

Abstract
DM catalyzes the exchange of peptides bound to Class II major histocompatibility complex (MHC) molecules. Because the dissociation and association components of the overall reaction are difficult to separate, a detailed mechanism of DM catalysis has long resisted elucidation. UV irradiation of DR molecules loaded with a photocleavable peptide (caged Class II MHC molecules) enabled synchronous and verifiable evacuation of the peptide-binding groove and tracking of early binding events in real time by fluorescence polarization. Empty DR molecules generated by photocleavage rapidly bound peptide but quickly resolved into species with substantially slower binding kinetics. DM formed a complex with empty DR molecules that bound peptide with even faster kinetics than empty DR molecules just having lost their peptide cargo. Mathematical models demonstrate that the peptide association rate of DR molecules is substantially higher in the presence of DM. We therefore unequivocally establish that DM contributes directly to peptide association through formation of a peptide-loading complex between DM and empty Class II MHC. This complex rapidly acquires a peptide analogous to the MHC class I peptide-loading complex.
Major histocompatibility complex (MHC)5 molecules are cell surface proteins that present peptides to antigen-specific receptors on T cells. The Class II MHC products are specialized in sampling endosomal compartments to acquire these peptides. Delivery of newly synthesized and assembled Class II MHC proteins to endo-lysosomal compartments is assured by means of the transient association of the MHC αβ heterodimer with the invariant chain, a protein endowed with both chaperone function and an address code (1, 2). In endosomal compartments, the invariant chain is destroyed, yielding a Class II MHC product occupied with an invariant chain-derived remnant, the CLIP peptide (3–5). The HLA-DM (DM) molecule, itself incapable of binding peptide, facilitates replacement of CLIP with antigenic peptides (6–10). The action of DM is not limited to the Class II MHC/CLIP complex and extends to Class II MHC-peptide complexes more generally. Such editing by DM favors presentation to CD4 T cells of those peptides that are most resistant to peptide displacement by DM (9, 11–17).
DM is a membrane-anchored heterodimer that belongs to the extended family of proteins with a MHC fold but lacks a functional peptide-binding groove (18, 19). Mutagenesis experiments identified lateral surfaces on DM and DR molecules that are involved in the interaction between the two proteins. On the DR side, these mutations span the entire length of the ectodomain and are localized to the α1 and β2 domains (20). On the DM side, an extended interaction surface has been mapped that also spans the entire length of the ectodomain (21). These data support a model in which lateral interactions between DM and DR molecules induce a conformational change that destabilizes the DR-bound peptide. It has been proposed that DM disrupts one or several hydrogen bonds between the peptide backbone and conserved DR residues, which could account for the fact that DM accelerates dissociation of peptides that are highly diverse in sequence (12, 19, 22). However, DR-peptide complexes vary substantially in susceptibility to DM and peptide, and DR residues along the entire length of the binding site can affect susceptibility to DM (23). These data suggest that DM causes a more global conformational change of the peptide-binding site.
High affinity peptides form stable, long-lived complexes with DR molecules that have a half-life of days to weeks (24). Purified, soluble DM greatly increases the rate at which labeled peptides dissociate from DR molecules at an acidic pH (6, 9, 12). In such experiments, DM obviously also accelerates the binding of labeled peptides to DR molecules, but it has been difficult to discern whether accelerated peptide binding is solely due to the increased availability of binding sites or also due to a direct contribution of DM to peptide association. The detailed mechanism of DM catalysis has resisted elucidation, partly because of this difficulty of measuring independently the contributions to peptide dissociation and peptide association. Isolating the contribution of DM to peptide association would require a homogenous population of empty Class II MHC molecules in a peptide-receptive state. In theory, empty Class II MHC molecules could be generated by dissociating bound peptide, but empty molecules tend to aggregate during the prolonged incubation periods that would be required. An alternative approach that has been pursued consists of the production of recombinant molecules refolded from denatured DR chains in the absence of peptide, but the majority of such molecules (~95%) bind peptides only slowly, apparently because conversion to more active conformer(s) is required (25, 26).
Because of these technical challenges, two different models have been proposed regarding the interaction of DM with DR molecules. According to the first model, DM interacts only with DR-peptide complexes but not empty DR molecules and catalyzes the interconversion between transient and stable DR-peptide complexes (26). The second model proposes that DM interacts with both DR-peptide complexes and empty DR molecules and that DM acts as a molecular chaperone that rescues empty DR molecules at an acidic pH (27, 28). Resolution of this issue requires a novel approach with which empty DR molecules can be generated rapidly and synchronously. An intriguing new development has been the generation of conditional ligands for Class I MHC molecules. Toebes et al. (29) synthesized photocleavable ligands for Class I MHC molecules and reconstituted the relevant subunits into an assembled Class I MHC complex. UV irradiation resulted in cleavage of the bound peptide and rapid departure of the cleavage fragments from the binding pocket.
Here, we apply this chemistry to generate empty Class II MHC molecules. The experiments make use of a human Class II MHC molecule, the expression of which predisposes its carrier to multiple sclerosis, HLA-DR2 (DRA, DRB1*1501), and a photocleavable peptide based on a T cell epitope of human myelin basic protein (MBP85–99) (30, 31). Rapid, early binding events were followed with a fluorescence polarization (FP) readout, in which the fraction of emitted fluorescent light that retains polarization is proportional to the amount of MHC-bound fluorescent reporter peptide, because of a slower tumbling speed of the MHC-peptide complex compared with free peptide (32). The presence of DM substantially accelerated peptide binding to such empty DR molecules under all of the reaction conditions tested. Mathematical models show excellent concordance with the experimental data for a model wherein DM contributes significantly to peptide association through formation of a complex with empty Class II MHC molecules. We propose that this DM-DR complex represents a peptide-loading complex that facilitates antigen presentation by rapid peptide capture, analogous to the MHC class I peptide-loading complex (33).
EXPERIMENTAL PROCEDURES
Peptide Synthesis and Photocleavage
Peptides were constructed manually with the aid of Fmoc (N-(9-fluorenyl)methoxycarbonyl)-based solid phase peptide chemistry using standard protocols. The photolabile 3-amino-3-(2-nitro)phenyl-propionic acid (Anp) residue was incorporated at the positions indicated in Fig. 1 as described previously (29). Details of the synthesis of MBP-C647 and MBP-N647 are provided in the supplemental information. Photocleavage was examined by preparing a stock solution of lyophilized peptide in Me2SO (10 mg/ml). This stock was diluted in H2O, and 200-µl aliquots were added to a 96-well plate on ice. Two UV sources were used, both of which induced efficient photolysis: a Stratalinker 2400 UV cross-linker (Stratagene, La Jolla, CA) fitted with lamps emitting at a wavelength of 365 nm and a dedicated long wavelength UV lamp (Blak Ray B100AP, UVP, Upland, CA; wavelength of 365 nm). Aliquots were taken at different time points to determine the kinetics of photolysis by liquid chromatography/mass spectrometry or matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry. For liquid chromatography/mass spectrometry analysis, an HP/Agilent 1100 HPLC system (UV detection at 214 nm) equipped with an analytical C18 column (2.1 mm × 50 mm, 3.5-µm particle size) was used in combination with buffers H2O, 0.085% trifluoroacetic acid (A) and MeCN, 0.085% trifluoroacetic acid (B), which was coupled to an LCT mass spectrometer (Waters, Milford, MA) with an electrospray interface. MALDI-TOF mass spectrometry was performed using a Waters MALDI Micromass spectrometer (Waters, Milford, MA) and 3,5-dimethoxy-4-hydroxycinnamic acid as a matrix.
FIGURE 1. Design of photocleavable peptides.

A, peptides based on the MBP85–99 sequence were synthesized to contain a nonproteinogenic amino acid (Anp, blue circles) that undergoes photocleavage when exposed to 365-nm UV light. MBP-488 is the reporter peptide used throughout, and it carries an Alexa-488 at the P5 Cys residue. MBP-C647 and MBP-N647 are peptides onto which Alexa-647 is appended C- or N-terminally to photocleavable MBP-P4* and were used to monitor the departure of cleavage fragments from DR2. B, model of MBP-P4* peptide and major cleavage products formed upon exposure to UV light. Cleavage occurs between the P3 and P4 positions, thus separating the major DR2 anchor residues (P1 Val and P4 Phe) (30).
Protein Expression and Purification
Soluble DR2 with covalently linked CLIP peptide (1 µm) was loaded with DNP-labeled photocleavable peptides (10 µm) following linker cleavage with thrombin (34) overnight at 30 °C in 50 mm sodium citrate, pH 5.2, 100 mm NaCl, 1% octylglucoside, protected from light. Free peptide was removed from the protein complex by HPLC gel filtration (Superose 6, GE Healthcare Bio-Sciences Corp, Piscataway, NJ). The fraction containing the DR2-peptide complex was then affinity-purified on an anti-DNP HPLC column (34). The yields of the purified complexes are listed in supplemental Table S1. Soluble DM was produced from stably transfected Drosophila melanogaster-derived S2 cells as described previously (9, 35).
FP Assay
Association of MBP-488 was monitored by FP as described previously (32). The assay was performed in black polystyrene 384-well flat bottom plates (Corning, Corning, NY) with a nonbinding surface, and the reactions were protected from external light and evaporation with an aluminum foil seal (Excel Scientific, Wrightwood, CA) when the plate was not in the reader. The data were acquired using a Victor3V plate reader (Perkin-Elmer Life Sciences) with 485/20 and 530/30 filter sets to track FP of the Alexa-488 (Invitrogen) label. Typically, the experiments were performed in citrate buffer (50 mm sodium citrate, 150 mm sodium chloride, pH 5.2) in a final volume of 40 µl. FP values were recorded at ambient temperature. The data were acquired from triplicate wells, and wells that contained only the fluorescent MBP-488 peptide were used to determine base-line FP values.
In experiments where FP was monitored in a cuvette, a QM-7 fluorometer was used (Photon Technology International, Lawrenceville NJ). DR2-MBP-P4* was UV-cleaved at the same conditions as above but 7.5-fold concentrated relative to the binding reaction. MBP-488 was added to a quartz cuvette containing 1300 µl of buffer, and the FP followed over time to generate a base line and stabilize the temperature to 20 °C before DR2 and DM addition. Photocleaved DR2 with or without DM was rapidly injected in a volume of 200 µl into the cuvette using a spring-loaded 200-µl Hamilton syringe (CR700-200, Hamilton Company, Reno, NV). A small opening in the cover of the reader allowed the injection to be performed while the FP was being measured. The reaction was under constant mixing with a magnetic stirrer. FP values were measured for 10 min after DR2 addition at a speed of 1 reading/s.
Binding of Photocleavable Peptides to DR2
Freshly cleaved DR2-CLIP complex (final concentration 100 nm) and MBP-488 (final concentration 10 nm) were added to citrate buffer containing 1% octylglucoside. Photocleavable DNP-labeled peptides were added to these reactions at different concentrations (3-fold dilution series, highest concentration of 250 µm) and incubated overnight at 37 °C. The amount of bound MBP-488 was then determined by FP, and the ability of the photocleavable peptides to compete for binding of MBP-488 to DR2 was compared with the MBP85–99 index peptide (supplemental Table S1).
Photocleavage and Exchange
In a typical experiment, a stock solution of DR2-MBP-P4* was deposited into several wells (maximum volume of 50 µl/well) of a 96-well V-bottom plate (Corning Inc., Corning, NY), placed on ice, and allowed to cool. In a separate 96-well plate (no UV control), the samples were shielded from UV light with aluminum foil but otherwise treated identically. The plates were then placed directly under a preheated UV lamp fitted with a UV source emitting at 365 nm (Blak Ray B100AP, UVP, Upland, CA). After 2–10 min of UV irradiation, the contents of wells were pooled (no signs of solvent evaporation were apparent at this point), and 30 µl were dispensed into appropriate wells of a 384-well plate, preloaded with MBP-488 alone or a mixture of DM and MBP-488. Following mixing, the 384-well plate was transferred to the FP reader. The FP values were measured at regular intervals, and the data was collected in triplicate.
Separation of Empty DR2 Molecules from Cleavage Fragments
DR2-MBP-P4* was photocleaved at a concentration of 3 µm as described above. 1 ml of the photocleaved product was loaded onto a HPLC gel filtration column (Superose 6) with 50 mm sodium citrate, 150 mm sodium chloride, pH 5.2, as the mobile phase to separate free peptide fragments from empty DR2 molecules. The protein concentration in collected fractions was determined using the Coomassie Plus protein assay reagent (Pierce). The purified molecules were then assayed by FP for their ability to bind MBP-488. In each experiment, the purification and reaction set-up time required 1.5–2 h starting at the end of photocleavage.
RESULTS
Design and Properties of Photocleavable Peptide Ligands
The structure of DR2 complexed with the MBP85–99 peptide (30) served as the point of departure for the design of photocleavable ligands. We applied the strategy first described by Schumacher and co-workers (29) for Class I MHC products and synthesized peptides that contained an Anp residue at the indicated positions (Fig. 1A). Peptide binding to Class II MHC molecules is stabilized by cooperative hydrogen bond networks and anchor residues nestled deep into the peptide-binding groove (36). A panel of peptides was synthesized that placed the Anp residue between the known anchor residues Val89 (P1) and Phe92 (P4) and critical hydrogen bonds such that, upon cleavage, two fragments would be generated, neither of which was expected to fulfill the criteria for strong binding on its own. We refer to Class II MHC molecules loaded with a photocleavable ligand as caged Class II MHC products. The peptides (MBP-P1* to MBP-P5*) were further fitted with a DNP group through a 4-aminobutyric acid spacer to allow affinity purification of the properly loaded DR2 complex. A model based on the crystal structure of MBP85–99 illustrates the close proximity of the nitro-substituent on the aromatic ring to the backbone of MBP-P4* (Fig. 1B), which accounts for the photochemical liberation of the N-terminal amide and C-terminal nitroso-fragments at the relatively long wavelength of 365 nm, at which proteins are essentially transparent.
Affinity of the Peptide Ligands for DR2 and Efficiency of Photocleavage
The peptide ligands were next used to generate complexes with DR2. A Chinese hamster ovary cell-derived DR2 preparation, in which the CLIP peptide was attached covalently to the DR2 β-chain, was exposed to thrombin, upon which the CLIP peptide could be readily exchanged in DM-independent fashion for other ligands. To avoid confounding effects of a mixture of DR2 loaded with MBP-P4*, DR2-CLIP, and empty DR2, a DNP moiety installed at the N terminus of the photocleavable peptide was used for retrieval of the correspondingly loaded DR2 molecules on an anti-DNP affinity matrix. This final purification step ensured that all DR2 molecules used as input for subsequent reactions were occupied homogeneously with the photocleavable peptide. We identified one MBP-derived ligand, MBP-P4*, for which the caged DR2 complex could be obtained in yields comparable with the DR2-MBP85–99 complex. The affinity of DR2 binding by photocleavable ligands (MBP-P1* to MBP-P5*) was explored by means of a competition assay (Fig. 2A and supplemental Table S1), confirming that the substitution of Phe92 of the MBP85–99 peptide (the P4 anchor in context of DR2) for Anp least affected the affinity (3-fold) for DR2. Therefore, the caged complex that contained MBP-P4*, which most closely matched the affinity of the MBP85–99 index peptide for DR2, was chosen as substrate for peptide exchange reactions in the following experiments.
FIGURE 2. Affinity of photocleavable peptides for DR2 and kinetics of MBP-P4* cleavage.

A, photocleavable peptides MBP-P1* to MBP-P5* were examined for their ability to compete with MBP-488 (10 nm) for DR2 binding (100 nm). MBP-P4* bound most tightly. B and C, photocleavage of MBP-P4* was rapid both in solution or bound to DR2. D, mass spectrometry of intact peptide (top panel, 2+ and 3+ ions) and photocleaved peptide (bottom panel, 1+ ions) showing major cleavage products of molecular weights 1002.6 (C-terminal fragment) and 1091.6 (N-terminal fragment).
Each of the photocleavable peptides was examined individually for its ability to undergo chromatic cleavage. Electron spray ionization mass spectroscopy was used to follow the disappearance of the dominant triple-charged ion species of the parent peptide. In the representative case of MBP-P4* (Fig. 2B), photocleavage was largely complete after 2 min of irradiation. Photocleavage proceeded with similar efficiency when the peptide was bound to DR2 (Fig. 2C). In addition to the expected reaction products of UV fragmentation that were identified with certainty (Fig. 2D), electron spray ionization mass spectroscopy as well as MALDI-TOF spectra showed several additional unidentified cleavage products, whereas the index MBP85–99 peptide was unaffected by UV irradiation (data not shown).
Functional Properties of DR2 Molecules Synchronously Liberated from Bound Peptide
The ability to quickly arrive at an empty state allowed us to examine the association of peptides with Class II MHC and revisit the role of DM in peptide loading. This method differs significantly from published procedures that use refolded empty Class II MHC as a starting point (37, 38). Such preparations contain a mixture of species that actively bind peptide, that slowly convert to an active form, or that are inactive. Furthermore, the conformations that may be accessed by a properly loaded Class II MHC molecule upon loss of its peptide ligand may well differ from those of Class II MHC products generated in the complete absence of a ligand.
Tracking of early stages of peptide association was important for examining the functional properties of DR2 molecules immediately following photocleavage of MBP-P4*. We used an Alexa-488-labeled version of the MBP85–99 peptide (MBP-488, Fig. 1A) as a fluorescent reporter peptide in the FP assay. In the absence of UV irradiation, the DR2-MBP85–99 complex was stable (Fig. 3B), showing no evidence for peptide exchange over the time course followed. The DR2-CLIP complex was less stable, with a slow increase in FP values over the 100-min observation window (Fig. 3C). In the absence of UV irradiation, the DR2-MBP-P4* complex showed peptide exchange behavior intermediate to that of the DR2-MBP85–99 and DR2-CLIP complexes (Fig. 3A, blue curve), consistent with their affinity rank order (MBP > MBP-P4* > CLIP).
FIGURE 3. Rapid binding of peptide to DR2 following photocleavage of MBP-P4*.

Different DR2-peptide complexes (150 nm) were left unilluminated (−UV) or exposed to UV light (+UV) in the presence of MBP-488 (30 nm) for 2 min. Association of MBP-488 was followed by FP after photocleavage. A, rapid association of MBP-488 occurs upon UV irradiation of DR2-MBP-P4*, but not in control reactions. B and C, high affinity MBP85–99 peptide could not be displaced (B), and slow binding to DR2 molecules with the lower affinity CLIP peptide (C) was observed. D, the rate of MBP-488 association was compared for reactions with and without DM. DM was present during photocleavage at an equimolar concentration to DR2, and MBP-488 was added after photocleavage.
When the identical assay was performed upon UV irradiation, neither peptide binding to the preformed DR2-CLIP nor to the DR2-MBP85–99 complex was affected. In contrast, we observed a steep increase in peptide association for the DR2-MBP-P4* complex upon UV exposure (Fig. 3A, red curve). In this experiment, the MBP-488 reporter peptide was included during photocleavage, and the reaction had already proceeded significantly when the first data points could be recorded, even though photocleavage was performed for 2 min on ice. Therefore, in the absence of DM, rapid peptide association occurs to what we propose are newly vacated DR2 molecules.
We then assessed the potential contribution of DM to MBP-488 association following photocleavage of the bound peptide. DM was added before photocleavage at an equimolar concentration relative to DR2 (150 nm), and MBP-488 was then added to measure peptide association. As shown above, the peptide binding reaction proceeded rapidly following photocleavage in the absence of DM, but DM further accelerated the rate of peptide association (Fig. 3D). There may be several explanations for this DM-dependent increase in the kinetics of peptide association: removal of residual uncleaved MBP-P4*, removal of cleavage fragments from the peptide-binding site, or, alternatively, a direct contribution of DM to peptide association.
The DR2 Conformer Created by Peptide Photocleavage Is Short-lived
Mass spectrometry showed that DR2-bound MBP-P4* was cleaved at a rate similar to that of free MBP-P4* (Fig. 2, B and C), and incomplete cleavage could thus not account for the increased rate of peptide association in the presence of DM. Acceleration of peptide association was observed even when photocleavage was extended to 10 min (Fig. 4B). Extension of photocleavage also demonstrated that the peptide-receptive DR2 conformer created by photocleavage was short-lived (Fig. 4A). During UV cleavage, significant quantities of MBP-488 rapidly bound to uncaged DR2 (Fig. 4A). In contrast, the reaction kinetics were substantially different when MBP-P4* was photocleaved in the absence of MBP-488. More than 50 min at a more favorable temperature (room temperature) were required to reach 100 mP, a reading reached in less than 10 min on ice when MBP-488 was available in the course of photocleavage in slight molar excess (220 nm MBP-488 to 160 nm DR2). Photolysis of MBP-P4* thus creates a peptide-receptive intermediate of DR2. This critical conformer is short-lived and converts to other species that bind peptide with substantially slower kinetics.
FIGURE 4. Photocleavage generates labile, short-lived DR species that rapidly bind peptide.

A, DR2-MBP-P4* (160 nm) was photocleaved for 10 min on ice in the presence (red line) or absence (blue line) of MBP-488 (220 nm). When peptide was present during photocleavage, the reaction proceeded rapidly (10 min on ice) to ~100 mP, whereas peptide association was slower when the peptide was added following photocleavage (<100 FP after 50 min). Time 0 is defined by the addition of MBP-488 following photocleavage (blue line). B, dose-dependent acceleration of peptide association to empty DR2. DM was present at different concentrations (150 nm to 5 µm) during photocleavage (10 min, 160 nm DR2-MBP-P4*), and MBP-488 was added (330 nm) after photocleavage. Saturation was not observed because of the lower affinity of soluble DM for soluble DR compared with full-length molecules (42).
DM Forms a Complex with Empty DR That Maintains Rapid Peptide Binding Properties over Extended Periods of Time
In the absence of DM, the initial rate of MBP-488 binding was slower when the photocleavage time was extended from 2 to 10 min (compare Fig. 3D and Fig. 4B). When DM was present during the 10-min photocleavage period, peptide binding was more rapid. Association kinetics of MBP-488 were dependent on the DM concentration (Fig. 4B), consistent with the modest affinity of soluble DM for DR (39). Only a fraction of DR molecules would be expected to be bound to DM at a given time at the DM concentrations used here.
To assess the effect of DM on the very early phase of the reaction, we used a cuvette-based system in which FP values could be tracked continuously. The reaction was initiated by the addition of DR2 and DM to a cuvette already containing MBP-488. Although DM clearly accelerated peptide association when added following photocleavage, a substantial initial burst of peptide binding was seen only when DM was present during photocleavage (Fig. 5A). We infer that formation of the DM-empty DR peptide-loading complex in this experimental setting is optimal when DR molecules have just lost their peptide.
FIGURE 5. Analysis of early, rapid binding events.

A, photocleaved DR2-MBP-P4* (160 nm) ± DM (160 nm) was injected into a cuvette containing 220 nm MBP-488 to monitor the early stages of the peptide binding reaction by FP, immediately upon the addition of DR2. When DM was present during photocleavage, a rapid burst of binding was observed. The rate of MBP-488 association was intermediate when DM was added following photocleavage. Association in the absence of DM was slower. B, incubation of DM with empty DR2 restores and maintains the highly active DR2 conformer. DR2-MBP-P4* was photocleaved for 10 min in the absence of DM. Association of MBP-488 was then compared between reactions with or without DM (1 µm). After different preincubation times (0–2 h), the DR/DM mix or DR alone was injected into a cuvette containing 50 nm MBP-488, and the FP values were monitored continuously.
How stable is the DM-empty DR peptide-loading complex? We uncaged DR2 molecules in the absence of DM, added DM, and then assessed peptide binding upon delayed addition of the MBP-488 peptide (Fig. 5B). DR2 molecules progressively lost activity with longer incubation times in the absence of DM, and only slow kinetics of peptide association were observed with 10-min to 2-h delays following photocleavage (Fig. 5B). In contrast, the rapid peptide binding properties of the DM-DR complex were maintained even with prolonged incubation times prior to the addition of peptide (11 min to 2 h) (Fig. 5B). The observed differences are inconsistent with the interpretation that DM merely removes residual uncleaved MBP peptide from DR2. Mass spectrometry shows that the vast majority of MBP-P4* is photocleaved even when bound to DR2. Maintenance of the rapid peptide binding properties of the DM-DR complex over extended periods of time is a remarkable trait in view of the rapid decay of active DR in the absence of DM.
The Contribution of DM to Peptide Association Is Not Due to Cleavage Fragments
Could retention of the peptide cleavage fragments affect the results of peptide binding measurements? We considered this explanation unlikely, given the strategic placement of the photocleavage site in the center of the peptide. We nevertheless designed three experiments to directly address this issue. We separated empty DR2 molecules from cleavage fragments by size exclusion chromatography and confirmed removal of fragments by monitoring absorbance of the DNP tag (350 nm) and mass spectroscopy (data not shown). We then compared the contribution of DM to peptide binding (Fig. 6, A and B) and obtained comparable results. Could the add-back of cleavage fragments affect the kinetics of peptide association in the presence or absence of DM? Even at a concentration equimolar to DR2 (150 nm), cleavage fragments did not affect the kinetics of peptide association in the presence or absence of DM and regardless of DR:DM ratios (Fig. 6, C and D). We conclude that the reaction is primarily dependent on the concentration of DM because of its low affinity for soluble DR2.
FIGURE 6. Peptide cleavage fragments do not account for DM contribution to peptide association.

DR2-MBP-P4* was photocleaved for 5 min, and peptide fragments were removed from DR2 on a HPLC gel filtration column. A and B, the contribution of DM (1 µm) to the kinetics of MBP-488 (1 µm) association was compared between reactions with photocleaved DR2-MBP-P4* (1 µm) from which cleavage fragments had been removed (B) or not removed (A) by Superose 6 size exclusion chromatography. C and D, cleavage fragments added back at different concentrations (D) were without effect on reactions with DR2 molecules not subjected to gel filtration (C) at concentrations of 150 nm DR2-MBP-P4*, 150 nm MBP-488, and 1 µm DM. D, photocleaved peptide fragments were added back at concentrations ranging from 15 nm to 150 nm (DR2 at 150 nm) following purification of DR2 by size exclusion chromatography. The cleavage fragments also did not affect association of MBP-488 in the absence of DM (−DM).
Finally, we synthesized two peptide derivatives based on the MBP-P4* peptide, but onto which an Alexa-647 fluorophore was installed at a single Cys residue either at the N or C terminus of the photocleavable peptide (Fig. 1A, MBP-N647 and MBP-C647, respectively). Without UV treatment, the complexes containing MBP-N647 and MBP-C647 were stable for at least 100 min (supplemental Fig. S1). Upon UV irradiation the decrease in FP is biphasic, with a rapid initial phase that cannot be captured because irradiation and FP measurements must necessarily be conducted in two different geometries. As soon as measurements can be made, the FP signal has already reached nearly plateau levels, with only a slow decay phase that follows for both MBP-N647 and MBP-C647 (supplemental Fig. S1A). In the same experiment, the association of MBP-488 was recorded. The low FP signal in the 488-nm channel in the absence of UV cleavage indicates that DR2 is firmly loaded with either MBP-N647 or MBP-C647, neither of which are displaced by the mere addition of free MBP-488 (supplemental Fig. S1B). Photolysis of these caged MHC molecules enables the loading of MBP-488 in a single phase kinetic fashion. Cleavage of the caged DR2 complex is therefore rapid, and retention of both N- and C-terminal fragments is significantly less than association of MBP-488 with DR2. DM-dependent removal of cleavage products therefore does not appear to be a rate-determining factor for MBP-488 binding.
Displacement of MBP-N647 and MBP-C647 by the addition of DM alone, in the absence of UV irradiation (supplemental Fig. S1C) shows the expected acceleration of exchange for MBP-C647 (compare with Fig. 3D). However, the DR2-MBP-N647 is less responsive to inclusion of DM. This suggests that the presence of the fluorophore at the N terminus of the photocleavable peptide may sterically hinder access of DM (20), whereas a C-terminal addition of a fluorophore does not.
Experimental Observations Are Integrated by a Mathematical Model of the Kinetic Processes
For a deeper mechanistic understanding of the role of DM in facilitating peptide association, we developed mathematical models of the pertinent processes (Fig. 7) to fit our experimental observations. Our kinetic data obtained with and with out DM can be used to examine the veracity of plausible models for the role of DM after peptide is released from the MHC-binding pocket upon UV irradiation. We developed mathematical models for different hypotheses, and calculations showed whether they do or do not compare well with the experimental data. This allowed us to eliminate certain models from further consideration and led us to a plausible model for how DM may function post-peptide removal. We began by considering whether the following hypothesis could fit the kinetic data when no DM was added during UV irradiation,
| (Eq. 1) |
where pMHC is the peptide-loaded DR, p*MHC is the species with the peptide photocleaved, eMHC is the empty DR molecule, and fpMHC is DR loaded with the peptide present in solution. The quantities labeled as ki are the rate coefficients characterizing the kinetics of the corresponding reactions. Ordinary differential equations describing the kinetics of this simple pathway were then developed (see supplemental information). A nonlinear regression scheme available as part of the MAT-LAB suite of programs was used to fit the kinetic constants in this model to the experimental data. This hypothesis could not fit the experimental data (not shown) because it cannot account for the second slow phase of peptide binding.
FIGURE 7. Model showing the rate constants used in mathematical models of peptide loading in the presence and absence of DM.

A, peptide-loaded MHC (pMHC), empty MHC (eMHC), and fluorescent peptide-loaded MHC (fpMHC) are shown in the absence of DM (top row) and interacting with DM (bottom row). Peptide photocleavage creates a short-lived MHC molecule with bound cleavage fragments (p*MHC), whereas incubation in the absence of peptide creates an inactive form (iMHC). eMHC remains in a peptide-receptive form only for short periods of time, but eMHC bound in the DM-eMHC complex (boxed) remains active for extended periods of time. B, the hypothesis represented by Equation 2 fits the experimental data without DM very well. The initial concentration of pMHC was 150 nm, and the amount of MBP-488 added after 10 min of UV was 200 nm. C, the model represented by Equation 3 fits the experimental data well in the presence of variable amounts of DM. The initial concentration of pMHC was 150 nm, and the amount of MBP-488 added after 10 min of UV was 200 nm. DM addition occurred at the same time as MBP-488 addition. A summary of the concordance between experimental data and the kinetic model for conditions not shown here is provided in the supplemental information. Because of uncertainties associated with the length of time required to add MBP-488 and to take the first reading, it was assumed that 30 s elapsed during this process for all DM experiments. Because the no DM data were read last, it was assumed that 2 min elapsed before the first reading. The values of the delay times were chosen to improve the quality of the fit. We also carried out the analyses assuming that the elapsed times were negligible. None of the qualitative conclusions changed upon making this assumption; only the quantitative comparisons with experimental data were less precise at short times.
This is not surprising, because this hypothesis does not account for the fact that DR can unfold or aggregate to form species that are not receptive to peptide binding. Thus, we examined the following hypothesis, which includes this feature.
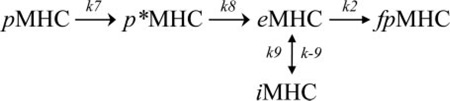 |
(Eq. 2) |
The symbols are the same as before, except that iMHC denotes the inactive conformer or aggregates of DR. It does not make sense to treat aggregates and unfolded conformers of DR as separate species because our experimental data cannot distinguish these species. Fitting this model to the experimental data without DM yields excellent fits as seen in Fig. 7B. The best fit parameter values were determined to be: k2 = 9.92 × 10−5 nm−1 min−1, k7 = 1.00 min−1, k8 = 1.00 min−1, k9 = 1.24 × 10−4 nm−1 min−1, k−9 = 1.39 × 10−4 min−1, and n = 2. n is the effective number of MHC molecules involved in the inactive form. A wide range of k9, k−9, and n values are capable of producing the same quality of fit. This emphasizes that the experimental data cannot distinguish between unfolding (n = 1) and aggregation (n > 1). The model is quite sensitive to the value of k2, because a value very close to 9.00 × 10−5 nm−1 min−1 is necessary to capture the behavior at the initial times.
Thus, we conclude that a key aspect of the mechanism is that the aggregated/unfolded DR molecules can slowly revert to the active conformer when peptide is present in excess. This is the origin of the second slow phase of peptide uptake in the data shown in Fig. 3D.
The kinetic parameters obtained by fitting this model (Equation 2) to the data without DM reflect reactive processes that should proceed in the same way if DM is added. So, we then fixed the values of these parameters to those described above and considered a model for the additional processes that can occur when DM is added. Our experimental findings showed that DM can restore a DR conformer that is peptide-receptive, and we model this by positing that DM binds DR molecules to create a species that can bind peptides. Then, by fitting the experimental data to the following model that includes these processes, we asked whether the DM-bound DR molecules could bind peptides more or less rapidly than the empty receptive DR molecules in the absence of DM.
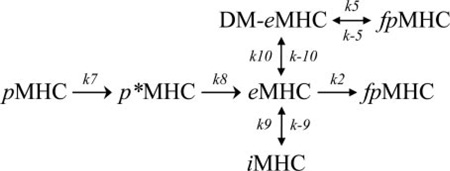 |
(Eq. 3) |
The model represented by Equation 3 fits the experimental data well in the presence of variable amounts of DM (Fig. 7C). The parameters, k2, k7, k8, k9, k−9, and n, were kept constant at values determined previously (Fig. 7B), and the new parameters fit the data best for the following values: k5 = 5.62 × 10−4 nm−1 min−1, k−5 = 6.65 × 10−6 nm−1 min−1, k10 = 0.59 nm−1 min−1, and k−10 = 177.57 min−1. Multiple combinations of parameter values are capable of producing the same fit; however, k5 must be at least two times greater than k2 to capture the observed experimental behavior. We also studied a model where DM could also bind to the inactive form of DR (iMHC) directly and create a stabilized peptide-receptive form. Even in this model, the experimental data can be fit only if k5 is greater than k2. Thus, it appears that DM-bound DR molecules bind peptide more rapidly than the receptive conformer of DR alone.
Of course, we are aware that our parameter sensitivity analysis and the space of models we have explored are not exhaustive. Nevertheless, the experimental data and the values of parameters obtained from the kinetic analyses suggest the following mechanistic picture. DM binds DR to stabilize it from unfolding and aggregating. The binding also probably leads to a conformational change in DR. The resulting conformation may be very favorable for peptide binding to the DR groove because it stabilizes the corresponding transition state. This may underlie the faster rate of peptide binding to the DM-stabilized DR compared with the receptive state of DR alone because peptide binding to the latter may proceed via an induced fit mechanism with associated free energy costs that are no longer relevant for DM-bound DR.
DISCUSSION
Peptide binding may at first glance appear to be a rather simple process, in which a peptide interacts with an available Class II MHC site. Closer inspection reveals the presence of multiple distinct species of different behavior and stability (12, 26, 40): (i) stable DR-peptide complexes from which peptides dissociate at very slow rates (half-life of days to weeks); (ii) a transition state induced by DM from which peptide dissociates rapidly; (iii) a labile empty DR conformer that rapidly binds peptide, but is short-lived; (iv) inactive and aggregated forms; and (v) DM-bound empty DR molecules that are stable and bind peptide rapidly.
Two different models of DM action have been proposed: either DM interacts with both empty and peptide-loaded species (27, 28) or only with peptide-loaded Class II MHC molecules (26). Pulse-chase experiments demonstrated a transient association of DR with DM. When invariant chain degradation was blocked by leupeptin, DM could be retrieved in complex with DR molecules together with a 21-kDa N-terminal fragment of the invariant chain. Furthermore, immunoprecipitation experiments showed that purified DM binds to DR/CLIP (27). Mass spectrometry-based analysis of bound peptides demonstrated that DM-DR complexes immunopurified from cells were largely devoid of peptides, suggesting that DM continues to interact with DR molecules following peptide dissociation. Furthermore, the presence of DM prevented aggregation of DR molecules at acidic pH. DM was therefore proposed to act as a chaperone that prevents inactivation of empty DR molecules (28). More recent experiments showed that DM interacts only with peptide-filled DR molecules and that DM catalyzes peptide exchange by facilitating a conformational change in the peptide-binding complex. This study (26) utilized empty DR molecules refolded from Escherichia coli inclusion bodies in the absence of peptide. DM had little or no effect on the conversion between peptide-receptive and -averse DR or on the rapid bimolecular binding reaction between empty DR and free peptide. The stabilization observed previously was attributed to an effect on the peptide binding reaction. DM would prevent inactivation indirectly through catalysis of productive binding in a reaction that competes with inactivation (26).
The dissection of key steps in peptide binding thus requires approaches with which critical, short-lived intermediates can be generated rapidly and synchronously. We created caged Class II MHC molecules from which peptide could be rapidly removed. Chinese hamster ovary cell-produced soluble DR2 molecules were converted, in DM-independent fashion, into DR2 complexes with photocleavable peptide and purified by means of an affinity tag attached to the peptide. This approach ensured that the input population of DR2 molecules was homogeneous with respect to peptide occupancy and ruled out DM contamination of the reactants. With the photocleavable Anp amino acid replacing the Phe92 residue at the P4 anchor position, MBP-P4* retained the highest affinity for DR2. This is a relatively minor change in the overall peptide topology, because the aromatic character of the amino acid side chain is preserved, and the P4 pocket is sufficiently large to accommodate a nitro-substituent on the phenyl side chain. When UV-irradiated, the caged complex is quickly converted into an uncaged peptide-receptive DR2 species. The peptide fragments themselves have negligible affinity for DR2, based on experiments that made use of N- or C-terminally derivatized peptides (MBP-C647 and MBP-N647), in which the departure of both of the anticipated cleavage products was monitored independently (supplemental Fig. S1) or when MBP-P4* fragments were added back to empty DR2 purified by size exclusion chromatography (Fig. 6D). The cleavage fragments had no effect on the progression of binding of MBP-488 to empty DR2, neither in the presence nor in the absence of DM.
Comparison of peptide binding reactions under different conditions allowed us to identify key factors that determined the magnitude of the DM contribution to peptide association. Acceleration of peptide association by DM was most pronounced with delayed addition of DM, such as experiments in which DM was added following photocleavage (Fig. 4), the duration of photolysis was extended from 2 to 10 min in the absence of DM (Fig. 5B), or cleavage fragments were removed by gel filtration chromatography (Fig. 6D). Thus, DM not only stabilizes an active, peptide-receptive DR2 conformer, but it can also restore such a conformer when the DR molecule lacks contact with peptide for extended periods of time. Restoration of the active conformation of DR by DM may be explained by two mechanisms: DM may bind to multiple DR species and cause the transition from a less active conformer to the active form, or it may bind to active DR species that are short-lived in its absence and indirectly favor the equilibrium toward the active conformer. Particularly notable in these experiments is that reactions with and without DM did not reach the same equilibrium, indicating that a substantial fraction of DR2 molecules is lost to the competing inactivation/aggregation reaction when the active conformer is not restored and maintained by DM. In the presence of DM, the active conformer is stable because near identical peptide association kinetics were observed when peptide addition was delayed for 2 h compared with reactions in which peptide was added directly after photocleavage (Fig. 5B).
Could the observed DM contribution be due to the removal of residual intact peptide or even cleavage fragments from the binding site? The following findings argue against this explanation: (i) Mass spectrometry demonstrated rapid cleavage of MBP-P4*, regardless of whether it was free in solution or DR2-bound. (ii) Monitoring of the DNP group based on its unique absorbance at 350 nm during separation of DR2 from cleavage fragments by size demonstrated removal of DNP-tagged peptide. (iii) The FP method measures the fraction of peptide that is DR2-bound versus peptide that remains free in solution. At equimolar quantities of peptide and DR, substantial changes in FP thus only occur when a considerable fraction of DR2 molecules bind peptide. (iv) The DM contribution to peptide association is more pronounced with longer photolysis times. If acceleration of peptide association were due to removal of residual peptide or peptide fragments, this effect should be diminished by longer photolysis times.
This study resolves the controversy of whether DM acts only on peptide-loaded DR species (26) or whether it also acts as a chaperone that stabilizes empty DR molecules (27, 28). We demonstrate that DM forms a complex with empty DR molecules that binds peptide with faster kinetics than DR molecules that just lost their peptide cargo, indicating that DM plays an important role in the peptide association stage. The differences in the conclusions reached by Zarutskie et al. (26) and this study are probably related to the DR preparations that were utilized. Zarutskie et al. (26) utilized empty DR1 molecules refolded in the absence of peptide from E. coli inclusion bodies. The majority of these refolded molecules (~95%) are in a peptide nonreceptive state, apparently resistant to the action of DM. In contrast, newly vacated DR molecules maintain the ability to interact with DM for at least some time.
We propose that DM forms a peptide-loading complex with empty DR that plays an important role in rapid peptide capture. Peptides are short-lived in cells because of proteolytic attack (41), and rapid binding may be essential for display of a diverse array of peptides, many of which contain cleavage sites for proteases present in the peptide-loading compartment.
Supplementary Material
Footnotes
This work was supported by National Institutes of Health Grants RO1 AI057493 (to K. W. W.) and PO1 AI045757 (to K. W. W. and H. L. P.).
The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and supplemental Fig. S1.
Supported by a postdoctoral fellowship from the Netherlands Organization for Scientific Research.
Supported by National Institutes of Health Grant P01 AI071195 and Director’s Pioneer Award 1DP1OD001022.
The abbreviations used are: MHC, major histocompatibility complex; FP, fluorescence polarization; Anp, 3-amino-3-(2-nitro)phenyl-propionic acid; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; HPLC, high pressure liquid chromatography; DNP, dinitrophenyl; MBP, myelin basic protein.
REFERENCES
- 1.Roche PA, Cresswell P. Nature. 1990;345:615–618. doi: 10.1038/345615a0. [DOI] [PubMed] [Google Scholar]
- 2.Bakke O, Dobberstein B. Cell. 1990;63:707–716. doi: 10.1016/0092-8674(90)90137-4. [DOI] [PubMed] [Google Scholar]
- 3.Riberdy JM, Newcomb JR, Surman MJ, Barbosa JA, Cresswell P. Nature. 1992;360:474–477. doi: 10.1038/360474a0. [DOI] [PubMed] [Google Scholar]
- 4.Ghosh P, Amaya M, Mellins E, Wiley DC. Nature. 1995;378:457–462. doi: 10.1038/378457a0. [DOI] [PubMed] [Google Scholar]
- 5.Wu S, Gorski J. Mol. Immunol. 1996;33:371–377. doi: 10.1016/0161-5890(95)00159-x. [DOI] [PubMed] [Google Scholar]
- 6.Denzin LK, Cresswell P. Cell. 1995;82:155–165. doi: 10.1016/0092-8674(95)90061-6. [DOI] [PubMed] [Google Scholar]
- 7.Busch R, Doebele RC, Patil NS, Pashine A, Mellins ED. Curr. Opin. Immunol. 2000;12:99–106. doi: 10.1016/s0952-7915(99)00057-6. [DOI] [PubMed] [Google Scholar]
- 8.Busch R, Rinderknecht CH, Roh S, Lee AW, Harding JJ, Burster T, Hornell TM, Mellins ED. Immunol. Rev. 2005;207:242–260. doi: 10.1111/j.0105-2896.2005.00306.x. [DOI] [PubMed] [Google Scholar]
- 9.Sloan VS, Cameron P, Porter G, Gammon M, Amaya M, Mellins E, Zaller DM. Nature. 1995;375:802–806. doi: 10.1038/375802a0. [DOI] [PubMed] [Google Scholar]
- 10.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco JJ, Mellins E. Nature. 1994;368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 11.Kropshofer H, Vogt AB, Moldenhauer G, Hammer J, Blum JS, Hammerling GJ. EMBO J. 1996;15:6144–6154. [PMC free article] [PubMed] [Google Scholar]
- 12.Weber DA, Evavold BD, Jensen PE. Science. 1996;274:618–620. doi: 10.1126/science.274.5287.618. [DOI] [PubMed] [Google Scholar]
- 13.Lich JD, Jayne JA, Zhou D, Elliott JF, Blum JS. J. Immunol. 2003;171:853–859. doi: 10.4049/jimmunol.171.2.853. [DOI] [PubMed] [Google Scholar]
- 14.Lovitch SB, Petzold SJ, Unanue ER. J. Immunol. 2003;171:2183–2186. doi: 10.4049/jimmunol.171.5.2183. [DOI] [PubMed] [Google Scholar]
- 15.Nanda NK, Sant AJ. J. Exp. Med. 2000;192:781–788. doi: 10.1084/jem.192.6.781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazarski CA, Chaves FA, Sant AJ. J. Exp. Med. 2006;203:1319–1328. doi: 10.1084/jem.20060058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katz JF, Stebbins C, Appella E, Sant AJ. J. Exp. Med. 1996;184:1747–1753. doi: 10.1084/jem.184.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fremont DH, Crawford F, Marrack P, Hendrickson WA, Kappler J. Immunity. 1998;9:385–393. doi: 10.1016/s1074-7613(00)80621-4. [DOI] [PubMed] [Google Scholar]
- 19.Mosyak L, Zaller DM, Wiley DC. Immunity. 1998;9:377–383. doi: 10.1016/s1074-7613(00)80620-2. [DOI] [PubMed] [Google Scholar]
- 20.Doebele CR, Busch R, Scott MH, Pashine A, Mellins DE. Immunity. 2000;13:517–527. doi: 10.1016/s1074-7613(00)00051-0. [DOI] [PubMed] [Google Scholar]
- 21.Pashine A, Busch R, Belmares MP, Munning JN, Doebele RC, Buckingham M, Nolan GP, Mellins ED. Immunity. 2003;19:183–192. doi: 10.1016/s1074-7613(03)00200-0. [DOI] [PubMed] [Google Scholar]
- 22.Narayan K, Chou CL, Kim A, Hartman IZ, Dalai S, Khoruzhenko S, Sadegh-Nasseri S. Nat. Immunol. 2007;8:92–100. doi: 10.1038/ni1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Belmares MP, Busch R, Wucherpfennig KW, McConnell HM, Mellins ED. J. Immunol. 2002;169:5109–5117. doi: 10.4049/jimmunol.169.9.5109. [DOI] [PubMed] [Google Scholar]
- 24.Lanzavecchia A, Reid PA, Watts C. Nature. 1992;357:249–252. doi: 10.1038/357249a0. [DOI] [PubMed] [Google Scholar]
- 25.Joshi RV, Zarutskie JA, Stern LJ. Biochemistry. 2000;39:3751–3762. doi: 10.1021/bi9923656. [DOI] [PubMed] [Google Scholar]
- 26.Zarutskie JA, Busch R, Zavala-Ruiz Z, Rushe M, Mellins ED, Stern LJ. Proc. Natl. Acad. Sci. U. S. A. 2001;98:12450–12455. doi: 10.1073/pnas.211439398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Denzin LK, Hammond C, Cresswell P. J. Exp. Med. 1996;184:2153–2165. doi: 10.1084/jem.184.6.2153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kropshofer H, Arndt SO, Moldenhauer G, Hammerling GJ, Vogt AB. Immunity. 1997;6:293–302. doi: 10.1016/s1074-7613(00)80332-5. [DOI] [PubMed] [Google Scholar]
- 29.Toebes M, Coccoris M, Bins A, Rodenko B, Gomez R, Nieuwkoop NJ, van de Kasteele W, Rimmelzwaan GF, Haanen JB, Ovaa H, Schumacher TN. Nat. Med. 2006;12:246–251. doi: 10.1038/nm1360. [DOI] [PubMed] [Google Scholar]
- 30.Smith KJ, Pyrdol J, Gauthier L, Wiley DC, Wucherpfennig KW. J. Exp. Med. 1998;188:1511–1520. doi: 10.1084/jem.188.8.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wucherpfennig KW, Sette A, Southwood S, Oseroff C, Matsui M, Strominger JL, Hafler DA. J. Exp. Med. 1994;179:279–290. doi: 10.1084/jem.179.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholson MJ, Moradi B, Seth NP, Xing X, Cuny GD, Stein RL, Wucherpfennig KW. J. Immunol. 2006;176:4208–4220. doi: 10.4049/jimmunol.176.7.4208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cresswell P, Ackerman AL, Giodini A, Peaper DR, Wearsch PA. Immunol. Rev. 2005;207:145–157. doi: 10.1111/j.0105-2896.2005.00316.x. [DOI] [PubMed] [Google Scholar]
- 34.Day CL, Seth NP, Lucas M, Appel H, Gauthier L, Lauer GM, Robbins GK, Szczepiorkowski ZM, Casson DR, Chung RT, Bell S, Harcourt G, Walker BD, Klenerman P, Wucherpfennig KW. J. Clin. Investig. 2003;112:831–842. doi: 10.1172/JCI18509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Busch R, Reich Z, Zaller DM, Sloan V, Mellins ED. J. Biol. Chem. 1998;273:27557–27564. doi: 10.1074/jbc.273.42.27557. [DOI] [PubMed] [Google Scholar]
- 36.Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 37.Altman JD, Reay PA, Davis MM. Proc. Natl. Acad. Sci. U. S. A. 1993;90:10330–10334. doi: 10.1073/pnas.90.21.10330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frayser M, Sato AK, Xu L, Stern LJ. Protein Expression Purif. 1999;15:105–114. doi: 10.1006/prep.1998.0987. [DOI] [PubMed] [Google Scholar]
- 39.Vogt AB, Kropshofer H, Moldenhauer G, Hammerling GJ. Proc. Natl. Acad. Sci. U. S. A. 1996;93:9724–9729. doi: 10.1073/pnas.93.18.9724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rabinowitz JD, Vrljic M, Kasson PM, Liang MN, Busch R, Boniface JJ, Davis MM, McConnell HM. Immunity. 1998;9:699–709. doi: 10.1016/s1074-7613(00)80667-6. [DOI] [PubMed] [Google Scholar]
- 41.Lennon-Dumenil AM, Bakker AH, Wolf-Bryant P, Ploegh HL, Lagaudriere-Gesbert C. Curr. Opin. Immunol. 2002;14:15–21. doi: 10.1016/s0952-7915(01)00293-x. [DOI] [PubMed] [Google Scholar]
- 42.Weber DA, Dao CT, Jun J, Wigal JL, Jensen PE. J. Immunol. 2001;167:5167–5174. doi: 10.4049/jimmunol.167.9.5167. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.