genetic algorithm (LGA) was employed to explore the optimal chemical space in the ATP binding site for each ligand.65 The docked compounds were subjected to 10 GA runs with a maximal energy evaluation number of 250000 and a maximum of 27000 generations. Default values were used for the other parameters and an RMSD tolerance of 2.0Å was defined during the cluster analysis.
Virtual screening
The SHAFTS program was employed to perform the 3D molecular similarity based virtual screening. It adopts a hybrid similarity metric of molecular shape and pharmcophore features for 3D similarity calculation and ranking, aiming to combine the strength of both pharmacophore matching and volumetric overlay.50 SHAFTS had demonstrated satisfactory active compounds enrichment and scaffold hopping capability against several representative kinases in retrospective virtual screening studies as well as recent prospective applications.51, 52 Taking the crystallographic conformation of Vemurafenib and SB-590885 in B-Raf/ligand complex as query templates, the conformational ensembles for each compound in the SPECS database were searched by SHAFTS, and only the best ranked conformer was reserved per molecule. The top 1000 molecules with similarity score >1.0 were reserved for each template. We employed the GLIDE program as the secondary filter to prioritize the compounds for experimental assessment. All of the candidate compounds derived from SHAFTS screening were docked into the ATP binding site of B-Raf without any constraint. The hit compounds were ranked based on G-score and the top ranking compounds were submitted for further docking validation with the extra precision (XP) mode to eliminate false positives as accomplished by more extensive sampling and advanced scoring, resulting in even higher enrichment. Finally, after visual inspection and chemical diversity analysis, the candidate compounds were purchased and subject to the in vitro kinase inhibition assay.
Cloning, protein expression and purification
The human B-Raf kinase domain and the V600E containing mutant (B-RafWT and B-RafV600E, residues 433–726) with an N-terminal 6X-His tag (MDRGSH6GS) to facilitate protein purification (in the presence of full-length mouse p50cdc37 to facilitate the proper folding of the B-Raf kinase) was expressed in Sf9 cells and purified to homogeneity as previously described.39 The protein was stored at 1.5 mg/mL at 4 °C until use. GST-MEK-His protein substrate for the B-Raf inhibitor screen was overexpressed at 37 °C in Escherichia coli BL21 (Gold) cells. The protein was stored at 10 mg/mL at −80°C until use.
In vitro B-Raf kinase assay
For IC50 calculations of inhibitors, an ELISA-based assay was employed as previously described39 at different inhibitor concentrations to generate a sigmoidal dose response curve using B-RafV600E or B-RafWT protein. All dose response measurements were carried out in duplicate or triplicate and IC50 values were derived from fitting the data to a sigmoidal dose response curve with a four-parameter logistic model using GraphPad Prism.
Melanoma proliferation assay
Human melanoma cell lines (A375, WM983B, 1205Lu, WM3918) were isolated as previously described69 and cultured in Dulbecco’s modified Eagle’s medium supplemented with 5% fetal bovine serum. Normal human primary fibroblasts (FF2508) were isolated from the human epidermis of neonatal foreskins and cultured as described.70, 71 Cells (5000/well) were seeded in 96-well plates and allowed to adhere overnight before treatment with each compound or the DMSO vehicle control for 72 h. Cells were then directly incubated with MTS substrate (CellTiter-96 Aqueous One Solution Cell Proliferation Assay, Promega). Absorbance was measured at 490 nm as per the supplier’s instructions and percent proliferation was normalized to the absorbance of vehicle-treated cells. For each experiment, cell line, and treatment, the absorbance values of at least 4 wells were used for data analysis and the experiment was conducted in triplicate.
Kinase profiling
Kinase profiling was performed at Shanghai ChemPartner Co., Ltd. according to the manufacturer’s protocols. ATP concentrations in the kinase assay were at the Km for each enzyme.
Chemistry
General methods
The reagents (chemicals) were purchased from Alfa Aesar, Sigma, Acros and Shanghai Chemical Reagent Company, and used without further purification. Column chromatography was performed with CombiFlash® Companion system (Teledyne Isco, Inc.). Nuclear magnetic resonance (NMR) spectra were performed on a Brucker AMX-400 and AMX-300 NMR (IS as TMS). Chemical shifts were reported in parts per million (ppm, δ) downfield from tetramethylsilane. Proton coupling patterns were described as singlet (s), doublet (d), triplet (t), quartet (q), multiplet (m), and broad (br). Low- and high-resolution mass spectra (LRMS and HRMS) were given with electric ionization (EI) produced by a Finnigan MAT-95. Melting points (Mp) are uncorrected and were measured in open capillary tubes, using a SGW X-4 melting point apparatus (−50–400 °C). An Agilent 1100 series HPLC with an Agilent Eclipse XDB-C18 (4.6 × 150 mm, 5 μm) reversed phase column was used for the HPLC analyses. The elution buffer was a mixture of H2O/MeCN = 40:60 (v/v) with 0.1% CF3COOH (v/v) as an additive. The retention time of each synthesized compound is given and the corresponding purity for each compound is proved to be >95% by both HPLC test and 1H-NMR Spectral Scan (see in the Supporting Information). The purity of compounds from the SPECS database is more than 90% and for most compounds greater than 95% (confirmed by SPECS, using NMR or LC MS; data available through the Web site).
General procedure for the synthesis of compounds
General procedure for the synthesis of compound 2 via the Suzuki reaction. To a solution of 5-formyl-2-furanboronic acid (5.0 mmol) in a mixture (7/3) of dry toluene and ethanol (10 mL), various substituted bromobenzene (1.0 equiv), Pd(PPh3)4 (0.1 equiv) and K2CO3 (2.0 equiv) was added. The mixture was stirred and heated at 100°C for 4 h under argon protection before it was cooled to room temperature. The organic solvents were removed under vacuum, and the residue was extracted with CH2Cl2 (3*50 mL) and washed with brine. The organic phase was finally dried over Na2SO4 and evaporated to dryness to give a crude compound that was purified through flash chromatography (ethyl acetate/petroleum ether) to yield compound 2.
-
General procedure for the synthesis of compound 4.
16 a-g & 17 a-f: a mixture of compound 2 (1.0 mmol), substituted 1-indanone or 2,3-dihydroindol-2-one (2.0 equiv) and NaOH (3.0 equiv) in MeOH was stirred and heated at room temperature for 3–4 h. Precipitation was obtained after filtration. The solid was re-crystallization with EtOH to yield pure product.
19 a-k72: a mixture of compound 2 (1.0 mmol), 2,4-thiazolidinedione (2.0 equiv) and β-alanine (2.0 equiv) in acetic acid was stirred and heated at 90°C for 2h before it was cooled to room temperature. Precipitation was obtained after filtration. The solid was washed with MeOH to yield pure product without further purification.
22 a-q73: a mixture of compound 2 (1.0 mmol) and 2-thiobarbituric acid (1.0 equiv) in acetic acid was stirred and heated at 90°C for 2h before it was cooled to room temperature. Precipitation was obtained after filtration. The solid was washed with MeOH to yield pure product without further purification.
2-[5-(2-nitro-phenyl)-furan-2-ylmethylene]-indan-1-one (16a)
1H NMR (400 MHz, DMSO) δ 8.00 (dd, J = 10.8, 8.3 Hz, 2H), 7.83 – 7.75 (m, 2H), 7.72 (d, J = 7.4 Hz, 1H), 7.70 – 7.60 (m, 2H), 7.50 (d, J = 7.4 Hz, 1H), 7.36 (s, 1H), 7.26 (q, J = 3.7 Hz, 2H), 3.95 (s, 2H). Mp 147–148 °C. LRMS (EI) m/z 331, M+. HRMS (EI) m/z calcd C20H13NO4 M+ 331.0845, found 331.0844.
2-[5-(3-trifluomethyl-phenyl)-furan-2-ylmethylene]-indan-1-one (16b)
1H NMR (400 MHz, DMSO) δ 8.19 – 8.17 (m, 2H), 7.77 – 7.71 (m, 5H), 7.50 – 7.48 (d, J = 3.7 Hz, 2H), 7.41 – 7.40(s, 1H), 7.25 – 7.24 (d, J = 3.7 Hz, 1H), 4.17 (s, 2H). Mp 171–173 °C. LRMS (EI) m/z 354, M+. HRMS (EI) m/z calcd C21H13F3O2 M+ 354.0868, found 354.0867.
2-[5-(3-trifluomethyl-phenyl)-furan-2-ylmethylene]-5-chloro-indan-1-one (16c)
1H NMR (400 MHz, DMSO) δ 8.28 (s, 1H), 7.89 (d, J = 8.4 Hz, 1H), 7.83 – 7.76 (m, 2H), 7.71 (s, 1H), 7.59 – 7.47 (m, 3H), 7.31 (d, J = 3.8 Hz, 1H), 4.17 (s, 2H). Mp 220–221 °C. LRMS (EI) m/z 388, M+. HRMS (EI) m/z calcd C21H12ClF3O2 M+ 388.0478, found 388.0475.
2-[5-(4-nitro-phenyl)-furan-2-ylmethylene]-indan-1-one (16d)
1H NMR (400 MHz, DMSO) δ 8.36 (d, J = 8.5 Hz, 2H), 8.14 (d, J = 8.6 Hz, 2H), 7.81 – 7.70 (m, 3H), 7.54 (d, J = 32.1 Hz, 2H), 7.43 (s, 1H), 7.31 (s, 1H), 4.22 (s, 2H). Mp 224–225 °C. LRMS (EI) m/z 331, M+. HRMS (EI) m/z calcd C20H13NO4 M+ 331.0845, found 331.0837.
2-[5-(4-chloro-phenyl)-furan-2-ylmethylene]-indan-1-one (16e)
1H NMR (400 MHz, DMSO) δ 7.89 (d, J = 8.6 Hz, 2H), 7.81 – 7.67 (m, 3H), 7.57 (d, J = 8.6 Hz, 2H), 7.53 – 7.46 (m, 1H), 7.38 (s, 1H), 7.29 (d, J = 3.6 Hz, 1H), 7.21 (d, J = 3.6 Hz, 1H), 4.15 (s, 2H). Mp 184–185 °C. LRMS (EI) m/z 320, M+. HRMS (EI) m/z calcd C20H13ClO2 M+ 320.0604, found 320.0610.
2-[5-(4-bromo-phenyl)-furan-2-ylmethylene]-indan-1-one (16f)
1H NMR (400 MHz, DMSO) δ 7.82 (d, J = 8.6 Hz, 2H), 7.76 (d, J = 7.6 Hz, 1H), 7.74 – 7.70 (m, 3H), 7.69 (s, 1H), 7.52 – 7.44 (m, 1H), 7.38 (s, 1H), 7.30 (d, J = 3.6 Hz, 1H), 7.21 (d, J = 3.6 Hz, 1H), 4.14 (s, 2H). Mp 190–192 °C. LRMS (EI) m/z 364, M+. HRMS (EI) m/z calcd C20H13BrO2 M+ 364.0099, found 320.0124.
2-[5-(2-chloro-5-trifluomethyl)-furan-2-ylmethylene]-indan-1-one (16g)
1H NMR (400 MHz, DMSO) δ 8.28 (s, 1H), 7.87 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 7.7 Hz, 2H), 7.72 (t, J = 7.4 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.50 (t, J = 5.3 Hz, 2H), 7.44 (s, 1H), 7.28 (d, J = 3.6 Hz, 1H), 4.14 (s, 2H). Mp 183–185 °C. LRMS (EI) m/z 388, M+. HRMS (EI) m/z calcd C21H12ClF3O2 M+ 388.0478, found 388.0482.
3-[5-(3-trifluomethyl-phenyl)-furan-2-ylmethylene]-1,3-dihydro-indol-2-one (17a)
1H NMR (400 MHz, DMSO) δ 10.73 (s, 1H), 8.46 (d, J = 2.0 Hz, 1H), 8.22 (s, 2H), 7.78 (d, J = 8.1 Hz, 2H), 7.58 (d, J = 3.7 Hz, 1H), 7.49 (d, J = 3.7 Hz, 1H), 7.40 (s, 1H), 7.32 (dd, J = 8.3, 2.1 Hz, 1H), 6.90 (d, J = 8.3 Hz, 1H). Mp 206–207 °C. LRMS (EI) m/z 355, M+. HRMS (EI) m/z calcd C20H12F3NO2 M+ 355.0820, found 355.0820.
3-[5-(3-trifluomethyl-phenyl)-furan-2-ylmethylene]-6-chloro-1,3-dihydro-indol-2-one (17b)
1H NMR (400 MHz, DMSO) δ 8.32 (d, J = 8.3 Hz, 1H), 8.13 (s, 2H), 7.76 (d, J = 5.8 Hz, 2H), 7.49 (d, J = 3.7 Hz, 1H), 7.39 (d, J = 3.7 Hz, 1H), 7.30 (s, 1H), 6.96 (dd, J = 8.3, 1.9 Hz, 1H), 6.87 (d, J = 1.9 Hz, 1H). Mp 226–228 °C. LRMS (EI) m/z 389, M+. HRMS (EI) m/z calcd C20H11ClF3NO2 M+ 389.0430, found 389.0427.
3-[5-(3-trifluomethyl-phenyl)-furan-2-ylmethylene]-5-chloro-1,3-dihydro-indol-2-one (17c)
1H NMR (400 MHz, DMSO) δ 8.45 (dd, J = 7.7, 3.9 Hz, 1H), 8.25 – 8.11 (m, 2H), 7.78 (d, J = 4.1 Hz, 2H), 7.53 (dd, J = 6.3, 3.8 Hz, 1H), 7.42 (dd, J = 6.2, 2.7 Hz, 1H), 7.39 – 7.27 (m, 2H), 7.02 (td, J = 7.6, 3.1 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H). Mp 246–248 °C. LRMS (EI) m/z 389, M+. HRMS (EI) m/z calcd C20H11ClF3NO2 M+ 389.0430, found 389.0432.
3-[5-(2-chloro-5-trifluomethyl)-furan-2-ylmethylene]-1,3-dihydro-indol-2-one (17d)
1H NMR (400 MHz, DMSO) δ 10.64 (s, 1H), 8.37 (dd, J = 8.8, 4.4 Hz, 2H), 7.91 (d, J = 8.4 Hz, 1H), 7.85 – 7.77 (m, 1H), 7.56 (d, J = 3.8 Hz, 1H), 7.47 (d, J = 3.8 Hz, 1H), 7.38 (s, 1H), 7.29 (d, J = 1.0 Hz, 1H), 6.90 (d, J = 7.8 Hz, 2H), 3.33 (s, 2H). Mp 207–208 °C. LRMS (EI) m/z 389, M+. HRMS (EI) m/z calcd C20H11ClF3NO2 M+ 389.0430, found 389.0430.
3-[5-(2-chloro-5-trifluomethyl)-furan-2-ylmethylene]-5-chloro-1,3-dihydro-indol-2-one (17e)
1H NMR (400 MHz, DMSO) δ 8.31 (d, J = 2.1 Hz, 1H), 8.26 (d, J = 1.7 Hz, 1H), 7.93 (dd, J = 12.2, 3.5 Hz, 1H), 7.85 (dd, J = 8.4, 2.2 Hz, 1H), 7.51 (dd, J = 11.4, 3.8 Hz, 2H), 7.43 (s, 1H), 7.30 (d, J = 2.1 Hz, 1H), 6.88 (d, J = 8.3 Hz, 1H). Mp 244–245 °C. LRMS (EI) m/z 423, M+. HRMS (EI) m/z calcd C20H10Cl2F3NO2 M+ 423.0041, found 423.0042.
3-[5-(2-chloro-5-trifluomethyl)-furan-2-ylmethylene]-6-chloro-1,3-dihydro-indol-2-one (17f)
1H NMR (400 MHz, DMSO) δ 10.80 (s, 1H), 8.32 (dd, J = 7.6, 5.1 Hz, 2H), 7.90 (t, J = 8.3 Hz, 1H), 7.85 (d, J = 2.2 Hz, 1H), 7.55 (d, J = 3.7 Hz, 1H), 7.50 (d, J = 3.8 Hz, 1H), 7.41 (s, 1H), 6.91 (d, J = 1.9 Hz, 1H), 6.87 (dd, J = 8.2, 1.9 Hz, 1H). Mp 286–287 °C. LRMS (EI) m/z 423, M+. HRMS (EI) m/z calcd C20H10Cl2F3NO2 M+ 423.0041, found 423.0042.
5-[5-(2-nitro-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19a)
1H NMR (400 MHz, DMSO) δ 12.51 (s, 1H), 8.00-7.98 (d, J = 8.0 Hz, 1H), 7.93-7.91 (d, J = 8.0 Hz, 1H), 7.81-7.79 (t, J = 8.0 Hz, 1H), 7.69-7.65 (t, J = 8.0 Hz, 1H), 7.61 (s, 1H), 7.25-7.24 (d, J = 4.0 Hz, 1H), 7.20-7.19 (d, J = 4.0 Hz, 1H). Mp 250–252 °C. LRMS (EI) m/z 316 M+. HRMS (EI) m/z C14H8N2O5S M+, calcd 316.0154, found 316.0153.
5-[5-(3-methoxy-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19b)
1H NMR (400 MHz, DMSO) δ 12.50 (s, 1H), 7.65 (s, 1H), 7.47-7.43 (t, J = 7.9 Hz, 1H), 7.42-7.39 (d, J = 7.9 Hz, 1H), 7.35 (s, 1H), 7.33-7.32 (d, J = 4.0 Hz, 1H), 7.24-7.23 (d, J = 4.0 Hz, 1H), 3.83 (s, 3H). Mp 222–223 °C. LRMS (EI) m/z 301 M+. HRMS (EI) m/z C15H11NO4S M+, calcd 301.0409, found 301.0409.
5-[5-(4-nitro-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19c)
1H NMR (400 MHz, DMSO) δ 12.53 (s, 1H), 8.31-8.29 (d, J = 8.1 Hz, 2H), 7.95-7.93 (d, J = 8.1 Hz, 2H), 7.59 (s, 1H), 7.51-7.50 (d, J = 4.0 Hz, 1H), 7.24-7.22 (d, J = 4.0 Hz, 1H)). Mp 330–331 °C. LRMS (EI) m/z 316 M+. HRMS (EI) m/z C14H8N2O5S M+, calcd 316.0154, found 316.0149.
4-{5-[(2,4-dioxo-thiazolidin-5-ylidene)methyl]-furan-2-yl}benzoic acid ethyl ester (19d)
1H NMR δ (400 MHz, DMSO) δ 12.52 (s, 1H), 8.07-8.04 (d, J = 8.6 Hz, 2H), 7.92-7.89 (d, J = 8.6 Hz, 2H), 7.64 (s, 1H), 7.43-7.42 (d, J = 3.6 Hz, 1H), 7.24-7.23 (d, J = 3.6 Hz, 1H), 4.35-4.30 (q, J = 7.0 Hz, 2H), 1.35-1.32 (t, J = 7.0 Hz, 3H). Mp 259–261 °C. LRMS (EI) m/z 343 M+. HRMS (EI) m/z C17H13NO5S M+, calcd 343.0514, found 343.0513.
5-[5-(4-chloro-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19e)
1H NMR (400 MHz, DMSO) δ 12.49 (b, 1H), 7.83-7.80 (d, J = 8.1 Hz, 2H), 7.61-7.57 (m, 3H), 7.32-7.30 (d, J = 3.8 Hz, 1H), 7.22-7.21 (d, J = 3.8 Hz, 1H). Mp 279–280 °C. LRMS (EI) m/z 305 M+. HRMS (EI) m/z C14H8ClNO3S M+, calcd 304.9913, found 304.9920.
5-[5-(4-methyl-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19g)
1H NMR (400 MHz, DMSO) δ 12.44 (s, 1H), 7.71-7.69 (d, J = 8.1 Hz, 2H), 7.61 (s, 1H), 7.34-7.32 (d, J = 8.1 Hz, 2H), 7.21-7.19 (m, 2H), 2.34 (s, 3H). Mp 262–264 °C. LRMS (EI) m/z 285 M+. HRMS (EI) m/z C15H11NO3S M+, calcd 285.0460, found 285.0466.
5-[5-(4-methoxy-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19h)
1H NMR (400 MHz, DMSO) δ 12.41 (s, 1H), 7.77-7.72 (d, J = 8.8 Hz, 2H), 7.61 (s, 1H), 7.20-7.18 (d, J = 3.6 Hz, 1H), 7.12-7.06 (m, 3H), 3.79 (s, 3H). Mp 272 °C. LRMS (EI) m/z 301 M+. HRMS (EI) m/z C15H11NO4S M+, calcd 301.0409, found 301.0404.
5-[5-(4-tert-butyl-phenyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19i)
1H NMR (400 MHz, DMSO) δ 12.44 (s, 1H), 7.75-7.72 (d, J = 8.1 Hz, 2H), 7.62 (s, 1H), 7.55-7.52 (d, J = 8.1 Hz, 2H), 7.21-7.19 (m, 2H), 1.29 (s, 3H). Mp 275–277 °C. LRMS (EI) m/z 327 M+. HRMS (EI) m/z C18H17NO3S M+, calcd 327.0929, found 327.0928.
5-[5-(2-naphthyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19j)
1H NMR (400 MHz, DMSO) δ 12.51 (s, 1H), 8.32 (s, 1H), 8.06-8.03 (m, 2H), 7.96-7.94 (d, J = 8.0 Hz, 2H), 7.67 (s, 1H), 7.60-7.54 (m,2H), 7.42-7.41 (d, J = 4.0 Hz, 1H), 7.29-7.28 (d, J = 4.0 Hz, 1H). Mp 246–247 °C. LRMS (EI) m/z 321 M+. HRMS (EI) m/z C18H11NO3S M+, calcd 321.0460, found 321.0462.
5-[5-(2-chloro-5-trifluomethyl)-furan-2ylmethylene]-thiazolidine-2,4-dione (19k)
1H NMR (400 MHz, DMSO) δ 12.57 (s, 1H), 8.12 (s, 1H), 7.85-7.83 (d, J = 8.5 Hz, 2H), 7.77-7.75 (d, J = 8.5 Hz, 2H), 7.66 (s, 1H), 7.52-7.51 (d, J = 3.8 Hz, 1H), 7.26-7.25 (d, J = 3.8 Hz, 1H). Mp 259 °C. LRMS (EI) m/z 373 M+. HRMS (EI) m/z C18H11NO3S M+, calcd 372.9787, found 372.9791.
5-[5-(2-nitro-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22a)
1H NMR (400 MHz, DMSO) δ 12.51 (s, 1H), 12.45 (s, 1H), 8.61-8.60 (d, J = 4.0 Hz, 1H), 8.09-8.03 (t, J = 8.3 Hz, 2H), 7.89 (s, 1H), 7.88-7.83 (t, J = 8.3 Hz, 1H), 7.78-7.74 (t, J = 8.3 Hz, 1H), 7.42-7.40 (d, J = 4.0 Hz, 1H). Mp 302–303 °C. LRMS (EI) m/z 343 M+. HRMS (EI) m/z C15H9N3O5S M+, calcd 343.0263, found 343.0272.
5-[5-(3-methoxy-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22b)
1H NMR (400 MHz, DMSO) δ 12.43 (s, 1H), 12.37 (s, 1H), 8.62-8.61 (d, J = 4.0 Hz, 1H), 8.11 (s, 1H), 7.57-7.51 (m, 3H), 7.46-7.42 (d, J = 8.4 Hz, 1H), 7.07-7.04 (d, J = 8.4 Hz, 1H), 3.83 (s, 3H). Mp 335–336 °C. LRMS (EI) m/z 328 M+. HRMS (EI) m/z C16H12N2O4S M+, calcd 328.0518, found 328.0515.
5-[5-(4-nitro-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22c)
1H NMR (400 MHz, DMSO) δ 12.50 (s, 1H), 12.44 (s, 1H), 8.60-8.58 (d, J = 4.0 Hz, 1H), 8.48-8.35 (d, J = 8.5 Hz, 2H), 8.24-8.21 (d, J = 8.6 Hz, 2H), 8.12 (s, 1H), 7.74-7.72 (d, J = 4.0 Hz, 1H). Mp 329–330 °C. LRMS (EI) m/z 343 M+. HRMS (EI) m/z calcd C15H9N3O5S M+ 343.0263, found 343.0272.
5-[5-(4-chloro-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22e)
1H NMR (400 MHz, DMSO) δ 12.44 (s, 1H), 12.38 (s, 1H), 8.61-8.60 (d, J = 4.0 Hz, 1H), 8.11 (s, 1H), 8.03-7.99 (d, J = 8.6 Hz, 2H), 7.63-7.60 (d, J = 8.6 Hz, 2H), 7.55-7.53 (d, J = 4.0 Hz, 1H). Mp 317–318 °C. LRMS (EI) m/z 332 M+. HRMS (EI) m/z C15H9ClN2O3S M+, calcd 332.0022, found 332.0018.
5-[5-(4-bromo-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22f)
1H NMR (400 MHz, DMSO) δ 12.44 (s, 1H), 12.38 (s, 1H), 8.59-8.58 (d, J = 4.0 Hz, 1H), 8.09 (s, 1H), 7.94-7.90 (d, J = 8.6 Hz, 2H), 7.75-7.72 (d, J = 8.6 Hz, 2H), 7.54-7.53 (d, J = 4.0 Hz, 1H). Mp 327–328 °C. LRMS (EI) m/z 376 M+. HRMS (EI) m/z C15H9BrN2O3S M+, calcd 375.9517, found 375.9519.
5-[5-(4-methyl-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22g)
1H NMR (400 MHz, DMSO) δ 12.35 (s, 1H), 12.28 (s, 1H), 8.62-8.61 (d, J = 4.1 Hz, 1H), 8.09 (s, 1H), 7.88-7.86 (d, J = 8.1 Hz, 2H), 7.43-7.42 (d, J = 4.1 Hz, 2H), 7.35-7.33 (d, J = 8.1 Hz, 1H), 2.36 (s, 3H). Mp 335 °C. LRMS (EI) m/z 312 M+. HRMS (EI) m/z C16H12N2O3S M+, calcd 312.0569, found 312.0572.
5-[5-(4-methoxy-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22h)
1H NMR (400 MHz, DMSO) δ 12.36 (s, 1H), 12.30 (s, 1H), 8.64-8.63 (d, J = 4.0 Hz, 1H), 8.07 (s, 1H), 7.95-7.90 (d, J = 8.5 Hz, 2H), 7.39-7.37 (d, J = 4.0 Hz, 1H), 7.11-7.06 (d, J = 8.5 Hz, 2H), 3.83 (s, 3H). Mp 346–347 °C. LRMS (EI) m/z 328 M+. HRMS (EI) m/z C16H12N2O4S M+, calcd 328.0518, found 328.0517.
5-[5-(4-tert-butyl-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22i)
1H NMR (400 MHz, DMSO) δ 12.40 (s, 1H), 12.35 (s, 1H), 8.64-8.63 (d, J = 4.1 Hz, 1H), 8.09 (s, 1H), 7.92-7.90 (d, J = 8.6 Hz, 2H), 7.56-7.53 (d, J = 8.6 Hz, 2H), 7.46-7.45 (d, J = 4.1 Hz, 1H), 1.29 (s, 9H). Mp 303–304 °C. LRMS (EI) m/z 354 M+. HRMS (EI) m/z C19H18N2O3S M+, calcd 354.1038, found 354.1036.
5-[5-(2-naphthyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22j)
1H NMR (400 MHz, DMSO) δ 12.45 (s, 1H), 12.39 (s, 1H), 8.69-8.68 (d, J = 4.0 Hz, 1H), 8.59 (s, 1H), 8.16 (s, 1H), 8.09-8.04 (m, 3H), 7.97 (s, 1H), 7.64-7.59 (m, 3H). Mp 352–354 °C. LRMS (EI) m/z 348 M+. HRMS (EI) m/z C19H12N2O3S M+, calcd 348.0569, found 348.0569.
5-[5-(2-chloro-5-trifluomethyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4, 6-dione (22k)
1H NMR (400 MHz, DMSO) δ 12.49 (s, 1H), 12.44 (s, 1H), 8.59-8.58 (d, J = 4.0 Hz, 1H), 8.45 (s, 1H), 8.19 (s, 1H), 7.90-7.85 (d, J = 8.5 Hz, 1H), 7.84-7.82 (d, J = 8.5 Hz, 1H), 7.70-7.69 (d, J = 4.0 Hz, 1H). Mp 360–362 °C. LRMS (EI) m/z 400 M+. HRMS (EI) m/z C18H6ClF3N2O3S M+, calcd 399.9896, found 399.9857.
5-[5-(3-trifluomethyl-phenyl)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6 -dione (22l)
1H NMR (400 MHz, DMSO) δ 12.46 (s, 1H), 12.41 (s, 1H), 8.62-8.61 (d, J = 4.0 Hz, 1H), 8.33 (s, 1H), 8.29-8.27 (d, J = 8.4 Hz, 1H), 8.16 (s, 1H), 7.83-7.85 (d, J = 8.4 Hz, 1H), 7.80-7.76 (t, J = 8.4 Hz, 1H), 7.70-7.69 (d, J = 4.0 Hz, 1H). Mp 302–303 °C. LRMS (EI) m/z 366 M+. HRMS (EI) m/z C16H19F3N2O3S M+, calcd 366.0286, found 366.0284.
4-[5-(4,6-dioxo-2-thioxo-tetrahydro-pyrimidin-5-ylidenemethyl)furan-2-yl]benzoic acid (22m)
1H NMR (400 MHz, DMSO) δ 12.46 (s, 1H), 12.40 (s, 1H), 8.62-8.61 (d, J = 4.1 Hz, 1H), 8.11-8.03 (m, 5H), 7.63-7.62 (d, J = 4.1 Hz, 1H). Mp >400 °C. LRMS (EI) m/z 342 M+. HRMS (EI) m/z C16H10N2O5S M+, calcd 342.0301, found 342.0311.
5-[5-(2-nitro-4-chloro)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-dione (22n)
1H NMR (400 MHz, DMSO) δ 12.49 (s, 1H), 12.43 (s, 1H), 8.59-8.58 (d, J = 4.0 Hz, 1H), 8.26 (s, 1H), 8.11-8.09 (d, J = 8.4 Hz, 1H), 7.94-7.92 (d, J = 8.4 Hz, 1H), 7.86 (s, 1H), 7.42-7.41 (d, J = 4.0 Hz, 1H). Mp 300–301 °C. LRMS (EI) m/z 377 M+. HRMS (EI) m/z C15H8ClN3O5S M+, calcd 376.9873, found 376.9884.
5-[5-(2-trifluomethyl-4-chloro)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4, 6-dione (22o)
1H NMR (400 MHz, DMSO) 12.49 (s, 1H), 12.43 (s, 1H), 8.62-8.60 (d, J = 4.0 Hz, 1H), 8.08-8.06 (d, J = 8.4 Hz, 1H), 8.03 (s, 1H), 8.00 (s, 1H), 7.94-7.92 (d, J = 8.4 Hz, 1H), 7.36-7.35 (d, J = 4.0 Hz, 1H). Mp 325–326 °C. LRMS (EI) m/z 400 M+. HRMS (EI) m/z C16H8ClF3N2O3S M+, calcd 399.9896, found 399.9857.
5-[5-(3-trifluomethyl-4-chloro)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4, 6-dione (22p)
1H NMR (400 MHz, DMSO) δ 12.47 (s, 1H), 12.43 (s, 1H), 8.57-8.56 (d, J = 4.0 Hz, 1H), 8.35 (s, 1H), 8.26-8.23 (d, J = 8.4 Hz, 1H), 8.12 (s, 1H), 7.89-7.87 (d, J = 8.4 Hz, 1H), 7.70-7.69 (d, J = 4.0 Hz, 1H). Mp 323–325 °C. LRMS (EI) m/z 400 M+. HRMS (EI) m/z C16H8ClF3N2O3S M+, calcd 399.9896, found 399.9869.
5-[5-(3,4-methylenedioxy)-furan-2ylmethylene]-2-thioxo-dihydro-pyrimidine-4,6-di one (22q)
1H NMR (400 MHz, DMSO) δ 12.39 (s, 1H), 12.33 (s, 1H), 8.63-8.62 (d, J = 4.0 Hz, 1H), 8.10 (s, 1H), 7.59-7.56 (m, 2H), 7.42-7.41 (d, J = 4.0 Hz, 1H), 7.11-7.09 (d, J = 8.5 Hz, 1H), 6.14 (s, 2H). Mp >400 °C. LRMS (EI) m/z 342 M+. HRMS (EI) m/z C16H10N2O5S M+, calcd 342.0310, found 342.0302.
Supplementary Material
Table 4.
Chemical structures and activities for the analogues of compound 17
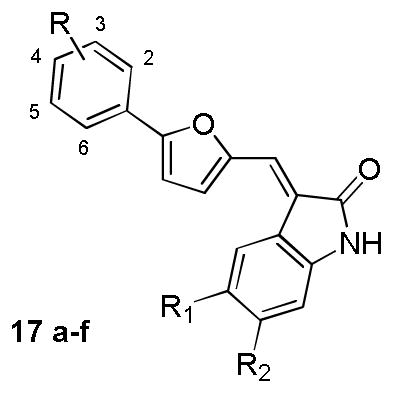
| |||||
|---|---|---|---|---|---|
| Compound | Substituent | Inhibition rate (%)a | |||
|
| |||||
| R | R1 | R2 | B-Raf | B-RafV600E | |
| 17a | 3-CF3 | H | H | n.a. n.d. |
34% n.d. |
| 17b | 3-CF3 | H | Cl | n.a. n.d. |
38% n.d. |
| 17c | 3-CF3 | Cl | H | 17% n.d. |
23% n.d. |
| 17d | 2-Cl,5-CF3 | H | H | n.a. n.d. |
23% n.d. |
| 17e | 2-Cl, 5-CF3 | H | Cl | n.a. n.d. |
34% n.d. |
| 17f | 2-Cl, 5-CF3 | Cl | H | n.a. n.d. |
34% n.d. |
For each compound, the values listed on the upper and bottom panel of the cell refer to the inhibition rate (%) at 2μM and 0.5μM, respectively.
n.a.= not active. n.d.=not determined.
Acknowledgments
We gratefully acknowledge financial support from National Institutes of Health (NIH) to RM and MH (CA114046). We also gratefully acknowledge financial support from the State Key Program of Basic Research of China grant to HJ (2009CB918502), the National Natural Science Foundation of China grants to CL (20972174 and 91029704), HJ (21021063) and HL (81025017), National S&T Major Projects to HL (2012ZX09103-101-072), National High Technology Research and Development Program of China (2012AA020302) and the Chinese Academy of Sciences (XDA01040305) grant to CL.
References
- 1.Seger R, Krebs EG. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 2.Robinson MJ, Cobb MH. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 3.Rubinfeld H, Seger R. Mol Biotechnol. 2005;31:151–174. doi: 10.1385/MB:31:2:151. [DOI] [PubMed] [Google Scholar]
- 4.Kim EK, Choi EJ. Biochim Biophys Acta. 2010;1802:396–405. doi: 10.1016/j.bbadis.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 5.Dhillon AS, Hagan S, Rath O, Kolch W. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 6.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, Kohno M. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- 7.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks N, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JWC, Leung SY, Yuen ST, Weber BL, Siegler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 8.Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Nature. 2002;418:934. doi: 10.1038/418934a. [DOI] [PubMed] [Google Scholar]
- 9.Rapp UR, Fensterle J, Albert S, Gotz R. Adv Enzyme Regul. 2003;43:183–195. doi: 10.1016/s0065-2571(03)00002-5. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee R, Bartlett JM, Krishna NS, Underwood MA, Edwards J. Prostate. 2005;64:101–107. doi: 10.1002/pros.20211. [DOI] [PubMed] [Google Scholar]
- 11.Damodar Reddy C, Marwaha S, Patti R, Raghunath M, Duhaime AC, Sutton L, Phillips PC. Anticancer Res. 2001;21:2733–2738. [PubMed] [Google Scholar]
- 12.Giuliani N, Lunghi P, Morandi F, Colla S, Bonomini S, Hojden M, Rizzoli V, Bonati A. Leukemia. 2004;18:628–635. doi: 10.1038/sj.leu.2403269. [DOI] [PubMed] [Google Scholar]
- 13.McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Wong EW, Chang F, Lehmann B, Terrian DM, Milella M, Tafuri A, Stivala F, Libra M, Basecke J, Evangelisti C, Martelli AM, Franklin RA. Biochim Biophys Acta. 2007;1773:1263–1284. doi: 10.1016/j.bbamcr.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steelman LS, Abrams SL, Shelton JG, Chappell WH, Basecke J, Stivala F, Donia M, Nicoletti F, Libra M, Martelli AM, McCubrey JA. Cell Cycle. 2010;9:1629–1638. doi: 10.4161/cc.9.8.11487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sebolt-Leopold JS, Herrera R. Nat Rev Cancer. 2004;4:937–947. doi: 10.1038/nrc1503. [DOI] [PubMed] [Google Scholar]
- 16.Roberts PJ, Der CJ. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 17.Gollob JA, Wilhelm S, Carter C, Kelley SL. Semin Oncol. 2006;33:392–406. doi: 10.1053/j.seminoncol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 18.Zebisch A, Troppmair J. Cell Mol Life Sci. 2006;63:1314–1330. doi: 10.1007/s00018-006-6005-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roskoski R., Jr Biochem Biophys Res Commun. 2010;399:313–317. doi: 10.1016/j.bbrc.2010.07.092. [DOI] [PubMed] [Google Scholar]
- 20.Leicht DT, Balan V, Kaplun A, Singh-Gupta V, Kaplun L, Dobson M, Tzivion G. Biochim Biophys Acta. 2007;1773:1196–1212. doi: 10.1016/j.bbamcr.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chong H, Vikis HG, Guan KL. Cell Signal. 2003;15:463–469. doi: 10.1016/s0898-6568(02)00139-0. [DOI] [PubMed] [Google Scholar]
- 22.Mercer KE, Pritchard CA. Biochim Biophys Acta. 2003;1653:25–40. doi: 10.1016/s0304-419x(03)00016-7. [DOI] [PubMed] [Google Scholar]
- 23.Pritchard CA, Samuels ML, Bosch E, McMahon M. Mol Cell Biol. 1995;15:6430–6442. doi: 10.1128/mcb.15.11.6430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mason CS, Springer CJ, Cooper RG, Superti-Furga G, Marshall CJ, Marais R. EMBO J. 1999;18:2137–2148. doi: 10.1093/emboj/18.8.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moodie SA, Paris MJ, Kolch W, Wolfman A. Mol Cell Biol. 1994;14:7153–7162. doi: 10.1128/mcb.14.11.7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wojnowski L, Stancato LF, Larner AC, Rapp UR, Zimmer A. Mech Dev. 2000;91:97–104. doi: 10.1016/s0925-4773(99)00276-2. [DOI] [PubMed] [Google Scholar]
- 27.Garnett M, Marais R. Cancer Cell. 2004;6:313. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 28.Wellbrock C, Hurlstone A. Biochem Pharmacol. 2010;80:561–567. doi: 10.1016/j.bcp.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 29.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, Jones CM, Marshall CJ, Springer CJ, Barford D, Marais R. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 30.Pratilas CA, Taylor BS, Ye Q, Viale A, Sander C, Solit DB, Rosen N. Proc Natl Acad Sci U S A. 2009;106:4519–4524. doi: 10.1073/pnas.0900780106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wellbrock C, Karasarides M, Marais R. Nat Rev Mol Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 32.Houben R, Becker JC, Kappel A, Terheyden P, Brocker EB, Goetz R, Rapp UR. J Carcinog. 2004;3:6. doi: 10.1186/1477-3163-3-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA, Wolff RK, Slattery ML. Cancer Res. 2005;65:6063–6069. doi: 10.1158/0008-5472.CAN-05-0404. [DOI] [PubMed] [Google Scholar]
- 34.Liu L, Cao Y, Chen C, Zhang X, McNabola A, Wilkie D, Wilhelm S, Lynch M, Carter C. Cancer Res. 2006;66:11851–11858. doi: 10.1158/0008-5472.CAN-06-1377. [DOI] [PubMed] [Google Scholar]
- 35.King AJ, Patrick DR, Batorsky RS, Ho ML, Do HT, Zhang SY, Kumar R, Rusnak DW, Takle AK, Wilson DM, Hugger E, Wang L, Karreth F, Lougheed JC, Lee J, Chau D, Stout TJ, May EW, Rominger CM, Schaber MD, Luo L, Lakdawala AS, Adams JL, Contractor RG, Smalley KS, Herlyn M, Morrissey MM, Tuveson DA, Huang PS. Cancer Res. 2006;66:11100–11105. doi: 10.1158/0008-5472.CAN-06-2554. [DOI] [PubMed] [Google Scholar]
- 36.Luo C, Xie P, Marmorstein R. J Med Chem. 2008;51:6121–6127. doi: 10.1021/jm800539g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ramurthy S, Subramanian S, Aikawa M, Amiri P, Costales A, Dove J, Fong S, Jansen JM, Levine B, Ma S, McBride CM, Michaelian J, Pick T, Poon DJ, Girish S, Shafer CM, Stuart D, Sung L, Renhowe PA. J Med Chem. 2008;51:7049–7052. doi: 10.1021/jm801050k. [DOI] [PubMed] [Google Scholar]
- 38.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, Fong D, Zhu YL, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim SH, Schlessinger J, Zhang KY, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Proc Natl Acad Sci U S A. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie P, Streu C, Qin J, Bregman H, Pagano N, Meggers E, Marmorstein R. Biochemistry. 2009;48:5187–5198. doi: 10.1021/bi802067u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bollag G, Hirth P, Tsai J, Zhang JZ, Ibrahim PN, Cho HN, Spevak W, Zhang C, Zhang Y, Habets G, Burton E, Wong B, Tsang G, West BL, Powell B, Shellooe R, Marimuthu A, Nguyen H, Zhang KYJ, Artis DR, Schlessinger J, Su F, Higgins B, Iyer R, D’Andrea K, Koehler A, Stumm M, Lin PS, Lee RJ, Grippo J, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, Chapman PB, Flaherty KT, Xu XW, Nathanson KL, Nolop K. Nature. 2010;467:596–599. doi: 10.1038/nature09454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Alcala AM, Flaherty KT. Clin Cancer Res. 2012;18:33–39. doi: 10.1158/1078-0432.CCR-11-0997. [DOI] [PubMed] [Google Scholar]
- 42.Zambon A, Niculescu-Duvaz I, Niculescu-Duvaz D, Marais R, Springer CJ. Bioorg Med Chem Lett. 2012;22:789–792. doi: 10.1016/j.bmcl.2011.11.060. [DOI] [PubMed] [Google Scholar]
- 43.Yang H, Higgins B, Kolinsky K, Packman K, Go Z, Iyer R, Kolis S, Zhao S, Lee R, Grippo JF, Schostack K, Simcox ME, Heimbrook D, Bollag G, Su F. Cancer Res. 2010;70:5518–5527. doi: 10.1158/0008-5472.CAN-10-0646. [DOI] [PubMed] [Google Scholar]
- 44.Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA, O’Dwyer PJ, Lee RJ, Grippo JF, Nolop K, Chapman PB. New Engl J Med. 2010;363:809–819. doi: 10.1056/NEJMoa1002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA. New Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Solit DB, Rosen N. New Engl J Med. 2011;364:772–774. doi: 10.1056/NEJMcibr1013704. [DOI] [PubMed] [Google Scholar]
- 47.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Q, Muegge I. J Med Chem. 2006;49:1536–1548. doi: 10.1021/jm050468i. [DOI] [PubMed] [Google Scholar]
- 49.Nicholls A, McGaughey GB, Sheridan RP, Good AC, Warren G, Mathieu M, Muchmore SW, Brown SP, Grant JA, Haigh JA, Nevins N, Jain AN, Kelley B. J Med Chem. 2010;53:3862–3886. doi: 10.1021/jm900818s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu XF, Jiang HL, Li HL. J Chem Inf Model. 2011;51:2372–2385. doi: 10.1021/ci200060s. [DOI] [PubMed] [Google Scholar]
- 51.Lu WQ, Liu XF, Cao XW, Xue MZ, Liu KD, Zhao ZJ, Shen X, Jiang HL, Xu YF, Huang J, Li HL. J Med Chem. 2011;54:3564–3574. doi: 10.1021/jm200139j. [DOI] [PubMed] [Google Scholar]
- 52.Bai F, Liu HY, Tong LJ, Zhou W, Liu L, Zhao ZJ, Liu XF, Jiang HL, Wang XC, Xie H, Li HL. Bioorg Med Chem Lett. 2012;22:1365–1370. doi: 10.1016/j.bmcl.2011.12.067. [DOI] [PubMed] [Google Scholar]
- 53.Glide, version 5.5. Schrodinger, LLC; New York, NY: 2009. [Google Scholar]
- 54.Hansen JD, Grina J, Newhouse B, Welch M, Topalov G, Littman N, Callejo M, Gloor S, Martinson M, Laird E, Brandhuber BJ, Vigers G, Morales T, Woessner R, Randolph N, Lyssikatos J, Olivero A. Bioorg Med Chem Lett. 2008;18:4692–4695. doi: 10.1016/j.bmcl.2008.07.002. [DOI] [PubMed] [Google Scholar]
- 55.Wenglowsky S, Ren L, Ahrendt KA, Laird ER, Aliagas I, Alicke B, Buckmelter AJ, Choo EF, Dinkel V, Feng BNA, Gloor SL, Gould SE, Gross S, Gunzner-Toste J, Hansen JD, Hatzivassiliou G, Liu BN, Malesky K, Mathieu S, Newhouse B, Raddatz NJ, Ran YQ, Rana S, Randolph N, Risom T, Rudolph J, Savage S, Selby LT, Shrag M, Song K, Sturgis HL, Voegtli WC, Wen ZY, Willis BS, Woessner RD, Wu WI, Young WB, Grina J. ACS Med Chem Lett. 2011;2:342–347. doi: 10.1021/ml200025q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zuccotto F, Ardini E, Casale E, Angiolini M. J Med Chem. 2010;53:2681–2694. doi: 10.1021/jm901443h. [DOI] [PubMed] [Google Scholar]
- 57.Hopkins AL, Groom CR, Alex A. Drug Discov Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 58.Ungnade HE, Roberts EM, Kissinger LW. J Phys Chem. 1964;68:3225–3228. [Google Scholar]
- 59.Morphy R. J Med Chem. 2010;53:1413–1437. doi: 10.1021/jm901132v. [DOI] [PubMed] [Google Scholar]
- 60.Cohen MS, Zhang C, Shokat KM, Taunton J. Science. 2005;308:1318–1321. doi: 10.1126/science1108367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kefford R, Arkenau H, Brown M, Millward M, Infante J, Long G, Ouellet D, Curtis M, Lebowitz P, Falchook G. J Clin Oncol. 2010;28:8503. [Google Scholar]
- 62.Jones G, Willett P, Glen RC. J Mol Biol. 1995;245:43–53. doi: 10.1016/s0022-2836(95)80037-9. [DOI] [PubMed] [Google Scholar]
- 63.Jones G, Willett P, Glen RC, Leach AR, Taylor R. J Mol Biol. 1997;267:727–748. doi: 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- 64.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 65.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 66.Huey R, Morris GM, Olson AJ, Goodsell DS. J Comput Chem. 2007;28:1145–1152. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 67.Jorgensen WL, Maxwell DS, TiradoRives J. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 68.Verdonk ML, Berdini V, Hartshorn MJ, Mooij WT, Murray CW, Taylor RD, Watson P. J Chem Inf Comput Sci. 2004;44:793–806. doi: 10.1021/ci034289q. [DOI] [PubMed] [Google Scholar]
- 69.Fang D, Nguyen TK, Leishear K, Finko R, Kulp AN, Hotz S, Van Belle PA, Xu X, Elder DE, Herlyn M. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 70.Hsu MY, Shih DT, Meier FE, Van Belle P, Hsu JY, Elder DE, Buck CA, Herlyn M. Am J Pathol. 1998;153:1435–1442. doi: 10.1016/s0002-9440(10)65730-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fukunaga-Kalabis M, Martinez G, Liu ZJ, Kalabis J, Mrass P, Weninger W, Firth SM, Planque N, Perbal B, Herlyn M. J Cell Biol. 2006;175:563–569. doi: 10.1083/jcb.200602132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson SL, Jung D, Forino M, Chen Y, Satterthwait A, Rozanov DV, Strongin AY, Pellecchia M. J Med Chem. 2006;49:27–30. doi: 10.1021/jm050892j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pomel V, Klicic J, Covini D, Church DD, Shaw JP, Roulin K, Burgat-Charvillon F, Valognes D, Camps M, Chabert C, Gillieron C, Francon B, Perrin D, Leroy D, Gretener D, Nichols A, Vitte PA, Carboni S, Rommel C, Schwarz MK, Ruckle T. J Med Chem. 2006;49:3857–3871. doi: 10.1021/jm0601598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.