

Abstract
195 Plasmodium falciparum merozoite surface protein-2 alleles collected in Tak Province, Thailand, in 1996 and 2006 revealed extremely limited sequence polymorphism except in the variable (V) region, which defines the two allelic families 3D7 and FC27. This pattern is most easily explained by repeated inter-allelic gene conversion events homogenizing alleles outside the V region. Comparison of synonymous and nonsynonymous differences in V regions within allelic families supported the hypothesis that amino acid sequence polymorphism in this region is selectively favored. The pattern of sequence differentiation supported the hypothesis that repeats in the V region have evolved by concerted evolution in the 3D7 family but not in the FC27 family. In the FC27 family two alleles of relatively high frequency were the most common V-region alleles in both 1996 and 2006, while 3D7 alleles constituted a significantly greater proportion of the sequences collected in 2006 (56.1%) than of those collected in 1996 (28.9%). These changes in the frequencies of 3D7 alleles may reflect increased intensity of selection on the P. falciparum population in Thailand as a result of effective control measures that have sharply reduced the incidence of malaria infection.
1. Introduction
In Plasmodium falciparum, the most virulent of the human malaria parasites, there are a number of highly polymorphic antigen-encoding loci; and at a number of these loci, there is evidence that the polymorphism is selectively maintained (Hughes 1990, 1992; Hughes and Hughes 1995; Verra and Hughes 2000). Interestingly, at a number of these polymorphic loci, alleles fall into a small number (often just two) of highly divergent and apparently ancient families (Roy et al. 2008). This phenomenon has been designated “allelic dimorphism” by Roy et al. (2008).
In the known cases of allelic dimorphism in P. falciparum, the sequences that characterize the divergent allelic families typically are found only in a certain portion of the gene. For example, there is a highly divergent region in the sequence of the P. falciparum pfmsp-1 gene encoding merozoite surface protein 1, at which the polymorphism is apparently very ancient, with an age estimated at as much as 35 million years (Hughes 1992). By contrast, in other regions of the pfmsp-1 gene, alleles are much more similar, suggesting that other portions of the sequence have been homogenized by repeated gene-conversion like events that have not affected the divergent portion of the gene (Hughes 1992). Similarly, alleles at the P. falciparum pfmsp-2 locus, encoding merozoite surface protein-2 (PfMSP-2), fall into two highly divergent families in a central repeat region; these families have been designated respectively 3D7 and FC27 after the names of the strains from which they were first described (Smythe et al. 1990).
The hypotheses that have been proposed to account for allelic dimorphism can be broadly classified as follows: (1) nonselective hypotheses, which rely on population processes such as introgression from another species or pseudo-allelism arising from the differential deletion of paralogs; and (2) selective hypotheses, which involve some form of balancing selection (Roy et al. 2008). It is known that balancing selection can maintain polymorphisms for long periods of time, even pre-dating speciation events (Takahata and Nei 1990). Thus, balancing selection can easily explain the extensive sequence divergence between the classes of alleles that occurs in allelic dimorphism (Hughes 1992). However, balancing selection must be of a somewhat unusual sort in order to maintain two discrete classes of alleles rather than a diverse array of alleles of intermediate frequencies (Roy et al. 2008).
In common with several other cell surface proteins of Plasmodium (Kemp et al. 1987), PfMSP-2 is characterized by arrays of repeated amino acid motifs; and these motifs differ substantially between the 3D7 and FC27 allelic families (Smythe et al. 1990; Fenton et al. 1991; Felger et al. 1997; Ferreira and Hartl 2007). A number of authors have suggested that Plasmodium amino acid repeats can evolve in a concerted fashion, whereby repeats within a given allelic sequence can come to resemble one another to a greater extent than they resemble repeats of other alleles (Ferreira and Hartl 2007; Hoffmann et al. 2006; Hughes 1991; Jongwutiwes et al. 1994; Rich and Ayala 2000). For example, in the circumsporozoite protein of P. falciparum, there are numerous examples of within-allele duplication of repeat blocks (Jongwutiwes et al. 1994). The hypothesis of concerted evolution is further supported by a highly skewed distribution of pairwise nucleotide sequence differences among repeat units in the genes encoding Plasmodium surface proteins, with a high frequency of repeat units that are identical at the nucleotide level within alleles (Hughes 2004). Concerted evolution leading to homogenization of amino acid repeat units occurs in many organisms (Hughes 1999, 2000); however, there is evidence that this process is unusually rapid in asexual stage surface antigens of malaria parasites, suggesting that natural selection arising from interactions with the immune system of the vertebrate host may favor homogenization of arrays (Hughes 2004).
In order to better understand the selective forces that may be acting on P. falciparum loci exhibiting allelic dimorphism, it is important to understand the patterns of occurrence of these alleles in natural populations. Partial pfmsp-2 sequences collected twenty-nine months apart in one region of Irian Jaya, Indonesia, surprisingly shared no alleles, even when the same patient was examined at both time points (Marshall et al. 1994; Eisen et al. 1998). Eisen et al. (1998) suggested that these results are consistent with the hypothesis that infection induces an allele-specific immunity, reducing the likelihood of reinfection by parasites expressing identical forms of MSP-2. This process in turn might give rise to a form of balancing selection at the pfmsp-2 locus, based on an advantage to parasites bearing alleles that have previously been rare in a given host population. However, the numbers of sequences in the two collections in Irian Jaya (12 and 17, respectively) were small, making it difficult to determine whether the observed differences between the time points simply reflected sampling error.
In the present study, we collected complete pfmsp-2 sequences from Tak Province in Thailand at two time points separated by 10 years. We completely sequenced the pfmsp-2 gene from a total of 195 isolates, 97 from 1996 and 98 from 2006. Over the period from 1965 to 2002, there was a 35-fold reduction in the incidence of P. falciparum malaria in Thailand, as a result of both vector control programs and the use of anti-malarial drugs (Zhou et al. 2005). This decline continued over the period from 1996 to 2006, when the number of reported cases of malaria in Thailand decreased more than 50%, from 148,610 to 65,804 (Source: Annual Statistics, Division of Vector-Borne Diseases, Ministry of Public Health, Thailand). A substantial reduction in infections of the vertebrate host is expected to cause a decrease in the parasite population size. Population reduction might affect allele frequencies at polymorphic loci in P. falciparum, as a result of both chance events and intensified competition within the parasite population, possibly leading to changes in the intensity and the direction of natural selection. In addition, we tested the hypothesis of concerted evolution on the repeat regions of pfmsp-2 sequences. In the case of the 3D7 family of alleles, repeat regions could not be aligned easily; therefore we developed innovative alignment-independent methods to measure the sharing of motifs within and between repeat arrays.
2. Methods
2.1 Parasite Populations
Blood samples were collected from P. falciparum infected patients in Tak Province, northern Thailand in 1996 and 2006. Genomic DNA was extracted by either using proteinase K digestion followed by phenol/chloroform extraction or using the QIAGEN DNA minikit (Hilden, Germany) following the manufacturer’s protocol. After the purification procedure, these DNA samples were stored at −30°C until use. We excluded P. falciparum isolates that contained multiple clone infections by genotyping and sequencing block 2 of the merozoite surface protein 1 locus. In addition, isolates giving more than one PCR bands or having superimposed signals in electropherogram of pfmsp-2 were also excluded from analysis. The ethical aspects of this study have been approved by the Institutional Review Board of Faculty of Medicine, Chulalongkorn University.
2.2. Amplification and Sequencing
The entire pfmsp-2 gene from each isolate was amplified by PCR using primers whose sequences were derived from the 5′ untranslated region, FMSP2F0: 5′-AAAGAATTGTATTTATTAATTCTTAAC-3′, and the 3′ untranslated region, FMSP2R0: 5′-CTCTTCATTTTAAAACATTGAC-3′, of the 3D7 sequence. Thermal cycling profiles contained a pre-amplification denaturation at 94°C, 2 min; 35 cycles of denaturation at 94°C, 40 s; annealing at 60°C, 30 s; extension at 72°C, 3 min, and post amplification extension at 72°C, 5 min. DNA amplification was performed by using a GeneAmp 9700 PCR thermal cycler (Applied Biosystems, Foster City, CA). Polymerisation was performed by using ExTaq DNA polymerase that possesses efficient 5′ → 3′ exonuclease activity to increase fidelity and no strand displacement (Takara, Japan). The size of PCR product was examined by electrophoresis in a 1% agarose gel and visualized under a UV transilluminator (Mupid Scope WD, Japan).
DNA sequences were determined directly from PCR-purified products and from both directions using the Big Dye Terminator v3.1 Cycle Sequencing Kit on an ABI3100 Genetic Analyzer (Applied Biosystems, USA). Overlapping sequences were obtained by using sequencing primers. Whenever singleton substitution occurred, sequence was re-determined using PCR products that were newly amplified from the same DNA template. Nucleotide sequences reported in this paper have been available in the GenBank databases under the accession numbers EU810410-EU810604.
2.3 Sequence Analysis
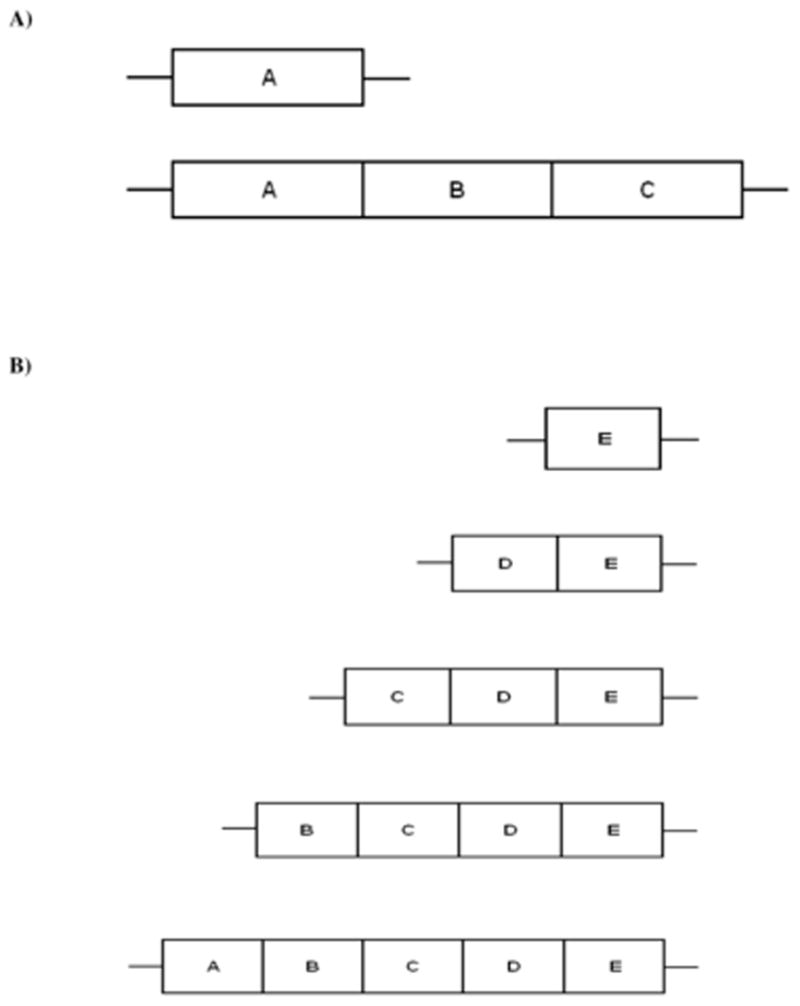
Sequences were aligned at the nucleotide sequence level using the CLUSTAL X program (Thompson et al. 1997). Previous analyses have shown that the PfMSP2 sequence can be divided into five domains that are distinct with regard to function and/or polymorphism (Smythe et al. 1990; Marshall et al. 1992): the signal peptide (SP); first conserved domain (C1); variable domain (V); second conserved domain (C2); and propeptide (Pro; Figure 1A). The V domain includes amino acid repeats, and we found that this region could not be easily aligned between the two major allelic groups (Fenton et al.1991; Ferreira and Hartl 2007), Thus we produced separate alignments for the two major allelic families, 3D7 (Supplementary Figure S1) and FC27 (Supplementary Figure S2); and we conducted sequence comparisons within the two major allelic groups but not between them. In the case of the 3D7 family, the alignment of the repeat regions was uncertain; thus, although an alignment of the repeats in this region is presented in Supplementary Figure S1, no analyses were based on this alignment.
Figure 1.

(A) Schematic representation of the major regions of the P. falciparum MSP-2 protein.: SP = signal peptide; C1 = conserved region 1; V = variable region; C2 = conserved region 2; PRO = propeptide. (B) Schematic representation of the V region of members of the 3D7 allelic family: NR = non-repeat; p-T = poly T. (C) Schematic representation of the V region of members of the FC27 allelic family: NR = non-repeat; R1 = repeat region 1; R2 = repeat region 2.
In the 3D7 family, there are two distinct repeat regions. The first is a region rich in the amino acids glycine, serine, and alanine. Felger et al. (1997) hypothesized that the ancestral repeat in this region was the 6-bp unit (hexamer) GGT GCT, encoding Gly-Ala. We therefore refer to these as the GA-rich repeats (Figure 1B and Supplementary Figure S1). An additional repeat region involves a variable number of copies of the amino acid threonine; we refer to this as the poly-T region (Figure 1B and Supplementary Figure S1).
In the FC27 allelic family, the V region contains two distinct repeat regions designated R1 and R2 (Felger et al. 1997; Figure 1C and Supplementary Figure S1). The R1 repeat is a 96-bp unit that was present in 1 or 3 copies in the sequences in our data set (Supplementary Figure S1). The R2 repeat is a 36-bp unit that was present in 1–5 copies in the sequences in our data set (Figure 1C and Supplementary Figure S2). In aligning the V regions, the repeat arrays were aligned separately, and the alignment corrected slightly by eye (Supplementary Figures S1 and S2).
In pairwise comparisons among a set of sequences all sites at which the alignment postulated a gap in any sequence were eliminated so that a comparable data set was used for each comparison. We estimated the number of synonymous substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) by Nei and Gojobori’s (1986). The standard errors of mean dS and dN (synonymous and nonsynonymous nucleotide diversity, respectively) were estimated by the bootstrap method (Nei and Kumar 2000). In comparisons of individual repeat units, we used the same method (Nei and Gojobori 1986) to compute the uncorrected number of synonymous differences per synonymous site (pS) and the number of nonsynonymous differences per nonsynonymous site (pN). The correction for multiple hits was not used in the latter case because the correction formula may not be applicable to the short repeat sequences.
Because alignment of the GA-rich repeats was uncertain, we did not compute any evolutionary distances based on the alignment. Following Felger et al.’s (1997) hypothesis that the basis repeat unit is the hexamer GGT GCT, we analyzed non-overlapping nucleotide hexamers in the GA-rich repeat region. (Note that the total number of nucleotides in the GA-rich region was evenly divisible by 6 in the case of each 3D7 family sequence in our data set.) Let pij and pkj represent the proportion of the jth hexamer in array i and array k, respectively. Then we define the coefficient of identity (CI) as follows:
| (1) |
CI represents the probability that a hexamer drawn at random from array i will be identical to a hexamer drawn at random from array k. Similarly, the probability that two hexamers drawn at random from array i will be identical is computed as follows:
| (2) |
In order to test hypotheses regarding pairwise comparisons of repeat units, we used randomization tests. These involved creating 1000 pseudo-data sets by sampling (from replacement) in the original data.
3. Results
3.1 Domains and Families
We examined nucleotide sequence polymorphism in 195 pfmsp-2 sequences from Thailand, 97 from 1996 and 98 from 2006. In the present data set, the SP regions of the gene (Figure 1) contained 20 codons in all alleles, and there were no synonymous or nonsynonymous nucleotide differences among the 195 sequences. Likewise, the C1 region (23 codons in all alleles) and the Pro region (24 codons in all alleles) showed no synonymous or nonsynonymous nucleotide differences among the 195 sequences.
The V region defines the two major allelic families 3D7 and FC27. In our data set, the V region of 3D7 family sequences ranged in length from 155–208 amino acid residues (median = 178 residues), while in FC27 family members the V region ranged from 127–179 residues (median = 139 residues). In the 1996 collection, there were 28 sequences (28.9%) from the 3D7 family and 69 sequences (71.1%) from the FC27 family. By contrast, in the 2006 collection, there were 55 sequences (56.1%) from the 3D7 family but only 43 (43.9%) from the FC27 family. The difference between the two years of collection with respect to the proportions of the two allelic families was highly significant (χ2 = 14.8; 1 d.f.; P < 0.001).
3.2. V region polymorphism
In the non-repeat portions of the V region, we computed the number of synonymous substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) in pairwise comparisons among alleles within the two allelic families (Table 1). In the 3D7 family, mean dN was significantly greater than mean dS in both 1996 and 2006 (Table 1). Likewise, in the 3D7 family, in comparisons between the two years, mean dN was significantly greater than mean dS (Table 1). This result provided evidence of natural selection acting to diversify the non-repeat portion of the V region in the 3D7 allelic family. In the FC27 family, mean dN was greater than mean dS in both 1996 and 2006 and in the comparison between the two years; but the difference was not significant (Table 1).
Table 1.
Mean numbers of synonymous (dS) nonsynonymous (dN) substitutions per site (± S.E.) in pairwise comparisons of non-repeat portions of the V regions within allelic families.
| Allelic family | Comparison | dS | dN |
|---|---|---|---|
| 3D7 | 1996 | 0.0021 ± 0.0021 | 0.0305 ± 0.0089a |
| 2006 | 0.0009 ± 0.0007 | 0.0273 ± 0.0088a | |
| 1996 vs. 2006 | 0.0015 ± 0.0011 | 0.0297 ± 0.0093a | |
| FC27 | 1996 | 0.0013 ± 0.0013 | 0.0118 ± 0.0053 |
| 2006 | 0.0024 ± 0.0023 | 0.0128 ± 0.0057 | |
| 1996 vs. 2006 | 0.0018 ± 0.0018 | 0.0123 ± 0.0055 |
Tests of the hypothesis that mean dS equals mean dN:
P < 0.01 (Z-test).
In 1996, there were 13 distinct allelic sequences in the V region belonging to the 3D7 family. The median population frequency of these 13 3D7 alleles was 0.021 (range 0.010–0.062). The same year there were 12 distinct allelic sequences in the V region belonging to the FC27 family, with a median population frequency of 0.015 (range 0.010–0.330). Although the 3D7 and FC27 families did not differ with respect to median allele frequency, the FC27 family included the two allelic forms with the highest frequencies (0.330 and 0.227, respectively) in the 1996 sample.
In 2006, there were 29 distinct allelic V region sequences in the 3D7 family, with median population frequency of 0.010 (range 0.010–0.082). In the same year’s collection, there were 9 distinct allelic V region sequences in the FC27 family, with median population frequency of 0.031 (range 0.010–0.204). In 2006, median allelic frequency differed significantly between the two families (Mann-Whitney test; P = 0.003).
Of 34 allelic V region sequences belonging to the 3D7 family, only 8 (23.5%) were found in both years’ collections, while 7 (50.0 %) of 14 allelic V region sequences in the FC27 family were found in both years’ collections. In the case of the FC27 family, there was a significant positive correlation between allelic frequencies in the two years (Figure 2; rS = 0.963; P < 0.001). A major factor in the latter correlation was that the most common V region allele in 1996 (frequency 0.330), which belonged to the FC27 family, was also the most common alleles (frequency 0.204) in 2006 (Figure 2). The second most common V region allele in 1996 (frequency 0.227), which was also a FC27 family member, likewise had the second highest allelic frequency in 2006 (Figure 2). In the case of the 3D7 family, there was a positive correlation between allelic frequencies in the two years, but it was not significant (Figure 2; rS = 0.661: n.s.).
Figure 2.
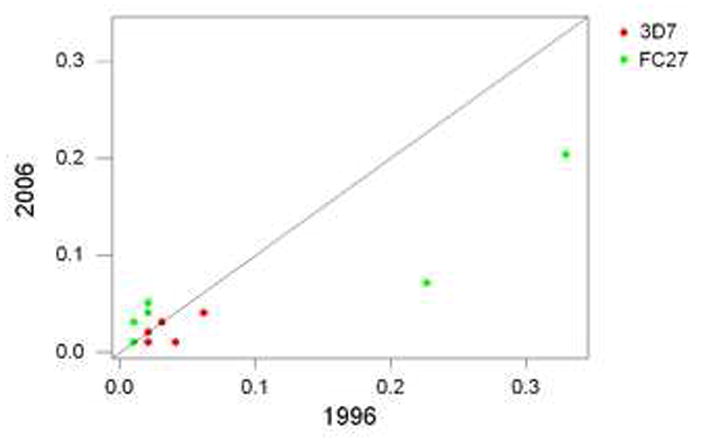
Plot of V-region allele frequencies in 2006 vs. 1996, for alleles in the two allelic families (3D7 and FC27) that were present in both years. In the case of the 3D7 family, there was not a significant correlation between frequencies in the two years (rS = 0.661: n.s.). In the case of the FC27 family, there was a highly significant correlation between frequencies in the two years (rS = 0.963; P < 0.001).
3.3. FC27 Family Repeats
The V-region repeats in the FC27 family are found in two regions designated R1 and R2 (Felger et al. 1997). Alignment of the R1 region identified three repeat types, on the basis of sequence similarity, which we designated A-C (Figure 3A and Supplementary Figure S2). In our data set, all but two FC27 sequences had only one copy of the 32-codon R1 repeat unit, which was of type A (Figure 3A and Supplementary Figure S2). Each of the other two sequences had two additional repeats (types B and C; Figure 3A and Supplementary Figure S2). In the case of the R2 repeats, the alignment identified on the basis of sequence similarity five different repeat types, which we designated A-E (Figure 3B and Supplementary Figure S2). Our sequences included between 1 and 5 repeat units, but all of them included a repeat unit of type E (Figure 3B).
Figure 3.
Schematic representation of the patterns observed in the alignment of repeat regions in the FC27 family: (A) the three repeat types (A–C) in the R1 repeats; and (B) the five repeat types (A–E) in the R2 repeats.
Analyses of the pattern of pairwise nucleotide difference among repeat units can provide evidence regarding the hypothesis of concerted evolution (Hughes 1991, 2004). In the case of the R1 repeats, we compared repeats within repeat types A, B, and C (Figure 3A) and between different repeat types, both within the same haplotype (i.e., the same individual sequence) and between haplotypes. No synonymous differences were found in the R1 repeats (Table 2). However, at nonsynonymous sites, repeats belonging to different repeat types were much more divergent than were repeats belonging to the same repeat type (Table 2). Mean pN between different repeat types within the same haplotype was nearly 20 times as great as that between repeats belonging to the same type; and the difference was highly significant (Table 2). Likewise, mean pN between different repeat types in different haplotypes was nearly 30 times as great as that between repeats belonging to the same type; and the difference was highly significant (Table 2). The fact that mean pN was significantly greater in comparisons between different repeat types than in comparisons within the same repeat type is evidence against the hypothesis of concerted evolution of the R1 repeats.
Table 2.
Mean numbers of synonymous (pS) nonsynonymous (pN) differences per site (± S.E.) in pairwise comparisons of individual repeat units belonging to the FC27 allelic family.
| Repeat Region | Comparison (no.) | pS | pN |
|---|---|---|---|
| R1 | Same repeat type (6218) | 0.0000 ± 0.0000 | 0.0005 ± 0.0001 a |
| Different repeat type | |||
| Same haplotype (6) | 0.0000 ± 0.0000 | 0.0091 ± 0.0027 a,b | |
| Different haplotype (446) | 0.0000 ± 0.0000 | 0.0138 ± 0.0001 a,b | |
| R2 | Same repeat type (24418) | 0.0000 ± 0.0000 | 0.00185 ± 0.0002 a |
| Different repeat type | |||
| Same haplotype (724) | 0.2984 ± 0.0118 b | 0.2920 ± 0.0117 b | |
| Different haplotype (79969) | 0.0000 ± 0.0000 b | 0.0000 ± 0.0000 b |
Tests of the hypothesis that mean pS equals mean pN:
P < 0.001 (randomization test).
Tests of the hypothesis that pS or pN equals the corresponding value for the same repeat type:
P < 0.001 (randomization test).
In analyses of the R2 repeats, mean pS and mean pN were both significantly lower in the case of comparisons of the same repeat type than in comparisons of different repeat types, whether or not the latter were of the same of different haplotypes (Table 2). In the case of the R2 repeats, mean pN between different repeat types from the same haplotype was over 15 times as great as mean pN for comparisons of the same repeat type (Table 2). Similarly, mean pN between different repeat types from different haplotypes was over 16 times as great as mean pN for comparisons of the same repeat type (Table 2). The fact that both mean pS and mean pN were significantly greater in comparisons between different repeat types than in comparisons within the same repeat type is strong evidence against the hypothesis of concerted evolution of the R2 repeats.
In the case of the R1 repeats, mean pN was significantly greater than mean pS in comparisons among repeats of the same type or of different types, whether belonging to the same or different haplotypes (Table 2). This provides evidence that natural selection has acted to diversify the R1 repeats at the amino acid level, both within and between repeat types. In the case of the R2 repeats, mean pN was significantly greater than mean pS in comparisons among repeats of the same type (Table 2). Thus, although there was evidence of natural selection favoring amino acid differences within R2 repeat types, However, there was not a significant difference between mean pS and mean pN in comparisons between repeats of different type, whether in the same haplotype or different haplotypes (Table 2).
3.4. 3D7 Family Repeats
The GA-rich repeat region ranged from 34 to 90 codons in length; thus each array of GA-rich repeats contained 17-45 2-codon units (nucleotide hexamers). The hexamers showed just 14 sequences out of the 3721 possible amino acid encoding hexamers (Table 3). The most abundant hexamer was GGT GCT (37.4%), and the second most abundant was AGT GCT (18.2%; Table 3). By contrast, GGC GCT and GAT GCT were each found just once (0.004%; Table 3). The GGC codon in the former hexamer was the only one of 4850 codons in the GA-rich repeats that did have T in the third position.
Table 3.
Nucleotide hexamer sequences in the GA-rich repeat region of 3D7 family pfmsp-2, and mean coefficient of identity (CI) between hexamer units.
| Hexamer repeat unit | Number | % of total |
|---|---|---|
| AAT CCT | 8 | 0.3 |
| AGT GCT | 441 | 18.2 |
| AGT GGT | 256 | 10.1 |
| CGT AAT | 14 | 0.6 |
| CGT GAT | 49 | 1.9 |
| GAT GCT | 1 | 0.04 |
| GGC GCT | 1 | 0.04 |
| GGT AAT | 33 | 1.4 |
| GGT ACT | 7 | 0.3 |
| GGT GAT | 27 | 1.1 |
| GGT GCT | 906 | 37.4 |
| GGT GGT | 159 | 6.6 |
| GCT TCT | 268 | 11.1 |
| GCT TCT | 255 | 10.5 |
| Mean CI (± S.E.) | ||
| Same haplotype | 0.315 ± 0.012 | |
| Different haplotype | 0.214 ± 0.002a | |
Test of the hypothesis that mean CI for comparisons of different haplotypes equals that for comparisons of the same haplotype: P < 0.001 (randomization test).
Because of the irregular nature of the GA-rich repeats, we compared hexamers within and between haplotypes using the coefficient of identity (CI), an estimate of the likelihood of drawing two identical hexamers at random. Mean CI within haplotypes (0.315) was significantly greater than that in comparisons between different haplotypes (0.214; Table 3). This result indicates that hexamers were more likely to be identical within a haplotype than between haplotypes, supporting the model of concerted evolution.
In the poly-T region, of 751 Thr-encoding codons, 361 (48.1%) were ACT codons; 195 (26.0%) were ACA codons; and 195 (26.0%) were ACC codons. Examination of the pattern of occurrence of these codons showed that in every case the poly-T region consisted of the sequence two to four repeats of the sequence ACT ACC ACA; after these repeats, the poly-T region ended in each case with the sequence ACT ACT (Supplementary Figure S1). The variable number of occurrences of the repeat unit ACT ACC ACA suggested that this sequence has been duplicated independently in different haplotypes, consistent with a model of concerted evolution.
3.5. C2 Region Polymorphism
The C2 region contained 48 amino acid residues (144 nucleotides) in all sequences. There were only three polymorphic nucleotide sites in our data set: T:C at site 96; A:C at site 127; and A:G at site 140. Only the latter two polymorphisms involved amino acid changes: Thr:Pro at amino acid residue 43 and Ser:Asn at amino acid residue 47. Combinations of the variants at these three sites yielded five distinct haplotypes in the C2 region, each of which was separated from one or two of the others by a single mutational step (Figure 4). There was not a strict association between a given C2 haplotype and V region families (Figure 4). The haplotype C96/A127/A140 was by far the most commonly observed (96.4%) in sequences with V regions belonging to the FC27 family, but the same haplotype was also seen in a substantial majority (69.9%) of sequences whose regions belonged to the 3D7 family (Figure 4). By contrast, the second most common haplotype (15.7%) in sequences with 3D7 family V regions (C96/C127/G140) was not found in any sequences with FC27 family V regions (Figure 5). Likewise, the third most common haplotype (13.3%) in sequences with 3D7 family V regions (C96/C127/A140) was found in only one sequence (0.9%) with an FC27 family V region (Figure 4). Of the four haplotypes other than the most common haplotype (C96/C127/G140), two were in both V region families, while two were found in only one of the two V region families (Figure 4).
Figure 4.
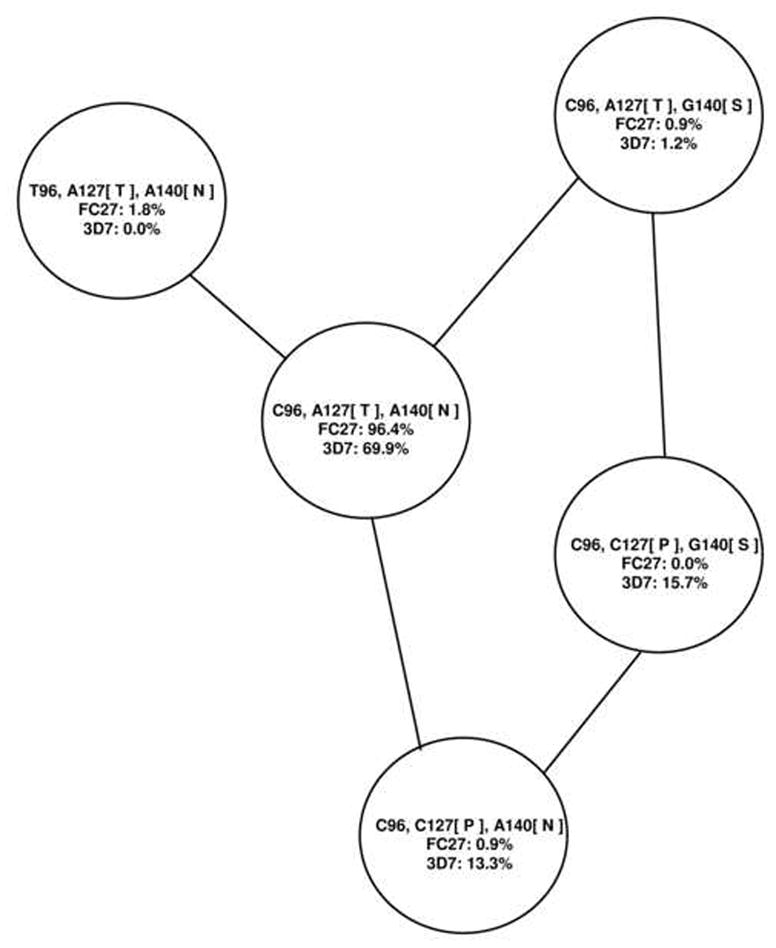
Haplotypes in the C2 region of pfmsp-2. Lines connecting haplotypes correspond to a single point-mutational step. The percentage of occurrence of each haplotype with 3D7 and FC27 family V regions is also shown.
4. Discussion
Polymorphism in 195 pfmsp-2 alleles collected in Thailand in 1996 and 2006 revealed a pattern of nucleotide sequence polymorphism reminiscent of that first described for pfmsp-1 (Hughes 1992); namely, a single gene contains regions of high sequence polymorphism as well as those of limited polymorphism or no polymorphism at all. The SP, C1, and Pro regions were completely identical at the amino acid level among all sequences, and the C2 region showed very limited polymorphism. By contrast, in the V region, as reported previously, sequences fell into two highly divergent allelic families (3D7 and FC27) between which V region sequences could not be aligned. As with pfmsp-1 (Hughes 1992), the simplest explanation for this pattern is repeated interallelic recombination, possibly involving a “gene-conversion-like” mechanism whereby portions of the gene outside the V region have been homogenized among alleles.
The pattern of limited polymorphism observed in the C2 region supports the hypothesis of homogenization by repeated gene conversion. In this region, a single haplotype (C96/A127/A140) is by far the most common in sequences of both the 3D7 and FC27 families, suggesting fairly recent homogenization of the two families in this region. Other haplotypes in the C2 region have presumably arisen since that homogenization by one or two point mutations (Figure 4). The fact that two of four minority haplotypes were found in association with both of the V region families can be attributed either to parallel point substitutions within the two V region families or to ongoing gene conversion in the C2 region between the two families.
By contrast, the V regions showed a much higher level of divergence both in repeat and non-repeat portions. There was evidence that this polymorphism is selectively maintained in both the 3D7 and FC27 allelic families. In the 3D7 family, the non-repeat portions of the V region showed significantly greater nonsynonymous than synonymous nucleotide diversity (Table 1), strongly supporting the hypothesis that balancing selection acts to favor amino acid diversity in this region. In the FC27 family, there was not a significant difference between synonymous and nonsynonymous nucleotide diversity in the non-repeat portion of the V region (Table 1). Nonetheless, there was evidence that natural selection has favored amino acid changes in the repeat regions of FC27 family alleles. In both the R1 and R2 repeat arrays, corresponding repeat units showed an excess of nonsynonymous nucleotide differences (Table 2), supporting the hypothesis that natural selection acts to favor amino acid differences in these arrays.
It has been proposed that Plasmodium amino acid repeats can evolve in a concerted fashion, whereby repeats within a given allelic sequence can come to resemble one another to a greater extent than they resemble repeats of other alleles (Hoffmann et al. 2006; Hughes 1991, 2004; Jongwutiwes et al. 1994; Rich and Ayala 2000), and evidence in support of this hypothesis has been provided in the case of certain loci (Hughes 2004; Jongwutiwes et al. 1994). In the case of pfmsp-2, it seems certain that concerted evolution has occurred in the past; otherwise, it is difficult to explain the existence of completely different types of repeats in the 3D7 and FC27 families (Felger et al. 1997; Ferreira and Hartl 2007). However, our analyses showed a striking difference between the two allelic families with respect to the evidence for recent concerted evolution within each family. In the FC27 family, in both R1 and R2 repeat regions, sequence alignment identified repeat units with significantly greater similar to the corresponding block in other sequences than to other blocks within the same sequence, exactly the opposite of the prediction of concerted evolution. By contrast, there was evidence of concerted evolution in the repeats of the 3D7 family. In the GA-rich repeat region, identical repeat units (nucleotide hexamers) were significantly more likely to be found within the same haplotype than between haplotypes, as predicted under concerted evolution. Likewise, the poly-T repeats of the 3D7 family, although encoding a single amino acid, showed a repeated pattern of codon usage suggestive of internal duplication.
Our results showed evidence of positive selection on the V regions of both allelic families, but the nature of this selection remains poorly understood. One hypothesis to explain this polymorphism would be simple overdominance (heterozygote advantage; Maruyama and Nei 1981). In the case of malaria parasites, heterozygote advantage might occur at the level of the zygote if a heterozygous infection is advantageous to the parasite. Heterozygote advantage would be an expected consequence if hosts develop immunity that is specific to the V region family (Eisen et al. 1998). A heterozygote would always have an advantage over a homozygote in infecting a previously infected host because the heterozygote would be able to infect a host previously exposed to either homozygote, whereas a homozygote would only be able to infect a host previously exposed to the other homozygote.
Our results showed clear differences between the two V region families with respect to the distribution of allelic frequencies. The 3D7 family included numerous subtypes of low to intermediate frequencies. In 3D7, the allelic frequencies in 1996 and 2006 were not correlated; this was an apparent consequence of the substantial sampling error associated with low allelic frequencies. By contrast, the FC27 family included two alleles of relatively high frequency in both 1996 and 2006 collections, suggesting a rather different pattern of selection within the FC27 family than within the 3D7 family.
One model of selection that might explain these differences would be a form of heterozygote advantage based on allele-specific immunity, in which the degree of advantage depends on the degree of amino acid sequence difference in the V region between the two alleles. Under this model, the greatest advantage would accrue to a heterozygote bearing alleles from the two different V-region families; but there would also be some advantage to a heterozygote bearing divergent alleles belonging to the same family. Furthermore, the latter advantage might be expected to be greater in the case of 3D7 alleles than in the case of FC27 alleles because of the more divergent V region sequences of the former family (Table 1). Escalante et al. (2004) argued that heterozygote advantage is impossible in Plasmodium because the stages infecting the vertebrate host are haploid. However, since Plasmodium species are obligately sexually reproducing diploids, natural selection acts at the level of the diploid zygote. It will be to the zygote’s advantage if heterozygosity at a given locus favors successful evasion of the vertebrate host’s immune response. In any event, the details of our proposed model of selection at this locus need to be examined further by computer simulation.
In addition to the insights into the action of natural selection at the pfmsp-2 locus, our data provided evidence regarding changes over time in allelic frequencies. Unlike the results of Eisen et al. (1998), we found a number of V-region alleles in samples from the same region of Thailand collected 10 years apart; most notably the two most common V-region alleles of the FC27 family. There were differences between the two years, but many of these differences can be attributed to the sampling error expected when dealing with a limited sample of low-frequency alleles. The fact that the proportion of V-region alleles that were collected in both years was lower in the 3D7 family than in the FC27 family supports a substantial role for sampling error because the allelic frequencies of individual alleles tended to be lower in the 3D7 family.
There was a difference in the proportion of 3D7 family sequences between 1996 (28.9%) and 2006 (56.1%). This difference seemed substantial enough to suggest that it could not be attributed to sampling error alone and suggests the possibility that natural selection favored certain 3D7 sequences in the decade between 1996 and 2006. This decade corresponded to a period of rapid decline in the number of malaria infections in Thailand, a process in which antimalarial drugs played a key role (Zhou et al. 2005). Successful antimalarial drug use, by limiting the number of hosts available for infection, may have intensified natural selection at the pfmsp-2 locus.
Supplementary Material
Acknowledgments
We are grateful to all patients who donated their blood samples for this study and to Thongchai Hongsrimuang and the staff of the Bureau of Vector Borne Disease, Department of Disease Control, Ministry of Public Health, Thailand for assistance in field work. This research was supported by grant from the National Research Council of Thailand and the Thai Government Research Budget to C.P and S.J. and grant GM43940 from the National Institutes of Health to A.L.H.
Abbreviations
- C1
conserved region 1
- C2
conserved region 2
- CI
coefficient of identity
- dN
number of nonsynonymous substitutions per nonsynonymous site
- dS
number of synynonymous substitutions per synonymous site
- PfMSP-2
Plasmodium falciparum merozoite surface protein-2
- pfmsp-2
Plasmodium falciparum merozoite surface protein-2 gene
- pN
number of nonsynonymous differences per nonsynonymous site
- Pro
propeptide
- pS
number of synynonymous differences per synonymous site
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Eisen D, Billman-Jacobe H, Marshall VF, Fryauff D, Coppel RL. Temporal variation of the merozoite surface protein-2 gene of Plasmodium falciparum. Infect Immun. 1998;66:239–246. doi: 10.1128/iai.66.1.239-246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escalante
- Felger I, Marshall VM, Reeder JC, Hunt JA, Mgone CS, Beck HP. Sequence diversity and molecular evolution of the merozoite surface antigen 2 of Plasmodium falciparum. J Mol Evol. 1997;45:154–160. doi: 10.1007/pl00006215. [DOI] [PubMed] [Google Scholar]
- Fenton B, Clark JT, Anjam Khan CM, Robinson JV, Walliker D, Ridley R, Scaife JG, McBride JS. Structural and antigenic polymorphism of the 35- to 48-kilodalton merozoite surface antigens (MSA-2) of the malaria parasite Plasmodium falciparum. Mol Cell Biol. 1991;11:963–971. doi: 10.1128/mcb.11.2.963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira MU, Hartl DL. Plasmodium falciparum: worldwide sequence diversity and evolution of the malaria vaccine candidate merozoite surface protein-2 (MSP-2) Exp Parasitol. 2007;115:32–40. doi: 10.1016/j.exppara.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Hoffmann EH, Malafronte RS, Moraes-Ávila SL, Osakabe AL, Wunderlich G, Durham AM, Ribolla PE, del Portillo HA, Ferreira MU. Origins of sequence diversity in the malaria vaccine candidate merozoite surface protein-2 (MSP-2) in Amazonian isolates of Plasmodium falciparum. Gene. 2006;376:224–230. doi: 10.1016/j.gene.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Circumsporozoite protein genes of malaria parasites (Plasmodium spp.): evidence for positive selection on immunogenic regions. Genetics. 1991;127:345–353. doi: 10.1093/genetics/127.2.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. Positive selection and interallelic recombination at the merozoite surface antigen-1 (MSA-1) locus of Plasmodium falciparum. Mol Biol Evol. 1992;9:381–393. doi: 10.1093/oxfordjournals.molbev.a040730. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Concerted evolution of exons and introns in the MHC-linked tenascin-X gene of mammals. Mol Biol Evol. 1999;16:1558–1567. doi: 10.1093/oxfordjournals.molbev.a026068. [DOI] [PubMed] [Google Scholar]
- Hughes AL. Modes of evolution in the proteases and kringle domains of the plasminogen-prothrombin family. Mol Phyl Evol. 2000;14:469–478. doi: 10.1006/mpev.1999.0685. [DOI] [PubMed] [Google Scholar]
- Hughes AL. The evolution of amino acid repeat arrays in Plasmodium and other organisms. J Mol Evol. 2004;59:528–535. doi: 10.1007/s00239-004-2645-4. [DOI] [PubMed] [Google Scholar]
- Hughes MK, Hughes AL. Natural selection on Plasmodium surface proteins. Mol Biochem Parasitol. 1995;71:99–113. doi: 10.1016/0166-6851(95)00037-2. [DOI] [PubMed] [Google Scholar]
- Jongwutiwes S, Tanabe K, Hughes MK, Kanbara H, Hughes AL. Allelic variation in the circumsporozoite protein of Plasmodium falciparum from Thai field isolates. Am J Trop Med Hyg. 1994;51:659–668. doi: 10.4269/ajtmh.1994.51.659. [DOI] [PubMed] [Google Scholar]
- Kemp DJ, Coppel RL, Anders RF. Repetitive genes and proteins of malaria. Annu Rev Microbiol. 1987;41:181–208. doi: 10.1146/annurev.mi.41.100187.001145. [DOI] [PubMed] [Google Scholar]
- Marshall VM, Coppel RL, Anders RF, Kemp DJ. Two novel alleles within subfamilies of the merozoite antigen 2 (MSA-2) of Plasmodium falciparum. Mol Biochem Parasitol. 1992;50:181–184. doi: 10.1016/0166-6851(92)90255-i. [DOI] [PubMed] [Google Scholar]
- Marshall VM, Anthony RL, Bangs MJ, Purnomo, Anders RF, Coppel RL. Allelic variants of the Plasmodium falciparum merozoite surface antigen 2 (MSA-2) in a geographically restricted area of Irian Jaya. Mol Biochem Parasitol. 1994;63:13–21. doi: 10.1016/0166-6851(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Maruyama T, Nei M. Genetic variability maintained by mutation and overdominant selection in finite populations. Genetics. 1981;98:441–459. doi: 10.1093/genetics/98.2.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford University Press; New York: 2000. [Google Scholar]
- Rich SM, Ayala F. Population structure and recent evolution of Plasmodium falciparum. Proc Natl Acad Sci USA. 2000;97:6994–2001. doi: 10.1073/pnas.97.13.6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy SW, Ferreira MU, Hartl DL. Evolution of allelic dimorphism in malarial surface antigens. Heredity. 2008;100:103–110. doi: 10.1038/sj.hdy.6800887. [DOI] [PubMed] [Google Scholar]
- Smythe JA, Peterson MG, Coppel RL, Saul AJ, Kemp DJ, Anders RF. Structural diversity in the 45-kilodalton merozoite surface antigen of Plasmodium falciparum. Mol Biochem Parasitol. 1990;39:227–234. doi: 10.1016/0166-6851(90)90061-p. [DOI] [PubMed] [Google Scholar]
- Takahata N, Nei M. Allelic genealogy under overdominant and frequency dependent selection and polymorphism of major histocompatibility complex loci. Genetics. 1990;124:967–978. doi: 10.1093/genetics/124.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Diggins DG. The CLUSTAL X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997;25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verra F, Hughes AL. Evidence for ancient balanced polymorphism at the Apical Membrane Antigen-1 (AMA-1) locus of Plasmodium falciparum. Mol Biochem Parasitol. 2000;105:149–153. doi: 10.1016/s0166-6851(99)00162-0. [DOI] [PubMed] [Google Scholar]
- Zhou G, Sirichaisinthop J, Sattabongkot J, Jones J, Bjørnstad ON, Yan G, Cui L. Spatio-temporal distribution of Plasmodium falciparum and P. vivax malaria in Thailand. Am J Trop Med Hyg. 2005;72:256–262. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.