

Abstract

The synthesis of the novel Lewis acid, aluminum tris(2,6-di-2-naphthylphenoxide) (ATNP), and its use in the vinylogous aldol reaction between methyl crotonate and enolizable aldehydes are described. ATNP is related to Yamamoto’s Lewis acid, aluminum tris(2,6-diphenylphenoxide) (ATPH), but the 2-naphthyl groups more effectively block the α-position of aldehydes, enabling the selective enolization of crotonate esters in the presence of enolizable aldehydes. Vinylogous aldol reactions then proceed smoothly and in high yields with a variety of substrates.
The aldol reaction is a versatile carbon-carbon bond forming transformation that is widely used and of great importance in organic synthesis.1 The extension of this reaction to its vinylog (i.e., the vinylogous aldol reaction) is well known, and in its simplest form, proceeds via a dienolate and an aldehyde.2 The product, a δ-hydroxy-α,β-unsaturated carbonyl compound, is rich in functionality, and as this bond construction occurs in an uncommon position (between the γ- and δ-carbons of the product), it offers novel strategic disconnections in synthetic planning. In recent years, the reaction has gained significant attention from the organic community, and an important advance was described by Hisashi Yamamoto with the use of the very bulky Lewis acid, aluminum tris(2,6-diphenylphenoxide) (ATPH 1, Scheme 1).3 ATPH is used in reactions of lithium enolates and aldehydes, and it is thought to bind to both the dienolate and the aldehyde components of the reaction. Due to its bulk, it prevents reaction at the α-carbon of the dienolate, thereby forcing the reaction to occur at the distal γ-carbon (Scheme 1). Remarkably, Yamamoto has used ATPH to direct reactivity to the terminal carbon in substrates as large as hexaenolates derived from pentaenoates.3b
Scheme 1.

Yamamoto Vinylogous Aldol Reaction with ATPH
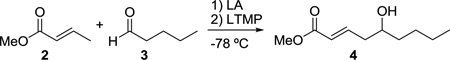
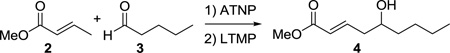
An important feature of the Yamamoto protocol is that the components must all be combined prior to the addition of the base; attempts to conduct the reaction in a stepwise fashion wherein the enolate is first formed then the aldehyde added provide significantly diminished yields.3b,4 Because this reaction is not amenable to a two-step enolization/aldehyde addition protocol, if the aldehyde component is enolizable, there must be selectivity in the initial deprotonation step; otherwise, a mixture of enolates will result, providing a mixture of products. While Yamamoto has described notable selectivity with many substrate combinations, in the case of crotonate esters, the reaction provides low yields when the aldehyde is enolizable and unhindered at the α-carbon. For example, attempted vinylogous aldol reaction between methyl crotonate (2) and valeraldehyde (3) provides the desired product in a yield of only 22% (Table 1, entry 1).3b This significantly limits the scope of this reaction, and we recently had a need to apply this method to an unbranched enolizable aldehyde. We have, therefore, devised a Lewis acid to overcome this limitation, and describe our results herein.
Table 1.
Vinylogous Aldol Reaction of Enolizable Aldehydes with Various Known Lewis Acidsa
 | |||
|---|---|---|---|
| entry | Lewis acid | LA structure |
yield (%) |
| 1 | ATPH, 1 | 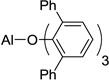 |
22 |
| 2 | MAD, 5 |  |
12 |
| 3 | MABR, 6 |  |
21 |
| 4 | MAT, 7 | 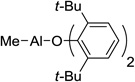 |
15 |
| 5 | ATD, 8 | 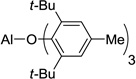 |
30 |
| 6 | Me-ATPH, 9 | 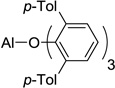 |
-b |
See text for reaction conditions.
A complex mixture was observed.
In order to use enolizable aldehydes in this reaction, we required a Lewis acid that upon binding to an aldehyde renders it immune to enolization by bulky kinetic bases such as LDA or LTMP. We, therefore, studied a number of Lewis acids as surrogates for ATPH in a model vinylogous aldol reaction between 2 and 3 in search of one possessing the right combination of tight binding and bulk (Table 1). Under standard reaction conditions (addition of a cooled solution of LTMP (2.3 equivs) in THF to a toluene solution of the Lewis acid (3.3 equivs), ester (2 equivs) and aldehyde at −78 °C), none of the known bulky Lewis acids that we studied, including those devised by Yamamoto (MAD5 5, MABR6 6, MAT7 7, ATD8 8, Me-ATPH9 9), were successful (Table 1).
We wished to maintain the major design features of ATPH and sought a Lewis acid with comparable bulk in the vicinity of the aluminum, but with extended “reach” such that, upon binding to an aldehyde, there is greater hindrance of the protons on the α-carbon and limited accessibility to base. We, therefore, considered replacing the phenyl groups of ATPH with 2-naphthyl groups and devised aluminum tris(2,6-di-2-naphthylphenoxide) (ATNP, 10, Scheme 2) wherein the naphthyl groups are expected to extend further forward toward the α-carbon of the aldehyde.
Scheme 2.

Synthesis of ATNP and its use in a Vinylogous Aldol Reaction with an Enolizable Aldehyde
To examine this hypothesis, we modeled the ATNP/valeraldehyde complex. We began with the crystal structure of Yamamoto’s ATPH/methyl crotonate complex,3d,10 added the corresponding 2-naphthyl groups, and replaced the ester with valeraldehyde. We then adjusted the complexation bond lengths and bond angles to the known values for ATPH/aldehyde complexes.3d Energy minimization of this structure (MM2 force field, see Supporting Information) provided the model of ATNP bound to valeraldehyde shown in Figure 1. This structure reveals that the additional reach of the 2-naphthyl groups can effectively block the enolizable protons of the aldehyde.
Figure 1.

Model of ATNP bound to valeraldehyhde. In this model, the aluminum atom (light blue) of ATNP is shown coordinated phenoxide oxygens (red) as well as to the oxygen of valeraldehyde (carbons shown in yellow). The distal phenyl rings of the 2-naphthyl groups are shown in green, and can be seen blocking the enolizable protons (highlighted in blue). The aldehydic proton is also shown in blue; all other hydrogens have been omitted for clairity.
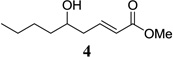
As in the case of ATPH, ATNP is prepared in situ immediately prior to use by stirring the corresponding phenol with trimethylaluminum (Scheme 2). The precursor to ATNP, phenol 13, can, in turn, be prepared by a one-step Suzuki cross-coupling using RuPhos11 as the ligand in excellent yield (>95%) from 2,6-dibromophenol (11) and 2-naphthylboronic acid (12), both of which are commercially available. Gratifyingly, with ATNP using the conditions described in Scheme 1, the vinylogous aldol reaction of methyl crotonate (2) and valeraldehyde (3) proceeds cleanly to provide the desired γ-adduct (4) in 82% yield (Scheme 2). This is a substantial improvement over the 22% yield observed by Yamamoto using ATPH.
With ATNP identified, we wished to optimize the reaction conditions and minimize the loading of methyl crotonate. Typically, two equivs of the crotonate are used by Yamamoto with approximately equal amounts of LTMP, and we wished to optimize this reaction using lower loadings of crotonate, which would require a corresponding decrease in LTMP and ATNP. We find that a modest decrease in yield is observed with the use of 1.5 equivs of crotonate (76%, Table 2, entry 2) and a more significant decrease with 1.1 equivs (60%, Table 2, entry 3). Neither slow addition of the base nor changing the order of addition such that the ATNP complexed substrates are added to the LTMP were beneficial (Table 2, entries 4, 5 and 6). Finally, the reaction requires lower temperature to proceed cleanly; at −20 °C, the reaction produces greater amounts of byproducts and proceeds in 54% yield (Table 2, entry 7).
Table 2.
Optimization of Reaction Conditions
 | ||||||
|---|---|---|---|---|---|---|
| entry | ester (equiv) |
ATNP (equiv) |
LTMP (equiv) |
mode of additiona |
temp (°C) |
yield (%) |
| 1 | 2 | 3.3 | 2.3 | std | −78 | 82 |
| 2 | 1.5 | 2.7 | 1.7 | std | −78 | 76 |
| 3 | 1.1 | 2.3 | 1.3 | std | −78 | 60 |
| 4 | 1.1 | 2.3 | 1.3 | slow std | −78 | 55 |
| 5 | 1.1 | 2.3 | 1.3 | inverse | −78 | 44 |
| 6 | 2 | 3.3 | 2.3 | inverse | −78 | 61 |
| 7 | 2 | 3.3 | 2.3 | std | −20 | 54 |
Mode of addition refers to the manner in which the complex and base were combined; std = cooled solution of LTMP added rapidly by cannula to ATNP-complex; slow std = cooled solution of LTMP added dropwise by cannula to ATNP-complex; inverse = cooled solution of ATNP-complex added rapidly by cannula to LTMP.
Our optimized conditions are very similar to those of Yamamoto and consist of running the reaction at −78 °C using 2 equivs of the ester, 2.3 equivs of LTMP and 3.3 equivs of ATNP for every equiv of aldehyde. With these conditions identified, we studied the scope of the reaction with a variety of aldehydes. Our results are shown in Table 3.
Table 3.
Scope of the Vinylogous Aldol Reaction with ATNP
| entry | aldehyde | product | yield (%) dr |
|---|---|---|---|
| 1 | 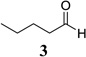 |
 |
82 |
| 2 | 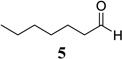 |
 |
76 |
| 3 | 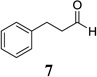 |
 |
82 |
| 4 | 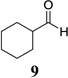 |
 |
96 |
| 5 | 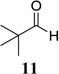 |
 |
97 |
| 6 | 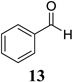 |
 |
97 |
| 7 | 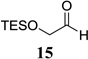 |
 |
77 |
| 8 | 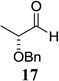 |
 |
77 2.4:1 dr |
| 9 | 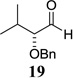 |
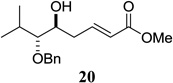 |
87 10:1 dr |
| 10 | 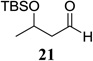 |
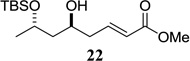 |
65 1.3:1 dr |
| 11 | 78 3.4:1 dr |
||
| 23 | 24 | ||
| 12 |  |
 |
43 |
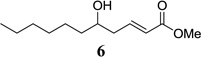
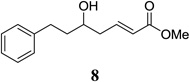
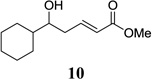
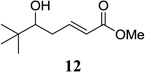
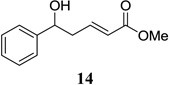
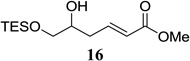
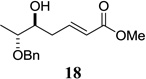
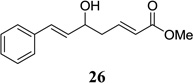
Other unbranched, enolizable, aliphatic aldehydes, such as heptanal (5) and hydrocinnamaldehyde (7), provided comparable results to valeraldehyde (76% and 82% yields, respectively, Table 3 entries 2 and 3). The α-branched aldehyde, cyclohexylcarboxaldehyde (9), was also an excellent partner and provided the product in 96% yield (Table 3, entry 4). The reaction proceeded cleanly using pivaldehyde (11), a highly congested non-enolizable aldehyde (97%, Table 3 entry 5), and as expected, benzaldehyde (13) was an efficient reaction partner and provided the product in excellent yield (97%, Table 3 entry 6). We also studied oxygenation at the α-and β-positions of the aldehyde and found in both cases that the reaction is tolerant of silyl as well as benzyl protecting groups. For example, the α-oxygentated substrate, triethylsiloxyacetaldehyde (15), provided the product in good yield (77%, Table 3, entry 7) while the α-benzylated substrates 17 and 19 also provided the products in yields of 77% and 87% (Table 3, entries 8 and 9, respectively). Compounds 17 and 19 also provided the opportunity to study asymmetric induction with stereogenicity at the α-position. We find that β-branching is required for high levels of selectivity; unbranched aldehyde 17 provided the product with a modest 2.4:1 dr while branched aldehyde 19 provided the product with a 10:1 dr (Table 3, entries 9 and 10, respectively). In both cases, the major isomer was the anti-diastereomer. In addition, we find that β-oxygenated substrates 21 and 23 provide the product in good chemical yield with no evidence of β-elimination, but with low to moderate levels of asymmetric induction (Table 3, entry 10, 65% yield, 1.3:1 dr; entry 11, 78% yield, 3.4:1 dr). Finally, cinnamaldehyde provided the desired product in modest yield due to competitive conjugate addition (Table 3, entry 12).12
In conclusion, we have described a new Lewis acid, ATNP, capable of promoting the selective enolization of methyl crotonate in the presence of enolizable aldehydes. The subsequent vinylogous aldol reaction proceeds in high yields with a variety of substrates including those that are hindered, α-branched, and oxygenated in the α-and β-positions. Additions to α-oxygenated substrates require β-branching for high levels of diastereoselectivity, and α,β-unsaturated aldehydes undergo competitive conjugate addition, thereby limiting the yield. We are currently studying the application of this reaction to complex molecule synthesis.
Supplementary Material
Acknowledgment
This work was supported in part by a generous grant from the National Institutes of Health (GM 48498).
Footnotes
Supporting Information Available. Experimental procedures and full characterization of products for the reactions described in Scheme 2 and Table 3, details of the structure determination of compounds 18, 20, 22, and 24, and computational data for the structure in Figure 1 are provided. The material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For recent reviews on the aldol reaction, see: Mahrwald R, editor. Modern Aldol Reactions. Wiley-VCH: Weinheim; 2004. Paterson I. Total Synthesis of Polyketides using Asymmetric Aldol Reactions. In: Christmann M, Brase S, editors. Asymmetric Synthesis. 2nd ed. Weinheim, Germany: Wiley-VCH Verlag GmbH & Co.; 2008. pp. 293–298.
- 2.For recent reviews on the vinylogous aldol reaction, see: Casiraghi G, Battistini L, Curti C, Rassu G, Zanardi F. Chem. Rev. 2011;111:3076. doi: 10.1021/cr100304n. Denmark SE, Heemstra JR, Beutner GL. Angew. Chem. Int. Ed. Engl. 2005;44:4682. doi: 10.1002/anie.200462338.
- 3.(a) Saito S, Shiozawa M, Ito M, Yamamoto H. J. Am. Chem. Soc. 1998;120:813. [Google Scholar]; (b) Saito S, Shiozawa M, Yamamoto H. Angew. Chem. Int. Ed. Engl. 1999;38:1769. doi: 10.1002/(SICI)1521-3773(19990614)38:12<1769::AID-ANIE1769>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]; (c) Saito S, Shiozawa M, Nagahara T, Nakadai M, Yamamoto H. J. Am. Chem. Soc. 2000;122:7847. doi: 10.1021/ja0205941. [DOI] [PubMed] [Google Scholar]; (d) Saito S, Nagahara T, Shiozawa M, Nakadai M, Yamamoto H. J. Am. Chem. Soc. 2003;125:6200. doi: 10.1021/ja0205941. [DOI] [PubMed] [Google Scholar]; (e) Takikawa H, Ishihara K, Saito S, Yamamoto H. Synlett. 2004:732. [Google Scholar]
- 4.Yamamoto’s procedure wherein formation of the enolate occurs in the presence of the aldehyde suggested to us that intramolecular reactions are feasible, and we have described the intramolecular vinylogous aldol reaction for the synthesis of up to 15-membered macrolides. See: Gazaille JA, Abramite JA, Sammakia T. Org. Lett. 2011;14:178. doi: 10.1021/ol202966m. Abramite JA, Sammakia T. Org. Lett. 2007;9:2103. doi: 10.1021/ol0704901.
- 5.Starowieyski KB, Pasynkiewicz S, Skowrońska-Ptasińska M. J. Organomet. Chem. 1975;90:C43. [Google Scholar]
- 6.Maruoka K, Nonoshita K, Banno H, Yamamoto H. J. Am. Chem. Soc. 1988;110:7922. [Google Scholar]
- 7.Maruoka K, Itoh T, Sakurai M, Nonoshita K, Yamamoto H. J. Am. Chem. Soc. 1988;110:3588. [Google Scholar]
- 8.Healy MD, Barron AR. Angew. Chem. Int. Ed. Engl. 1992;31:921. [Google Scholar]
- 9.Takikawa H, Ishihara K, Saito S, Yamamoto H. Synlett. 2004:732. [Google Scholar]
- 10.This structure is available from the Cambridge Crystallographic Data Centre CIF Depository.
- 11.Martin R, Buchwald SL. Acc. Chem. Res. 2008;41:1461. doi: 10.1021/ar800036s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Saito S, Yamamoto H. Chem. Comm. 1997:1585. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.