

Abstract
Venous thromboembolism (VTE) is major health problem and is sometimes complicated by lethal pulmonary embolism (PE). Disturbances of the coagulation and anticoagulation systems are important risk factors for VTE. Comparative studies suggest that coagulation and innate immunity have a shared evolutionary origin. It is therefore unsurprising that the immune and coagulation systems are linked, with many molecular components being important for both systems. Systemic inflammation modulates thrombotic responses by suppressing fibrinolysis, upregulating procoagulant, and downregulating anticoagulants, and autoimmune disorders such as systemic lupus erythematosus (SLE), inflammatory bowel disease (IBD), and Behçet’s syndrome have been linked to an increased risk of VTE. Recent reports have further shown that a majority of autoimmune and immune-mediated disorders are linked to an increased risk of venous thrombosis, PE, or VTE. For instance, a Swedish nationwide study found that the risk of PE was increased in the first year after hospitalization for 33 different autoimmune disorders. Especially high risks were noted for several autoimmune diseases such as immune thrombocytopenic purpura, polyarteritis nodosa, polymyositis/dermatomyositis, ulcerative colitis, and SLE. Another study from England, also based on hospitalization data, found that immune-mediated disorders were associated with an increased risk of VTE compared with other medical causes of hospitalization. Multiple mechanisms may operate and disease-specific factors, such as cardiolipin antibodies, have been identified. However, inflammation by itself appears to change the hemostatic balance in a thrombogenic direction. Recent epidemiological studies, together with previous experimental and clinical studies, indicate that autoimmune disorders should not only be viewed as inflammatory disorders, but also hypercoagulable disorders. Research to identify thrombotic risk factors, elucidate the mechanisms involved, and investigate prophylactic regiments is needed. The present review describes the epidemiological, clinical, and experimental evidence for the connection between VTE and autoimmune and immune-mediated disorders.
Keywords: Autoimmune diseases, immunology, inflammation, rheumatic diseases, inflammatory bowel diseases, venous thrombosis, venous thromboembolism, pulmonary embolism, blood coagulation disorders
Introduction
Pulmonary embolism (PE) and deep vein thrombosis (DVT) together constitute venous thromboembolism (VTE). VTE affects about 1-2 per 1,000 individuals per year [1,2]. PE is a potentially lethal complication of VTE with a mortality rate of >15% in the first 3 months after diagnosis [3,4]. According to Virchow’s triad, VTE results from stasis, changes in blood coagulability and alterations in the vessel wall (Figure 1) [5-8]. These changes, as well as hypercoagulable states, may be inherited and/or acquired and may act in concert (Table 1). Thus, VTE results from multiple interactions between acquired and inherited risk factors [9-11]. For familial thrombophilia – clustering of VTE in families – five major genetic risk factors have been identified: deficiencies of protein S, protein C, and antithrombin, factor V Leiden Gln506 (rs6025) and prothrombin G20210A (rs1799963) [12,13]. The list of new susceptibility loci for VTE is, however, growing fast (Table 1) [14]. Family history is the sum of the interactions of familial genetic and environmental causes [15-18]. However, family history is not a binary trait [17,18]. The risk is dependent on the number of affected relatives [17,18]. A large number of acquired risk factors for VTE have been identified and include age, previous episode of VTE, immobilization, surgery, trauma, pregnancy, puerperium, lupus anticoagulant, cardiolipin antibodies, malignant disease, hormone replacement therapy (HRT), and oral contraceptives [1,2,9-11]. The list of known acquired risk factors has also grown in recent years. Socioeconomic factors are associated with an increased risk not only of atherosclerosis, but also for VTE [19,20]. Infections have also been linked to an increased VTE risk, probably due to inflammation [21-23], though endotoxins have specific effects in severe infections. Inflammation may also explain the increased risk of VTE in patients with autoimmune and immune-mediated disorders, which this article will review (Table 1).
Figure 1.
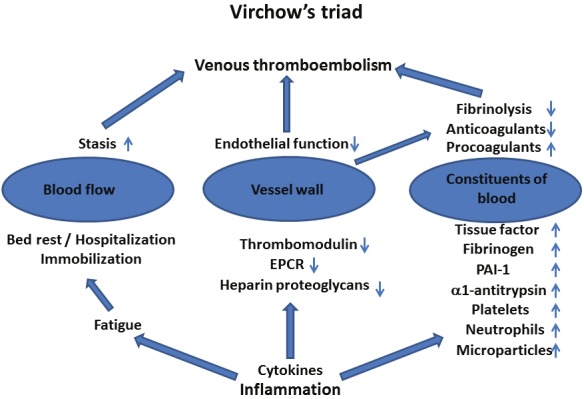
Virchow’s triad and some of the extensive inflammatory changes that may contribute to the development of venous thromboembolism. PAI-1=plasminogen activator inhibitor 1, EPCR=endothelial protein C receptor.
Table 1.
Inherited and acquired risk factors for hypercoagulable states.
| Inherited risk factors | Acquired risk factors |
|---|---|
| Established susceptibility loci for VTE | Advanced age |
| Antithrombin deficiency (SERPINC1) | Antiphospholipid antibodies |
| Cystathione beta-synthase deficiency (CBS) | Heart disease |
| Factor V Leiden Gln506 (rs6025)* (F5) | Hormone replacement therapy |
| Prothrombin G20210A (rs1799963) (F2) | Hyperviscosity |
| Protein C deficiency (PROC) | Infections |
| Protein S deficiency (PROS1) | Inflammatory disease |
| ABO blood group (ABO) | Immobilization (long bed rest or air travel, plaster cast) |
| Emerging susceptibility loci for VTE | Malignancy and chemotherapy |
| Complement component 4 binding protein, alpha and beta (CBPA/C4BPB) | Lifestyle factors (smoking, sedentary life, diet, low socioeconomic status, neighborhood deprivation) |
| Coagulation factor XI (F11) | Myeloproliferative disorders |
| Fibrinogen gamma chain (FGG) | Nephrotic syndrome |
| Von Willebrand factor (vWF) | Obesity |
| Glycoprotein VI (GP6) | Oral contraceptives |
| Human immunodeficiency virus type I enhancer binding protein 1 (HIVEP1) | Pregnancy and puerperium |
| Kininogen 1 (KNG1) | Trauma and surgery |
| Syntaxin binding protein 5 (STXBP5) | Varicose veins |
| Tandem C2 domains, nuclear (TC2N) | Venous catheters |
| Family history of VTE (sum of the effects of and interactions between familial genetic and environmental factors) | |
The major genetic cause of activated protein C (APC) resistance.
It has long been known that certain autoimmune disorders, such as systemic lupus erythematosus (SLE) [24-26], inflammatory bowel disease (IBD) [27,28] and Behçet’s syndrome [29], are linked to an increased risk of VTE. The list of autoimmune and immune-mediated disorders linked to an increased risk of VTE has grown longer and longer, and now also includes rheumatoid arthritis (RA) [30], celiac disease [31], hyperthyroidism [32], and Wegener’s granulomatosis [33]. However, two recent reports have linked a large number of autoimmune disorders/immune-mediated disease to an increased risk of PE and VTE [34,35]. Inflammation is a common feature of these disorders. The epidemiological and experimental evidence and clinical implications of the association between autoimmune/immune-mediated disorders and VTE will be discussed.
Inflammation, hypercoagulability and autoimmune/immune-mediated disorders
Inflammation is a common feature of many autoimmune and immune-mediated disorders [36]. Comparative studies indicate that coagulation and innate immunity have a common evolutionary origin [37,38]. It is therefore not surprising that the immune system and coagulations system are linked, with many molecular components being important for both systems [23,39-42]. Inflammatory pathways and coagulation are integrated by extensive crosstalk and a tendency to function in concert [23,39-42]. They comprise a large number of cellular and molecular factors, which interact in a very complex manner [23,39-42]. Some of the central features of the hypercoagulability induced by inflammation are cytokine induction of tissue factor (TF) expression, endothelial dysfunction, inhibition of the protein C system and inhibition of fibrinolysis (increased plasminogen activator inhibitor 1 levels) (Figure 1) [23,39-42]. Other important players that contribute to inflammation-induced hypercoagulability are platelets, microparticles (MP), neutrophils, thrombin and protease-activated receptors, fibrinogen, α1-antitrypsin, heparin proteoglycans and the contact system (Factor XII and the kallikrein-kinin system) [23,39-42].
Especially interesting are the effects of inflammation on the protein C system. Genetic defects affecting the protein C system (deficiencies in protein S and protein C, and the factor V Leiden Gln506 (rs6025) mutation associated with activated resistance to protein C) are the most common inherited risk factors for venous thrombosis [5,12]. Complete lack of protein C or protein S results in a fatal microvascular thrombotic disease (purpura fulminans) in the neonatal period [5]. This indicates that the protein C system is vitally important to keep the blood in a fluid state [5].
Protein C pathway
The anticoagulant protein C system regulates the activity of the coagulation factors VIIIa and Va, cofactors in the activation of factor X and prothrombin, respectively [5]. Protein C is activated on the endothelium by the thrombin–thrombomodulin–EPCR (endothelial protein C receptor) complex [5]. Activated protein C (APC)-mediated cleavage of factors VIIIa and Va occurs on negatively-charged phospholipid membranes and involves protein cofactors, protein S and factor V [5]. APC also has anti-inflammatory and anti-apoptotic activities that involve binding of APC to EPCR and cleavage of protease-activated receptor-1 (PAR-1) [43,44]. Thus, inhibition of the protein C system may augment the inflammatory response.
Tumor necrosis factor (TNF)-α and other inflammatory mediators can down-regulate EPCR and thrombomodulin (TM), while interleukin (IL)-6 can depress levels of protein S in experimental animals [43,44]. Neutrophils may also decrease TM activity through cleavage of TM by elastase or oxidation of methionine on TM by reactive oxygen species [43,44].
Another link between the protein C system and the complement pathway is the high-affinity interaction between the anticoagulant vitamin K -dependent protein S and the complement regulator C4b-binding protein (C4BP) [45]. Free protein S is a cofactor for APC in the inactivation of factor Va and factor VIIIa [5]. Approximately 70% of total protein S circulates in complex with C4BP; the remainder is free [45]. The free form is mainly responsible for the anticoagulant activity of protein S. The high-affinity binding of protein S to C4BP may direct C4BP to negatively-charged phospholipid membranes, thereby localizing complement regulatory activity to the membrane [45]. In inherited protein S deficiency, the tight binding of protein S to C4BP results in a pronounced and selective fall in the concentration of free protein S; the concentration of protein S in complex with C4BP is less affected [46]. C4BP is an acute-phase protein. During inflammation, levels of C4BP molecules lacking the protein S binding site (β chain) are selectively increased. This guarantees that free protein S levels during inflammation are normal [47]. However, an acquired decrease in protein S levels due to antiphospholipid antibodies against protein S has been reported in SLE patients [48-50] and may contribute to an increased risk of thrombosis. Non-phospholipid autoantibodies against protein S have also been reported to be associated with venous thrombosis [51,52].
Antiphospholipid antibodies, autoimmunity and VTE
Antiphospholipid antibodies target phospholipid-protein complexes [53-57] and have been reported to be risk factors for venous thrombosis [58-59]. These phospholipid-dependent autoantibodies include anticardiolipin antibodies, lupus anticoagulant and anti-β2 glycoprotein I antibodies. Primary antiphospholipid syndrome is an acquired condition that is characterized by venous or arterial thromboembolism, miscarriage and the presence of antiphospholipid antibodies [53-57]. Antibodies should be measurable on at least two occasions 12 weeks apart. However, absence of gold standards and marked heterogeneity in the autoantibodies make interpretation of laboratory results difficult [60]. Though antiphospholipid antibodies are a necessary feature of primary antiphospholipid syndrome, they are also common among patients with other autoimmune disorders. High prevalences of antiphospholipid antibodies have been observed in patients with SLE (33%) [61], primary systemic vasculitis (17%) [62], immune thrombocytopenic purpura (ITP) (25%) [63], thyroid disease (24%) [64], autoimmune hemolytic anemia (12%) [65] and RA (16%) [66]. Antiphospholipid antibodies may therefore be an important risk factor for thrombosis in several autoimmune disorders.
SLE and VTE
SLE is an autoimmune disease with a diverse array of clinical manifestations. Its incidence peaks between the age of 15 and 40 years, and it is more common in females than in males. There is strong evidence for an association between SLE and an increased risk of VTE [67,68]. VTE occurs with a higher frequency in SLE patients compared to the general population. Moreover, SLE is also associated with premature atherosclerosis. Antiphospholipid antibodies are important risk factors for VTE among SLE patients [67,68]. However, it not all SLE patients who develop thrombosis have antiphospholipid antibodies [61]. Other mechanisms such as inflammation, acquired protein S deficiency [48-50] and microparticles may also contribute to the thrombotic risk among SLE patients [42].
Thrombosis has been reported in about 10-26% of patients with SLE [69-75]. In one study, the risk of VTE was highest during the first 30 days after diagnosis of SLE [74]. However, in another study the risk of thrombosis remained elevated throughout the course of the disease [75]. In a large Swedish study of the risk of PE in patients hospitalized with 33 different autoimmune disorders, the risk for SLE was particularly high during the first year after diagnosis of an autoimmune disorder (Table 2), as compared to the general population [35]. In an English study of patients hospitalized with SLE, the risk of VTE was 3.7 times higher compared to a reference group of inpatients (Table 2) [34].
Table 2.
Risk of VTE among Swedish (only PE studied) and English patients hospitalized with autoimmune and immune-mediated diseases: the results of two follow-up studies.
| Autoimmune/immune-mediated disease | Swedish PE study 1964-2008 Zöller et al [35]* | English VTE study 1999-2008 Ramagopalan et al [34]** | |
|---|---|---|---|
|
| |||
| SIR (1-year follow-up) | SIR (follow-up 1964-2008) | Rate ratio (follow-up 1999-2008) | |
| Chorea minor | 16.7 | 1.8 | nd |
| Polymyositis/dermatomyositis | 16.4 | 3.4 | 3.0 |
| Lupoid hepatitis | 13.3 | 1.5 | 2.0 |
| Polyarteritis nodosa | 13.3 | 2.6 | 3.5 |
| Discoid lupus erythematosus | 12.0 | 2.2 | nd |
| Autoimmune hemolytic anemia | 11.1 | 3.4 | 3.8 |
| Immune thrombocytopenic purpura | 10.8 | 2.2 | 2.1 |
| Ulcerative colitis | 10.3 | 2.0 | nd |
| Systemic lupus erythematosus | 10.2 | 2.2 | 3.7 |
| Rheumatic fever | 10.1 | 1.6 | nd |
| Reiter’s disease | 9.4 | 1.0 | nd |
| Behçet’s disease | 9.0 | 1.7 | nd |
| Sarcoidosis | 8.9 | 1.5 | nd |
| Crohn’s disease | 8.7 | 1.6 | nd |
| Polymyalgia rheumatica | 7.9 | 1.9 | nd |
| Addison’s disease | 7.7 | 2.5 | 2.1 |
| Multiple sclerosis | 7.7 | 1.9 | 2.1 |
| Sjögren’s syndrome | 7.4 | 2.2 | 2.0 |
| Primary biliary cirrhosis | 7.4 | 2.4 | 1.5 |
| Myasthenia gravis | 7.2 | 1.7 | 2.3 |
| Systemic sclerosis | 7.1 | 1.6 | 2.0 |
| Wegener’s granulomatosis | 6.6 | 1.4 | nd |
| Graves’ disease | 6.5 | 1.3 | 1.3 |
| Diabetes mellitus type I | 6.4 | 1.0 | 2.6 |
| Celiac disease | 6.3 | 1.5 | 1.2 |
| Rheumatoid arthritis | 6.0 | 1.9 | 1.7 |
| Hashimoto’s thyroiditis | 5.3 | 1.6 | 1.4 |
| Amyotrophic lateral sclerosis | 5.1 | 2.1 | nd |
| Localized scleroderma | 4.9 | 1.5 | nd |
| Psoriasis | 4.8 | 1.4 | 1.7 |
| Ankylosing spondylitis | 4.3 | 1.2 | 1.9 |
| Chronic rheumatic heart disease | 4.2 | 1.0 | nd |
| Pernicious anemia | 3.9 | 1.3 | 1.4 |
| Goodpasture’s syndrome | nd | nd | 2.8 |
| All | 6.4 | 1.6 | nd |
Reference group: general population;
Reference group: inpatients.
SIR=standardized incidence ratio, nd=not determined. Values shown in bold were significant at the 95% level.
IBD and VTE
Numerous case reports and case series have described VTE in patients IBD [76-79]. A large number of studies have confirmed that the risk of VTE is increased in patients with IBD [76-86]. One study also found increased mortality from PE in IBD patients [87]. Only one study failed to show an association between VTE and IBD [88], but this study lacked a formal control group. Thus, there is strong clinical and epidemiological evidence for an increased risk of VTE among IBD patients. In a large Swedish study, the risk of PE in patients hospitalized with ulcerative colitis or Crohn’s disease was particularly high during the first year after diagnosis (Table 2), as compared to the general population [35]. A higher risk of VTE with increased disease activity (8.4 times increased compared to controls) was reported in an English study by Grainge et al [85]. Hospitalized IBD patients are at higher risk of VTE than ambulatory IBD patients, particularly in the context of active disease, and should be the focus of strategies aimed at preventing VTE among IBD patients [89].
In human and experimental murine IBD there is support for downregulation of EPCR and TM [90]. TM and EPCR expression was reduced in IBD, suggesting downregulation of the protein C pathway. Moreover, recombinant APC has potent anti-inflammatory effects, downregulating cytokine-dependent cell adhesion molecule expression and chemokine production and inhibiting leukocyte adhesion. In murine colitis, administration of APC was effective in reducing weight loss, disease activity index and histological colitis scores, and inhibiting leukocyte adhesion [90]. In another study, Yoshida et al showed that elevated APC levels protected against thrombosis in a murine colitis model [91]. Thus, increasing protein C pathway activity may reduce both inflammation and the risk of thrombosis in patients with IBD [90,91].
RA and VTE
The link between RA and VTE has not been as well studied as the ones for SLE and IBD. RA does not appear to be an additional risk factor for VTE after total knee arthroplasty [92]. However, in a large US study, DVT was diagnosed in 79,000 of 4,818,000 (1.64%) patients with RA who did not undergo joint surgery, compared with 7,681,000 of 891,055,000 patients (0.86%) who did not have RA or undergo joint surgery (relative risk (RR) = 1.90) [93]. The RR of VTE (PE and/or DVT) in these patients was 1.99. In other studies from England [34], Sweden [35] and Denmark [94] based on hospitalized patients; RA was a risk factor for VTE (Table 2). In the Danish study, the risk of VTE was high in patients with juvenile RA (RR 3.0) [94]. Thus, it appears that RA is a risk factor for VTE in hospitalized medical patients. A heightened awareness of the risk of VTE would be appropriate for hospitalized patients with RA.
Epidemiological link between autoimmunity and VTE
There is accumulating clinical, epidemiological and experimental data that an increased risk of VTE may not be limited to autoimmune conditions like Behçet’s syndrome, SLE and IBD. The inflammation associated with many autoimmune/immune-mediated disorders may increase the risk of VTE in a number of autoimmune disorders. In a study from England, Ramagopalan et al investigated the risk of VTE among patients hospitalized for 23 immune-mediated disorders (Table 2) [34]. The reference group was hospitalized patients with other diagnoses, mainly minor medical conditions [34]. The authors found that all 23 autoimmune disorders they studied were associated with an increased risk of VTE (Table 2). They found that the risk of VTE was increased both during and after the first 90 days after diagnosis. In a large Swedish study by Zöller et al, risk of PE in patients hospitalized with 33 different autoimmune/immune-mediated disorders was investigated (Table 2) [35]. The reference group was the general population adjusted for age, sex, time period and 10 different comorbidities, and the risk of PE over time was determined. For all 33 studied autoimmune disorders, the risk of PE was very high during the first year after hospitalization (Table 2) [35]. The overall risk of PE was 6.4 times higher compared to the normal population during the first year after diagnosis. Risk of PE was especially high in patients with autoimmune disorders such as immune thrombocytopenic purpura (standardized incidence ratio (SIR=11), polyarteritis nodosa (SIR=13), polymyositis/dermatomyositis (SIR=16), IBD (SIR=10) and SLE (SIR=10) (Table 2) [35] and the overall risk during whole follow up time was increased for 30 of the 33 autoimmune disorders. However, the overall risk during whole follow up time was much lower than that during the first year. The higher initial risk is most likely due to a combination of severe inflammation related to high disease activity and immobilization. Other possible factors are treatment effects (corticosteroids may affect the coagulation system) and the possibility that those patients with VTE predisposition (thrombophilia) may develop PE during the first year after diagnosis. For instance, among patients taking oral contraceptives, the risk of VTE is highest during the first year of treatment [95].
Though designs of the Swedish and English studies are somewhat different, the risk estimates for the different autoimmune disorders assessed in both studies are correlated. The correlation between the 1-year follow up PE risks in the Swedish study and the VTE risks in the English study (first versus third column in Table 2) is significant (Pearson’s coefficient 0.675; p=0.001). Correlating the overall risk for the whole follow-up period in the Swedish study (second column in Table 2) with the risk estimates from English study gives a Pearson’s coefficient of 0.643 (p=0.002). A limitation of both the English and Swedish studies is that they included only hospitalized patients, and not outpatients [34,35]. Hospitalized patients are likely to have not only higher disease activity but also more comorbidities. Thus, the risk of VTE in outpatients with autoimmune diseases may be much lower. A Danish study of autoimmune skin disorders and connective tissue disorders based on both inpatients and outpatients by Johannesdottir et al tried to correct for comorbidities by adjusting for prescribed medicines [94]. Autoimmune skin disorders were not found to be risk factors for VTE (RR 1.0) [94]. However, autoimmune connective tissue disorders were associated with an increased risk of VTE (2.3-fold increased risk within 90 days) [94]. Further studies of outpatients with autoimmune diseases may therefore be warranted.
Is inflammation a link between VTE and atherosclerosis?
Several studies have found associations between VTE and atherosclerosis and its different thromboembolic manifestations, including myocardial infarction (MI) and coronary heart disease [96-100]. After the discovery of APC resistance by Dahlbäck et al [101] and the finding that factor V Leiden Gln506 (rs6025) [102,103], the mutation that causes APC resistance in the majority of cases [104], is associated with premature MI [105], a large number of studies on this topic have been published. However, association studies of hemostatic factors and MI and CHD have produced varying results [106]. Meta-analysis data suggest that factor V Leiden Gln506 (rs6025) and the prothrombin G20210A (rs1799963), important risk factors for VTE, are only weak risk factors for CHD (RR 1.17 and 1.31, respectively) [106]. Moreover, in a nationwide Swedish family study, family history of VTE was not a strong risk factor for CHD and myocardial infarction [107]. In another nationwide Swedish family study, family history of VTE was not a strong risk factor for ischemic stroke [108]. These results argue against the existence of common shared disease-causing mutations for CHD and ischemic stroke and VTE in the Swedish population. During recent years it has become clear that systemic inflammation can increase atherogenesis [109]. It is therefore interesting that 27 of 32 immune-mediated diseases investigated in a nationwide Swedish study were associated with an increased risk of CHD during the first year after hospitalization [110]. Thus, most immune-mediated diseases are linked both to VTE and CHD [34,35,110], which further confirms that inflammation is a link between VTE and atherosclerosis [111].
Treatment of inflammation and risk of VTE
Treating inflammation in autoimmune and immune-mediated diseases is probably the most important way of preventing VTE, and is of course mandatory for many autoimmune/immune-mediated disorders in order to reduce morbidity and mortality. Inflammatory pathways and coagulation are integrated by extensive crosstalk and tend to function in concert [39-42]; inflammation promotes a number of procoagulant changes in the coagulation and anticoagulation systems, as discussed above. Moreover, higher inflammatory disease activity has been linked to higher VTE risk [35,85,89]. It is therefore a reasonable hypothesis that efficient treatment of the inflammation associated with autoimmune disorders will reduce the risk of PE and VTE. In mouse models of colitis, treatment with APC reduced inflammation and protected against thrombosis [90,91].
A recent study found no difference in thrombotic risk between RA patients treated with anti-TNF therapy and those treated with non-biological disease-modifying antirheumatic drugs, suggesting that these newer agents are safe from a thrombotic perspective [112]. A special topic is the possible induction of procoagulant hemostatic changes by glucocorticoids [113]. Whether glucocorticoid use contributes to a hypercoagulable state, and thereby increases the thrombotic risk, is controversial [114]. A systematic review showed that the effects of glucocorticoids differ according to the clinical situation in which there is given, most likely as a result of their disease-modifying properties. The authors conclude that clinical outcome studies are needed to assess the risk-benefit of glucocorticoid use regarding thrombotic complications [114].
Prophylactic treatment with low-molecular weight heparin?
Three large well-controlled studies (MEDENOX, PREVENT and ARTEMIS) showed a consistent 50% reduction in VTE events in acutely ill medical patients treated with low-molecular weight heparin (LMWH) [115-117]. However, LWMH has failed to reduce mortality in acutely ill medical patients [118]. A systematic review concluded that heparin prophylaxis had no significant effect on mortality, but might reduce PE risk [119]. However, heparin also increased the risk of bleeding risk [119]. Benefits and risks did not differ according to the type of heparin used.
As randomized trials focused on autoimmune and immune-mediated diseases are lacking, decisions regarding prophylactic treatment in hospitalized patients have to be based on general published guidelines [120]. According to the Evidence-Based Clinical Practice Guidelines of the American College of Chest Physicians, decisions regarding prophylaxis in nonsurgical patients should be made after consideration of risk factors for both thrombosis and bleeding, clinical context, and patients’ values and preferences [120]. For acutely ill hospitalized medical patients at increased risk of thrombosis, these guidelines recommend anticoagulant thromboprophylaxis with LMWH, low-dose unfractionated heparin or fondaparinux, and advise against extended thromboprophylaxis beyond the period of patient immobilization or acute hospitalization [120]. For patients at low risk of thrombosis, they advise against the use of pharmacologic prophylaxis or mechanical prophylaxis [120]. For thrombosis-prone hospitalized patients who are bleeding or are at high risk of major bleeding, they suggest mechanical thromboprophylaxis [120].
Conclusions
The immune and coagulation systems are tightly linked. Accumulating epidemiological, clinical and experimental evidence shows that an increased risk of VTE is a feature of most autoimmune disorders and immune-mediated diseases. Mechanisms involved in this association include inflammation and antiphospholipid autoantibodies. Active untreated autoimmune disorders should be considered as hypercoagulable disorders and not only inflammatory disorders. Further studies are needed to evaluate potential thrombotic mechanisms and clinical risk factors, and prophylactic strategies.
Acknowledgements
The authors wish to thank the CPF’s Science Editor Stephen Gilliver for his useful comments on the text. This work was supported by grants to Bengt Zöller from the Swedish Heart-Lung Foundation and Region Skåne (REGSKANE-124611), and to Kristina and Jan Sundquist from the Swedish Research Council (2008-3110 and 2008-2638), the Swedish Council for Working Life and Social Research (2006-0386, 2007-1754 and 2007-1962 ), and the Swedish Research Council for Environment, Agricultural Sciences and Spatial Planning (2006-4255-6596-99 and 2007-1352).
References
- 1.Heit JA, Silverstein MD, Mohr DN, Petterson TM, Lohse CM, O’Fallon WM, Melton LJ 3rd. The epidemiology of venous thromboembolism in the community. Thromb Haemost. 2001;86:452–463. [PubMed] [Google Scholar]
- 2.White R. The Epidemiology of venous thromboembolism. Circulation. 2003;107:I4–I8. doi: 10.1161/01.CIR.0000078468.11849.66. [DOI] [PubMed] [Google Scholar]
- 3.Goldhaber SZ, Visani L, De Rosa M. Acute pulmonary embolism: clinical outcomes in the International Cooperative Pulmonary Embolism Registry (ICOPER) Lancet. 1999;353:1386–1389. doi: 10.1016/s0140-6736(98)07534-5. [DOI] [PubMed] [Google Scholar]
- 4.Goldhaber SZ, Elliott CG. Acute pulmonary embolism: part I: epidemiology, pathophysiology, and diagnosis. Circulation. 2003;108:2726–2729. doi: 10.1161/01.CIR.0000097829.89204.0C. [DOI] [PubMed] [Google Scholar]
- 5.Dahlbäck B. Advances in understanding pathogenic mechanisms of thrombophilic disorders. Blood. 2008;112:19–127. doi: 10.1182/blood-2008-01-077909. [DOI] [PubMed] [Google Scholar]
- 6.Martinelli I, Bucciarelli P, Mannucci PM. Thrombotic risk factors: basic pathophysiology. Crit Care Med. 2010;38:S3–S9. doi: 10.1097/CCM.0b013e3181c9cbd9. [DOI] [PubMed] [Google Scholar]
- 7.Esmon CT. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. 2009;23:225–229. doi: 10.1016/j.blre.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reitsma PH, Versteeg HH, Middeldorp S. Mechanistic view of risk factors for venous thromboembolism. Arterioscler Thromb Vasc Biol. 2012;32:563–568. doi: 10.1161/ATVBAHA.111.242818. [DOI] [PubMed] [Google Scholar]
- 9.Rosendaal FR. Venous thrombosis: a multicausal disease. Lancet. 1999;353:1167–1173. doi: 10.1016/s0140-6736(98)10266-0. [DOI] [PubMed] [Google Scholar]
- 10.Lijfering WM, Rosendaal FR, Cannegieter SC. Risk factors for venous thrombosis –current understanding from an epidemiological point of view. Br J Haematol. 2010;149:824–833. doi: 10.1111/j.1365-2141.2010.08206.x. [DOI] [PubMed] [Google Scholar]
- 11.Stein PD, Matta F. Epidemiology and incidence: the scope of the problem and risk factors for development of venous thromboembolism. Clin Chest Med. 2010;31:611–628. doi: 10.1016/j.ccm.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 12.Zöller B, García de Frutos P, Hillarp A, Dahlbäck B. Thrombophilia as a multigenic disease. Haematologica. 1999;84:59–70. [PubMed] [Google Scholar]
- 13.Rosendaal FR, Reitsma PH. Genetics of venous thrombosis. J Thromb Haemost. 2009;7(Suppl 1):301–304. doi: 10.1111/j.1538-7836.2009.03394.x. [DOI] [PubMed] [Google Scholar]
- 14.Germain M, Saut N, Greliche N, Dina C, Lambert JC, Perret C, Cohen W, Oudot-Mellakh T, Antoni G, Alessi MC, Zelenika D, Cambien F, Tiret L, Bertrand M, Dupuy AM, Letenneur L, Lathrop M, Emmerich J, Amouyel P, Trégouët DA, Morange PE. Genetics of venous thrombosis: insights from a new genome wide association study. PLoS One. 2011;6:e25581. doi: 10.1371/journal.pone.0025581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guttmacher AE, Collins FS, Carmona RH. The family history - more important than ever. N Engl J Med. 2004;351:2333–2336. doi: 10.1056/NEJMsb042979. [DOI] [PubMed] [Google Scholar]
- 16.Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. The value of family history as a risk indicator for venous thrombosis. Bezemer ID, van der Meer FJ, Eikenboom JC, Rosendaal FR, Doggen CJ. Arch Intern Med. 2009;169:610–615. doi: 10.1001/archinternmed.2008.589. [DOI] [PubMed] [Google Scholar]
- 17.Zöller B, Li X, Sundquist J, Sundquist K. Parental history and venous thromboembolism: a nationwide study of age-specific and sex-specific familial risks in Sweden. J Thromb Haemost. 2011;9:64–70. doi: 10.1111/j.1538-7836.2010.04107.x. [DOI] [PubMed] [Google Scholar]
- 18.Zöller B, Li X, Sundquist J, Sundquist K. Age-and gender-specific familial risks for venous thromboembolism: a nationwide epidemiological study based on hospitalizations in Sweden. Circulation. 2011;124:1012–1020. doi: 10.1161/CIRCULATIONAHA.110.965020. [DOI] [PubMed] [Google Scholar]
- 19.Zöller B, Li X, Sundquist J, Sundquist K. Socioeconomic and occupational risk factors for venous thromboembolism in Sweden: A nationwide epidemiological study. Thromb Res. 2012;129:577–582. doi: 10.1016/j.thromres.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 20.Zöller B, Li X, Sundquist J, Sundquist K. Neighborhood deprivation and hospitalization for venous thromboembolism in Sweden. J Thromb Thrombolysis. 2012 doi: 10.1007/s11239-012-0728-4. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 21.Smeeth L, Cook C, Thomas S, Hall AJ, Hubbard R, Vallance P. Risk of deep vein thrombosis and pulmonary embolism after acute infection in a community setting. Lancet. 2006;367:1075–1079. doi: 10.1016/S0140-6736(06)68474-2. [DOI] [PubMed] [Google Scholar]
- 22.Schmidt M, Horvath-Puho E, Thomsen RW, Smeeth L, Sørensen HT. Acute infections and venous thromboembolism. J Intern Med. 2012;271:608–618. doi: 10.1111/j.1365-2796.2011.02473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dahlbäck B. Coagulation and inflammation-- close allies in health and disease. Semin Immunopathol. 2012;34:1–3. doi: 10.1007/s00281-011-0298-0. [DOI] [PubMed] [Google Scholar]
- 24.Peck B, Hoffman GS, Franck WA. Thrombophlebitis in systemic lupus erythematosus. JAMA. 1978;240:1728–1730. [PubMed] [Google Scholar]
- 25.Cervera R, Khamashta MA, Font J, Sebastiani GD, Gil A, Lavilla P, Mejía JC, Aydintug AO, Chwalinska-Sadowska H, de Ramón E, Fernández-Nebro A, Galeazzi M, Valen M, Mathieu A, Houssiau F, Caro N, Alba P, Ramos-Casals M, Ingelmo M, Hughes GR European Working Party on Systemic Lupus Erythematosus. Morbidity and mortality in systemic lupus erythematosus during a 10-year period: a comparison of early and late manifestations in a cohort of 1,000 patients. Medicine (Baltimore) 2003;82:299–308. doi: 10.1097/01.md.0000091181.93122.55. [DOI] [PubMed] [Google Scholar]
- 26.Palatinus A, Adams M. Thrombosis in systemic lupus erythematosus. Semin Thromb Hemost. 2009;35:621–629. doi: 10.1055/s-0029-1242716. [DOI] [PubMed] [Google Scholar]
- 27.Di Fabio F, Lykoudis P, Gordon PH. Thromboembolism in inflammatory bowel disease: an insidious association requiring a high degree of vigilance. Semin Thromb Hemost. 2011;37:220–225. doi: 10.1055/s-0031-1273086. [DOI] [PubMed] [Google Scholar]
- 28.Murthy SK, Nguyen GC. Venous thromboembolism in inflammatory bowel disease: an epidemiological review. Am J Gastroenterol. 2011;106:713–718. doi: 10.1038/ajg.2011.53. [DOI] [PubMed] [Google Scholar]
- 29.Yazici H, Fresko I, Yurdakul S. Behçet’s syndrome: disease manifestations, management, and advances in treatment. Nat Clin Pract Rheumatol. 2007;3:148–155. doi: 10.1038/ncprheum0436. [DOI] [PubMed] [Google Scholar]
- 30.Matta F, Singala R, Yaekoub AY, Najjar R, Stein PD. Risk of venous thromboembolism with rheumatoid arthritis. Thromb Haemost. 2009;101:134–138. [PubMed] [Google Scholar]
- 31.Ludvigsson JF, Welander A, Lassila R, Ekbom A, Montgomery SM. Risk of thromboembolism in 14,000 individuals with coeliac disease. Br J Haematol. 2007;139:121–127. doi: 10.1111/j.1365-2141.2007.06766.x. [DOI] [PubMed] [Google Scholar]
- 32.Lin HC, Yang LY, Kang JH. Increased risk of pulmonary embolism among patients with hyperthyroidism: a 5-year follow-up study. J Thromb Haemost. 2010;8:2176–2178. doi: 10.1111/j.1538-7836.2010.03993.x. [DOI] [PubMed] [Google Scholar]
- 33.Merkel PA, Lo GH, Holbrook JT, Tibbs AK, Allen NB, Davis JC Jr, Hoffman GS, McCune WJ, St Clair EW, Specks U, Spiera R, Petri M, Stone JH Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med. 2005;142:620–626. doi: 10.7326/0003-4819-142-8-200505030-00011. [DOI] [PubMed] [Google Scholar]
- 34.Ramagopalan SV, Wotton CJ, Handel AE, Yeates D, Goldacre MJ. Risk of venous thromboembolism in people admitted to hospital with selected immune-mediated diseases: record-linkage study. BMC Med. 2011;9:1–8. doi: 10.1186/1741-7015-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zöller B, Li X, Sundquist J, Sundquist K. Risk of pulmonary embolism in patients with autoimmune disorders: a nationwide follow-up study from Sweden. Lancet. 2012;379:244–249. doi: 10.1016/S0140-6736(11)61306-8. [DOI] [PubMed] [Google Scholar]
- 36.Rahman P, Inman RD, El-Gabalawy H, Krause DO. Pathophysiology and pathogenesis of immune-mediated inflammatory diseases: commonalities and differences. J Rheumatol Suppl. 2010;85:11–26. doi: 10.3899/jrheum.091462. [DOI] [PubMed] [Google Scholar]
- 37.Krem MM, Di Cera E. Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem Sci. 2002;27:67–74. doi: 10.1016/s0968-0004(01)02007-2. [DOI] [PubMed] [Google Scholar]
- 38.Loof TG, Schmidt O, Herwald H, Theopold U. Coagulation systems of invertebrates and vertebrates and their roles in innate immunity: the same side of two coins? J Innate Immun. 2011;3:34–40. doi: 10.1159/000321641. [DOI] [PubMed] [Google Scholar]
- 39.Esmon CT, Esmon NL. The link between vascular features and thrombosis. Annu Rev Physiol. 2011;73:503–514. doi: 10.1146/annurev-physiol-012110-142300. [DOI] [PubMed] [Google Scholar]
- 40.Xu J, Lupu F, Esmon CT. Inflammation, innate immunity and blood coagulation. Hamostaseologie. 2010;30:5–6. 8–9. [PubMed] [Google Scholar]
- 41.Esmon CT. The interactions between inflammation and coagulation. Br J Haematol. 2005;131:417–430. doi: 10.1111/j.1365-2141.2005.05753.x. [DOI] [PubMed] [Google Scholar]
- 42.Ardoin SP, Shanahan JC, Pisetsky DS. The role of microparticles in inflammation and thrombosis. Scand J Immunol. 2007;66:159–165. doi: 10.1111/j.1365-3083.2007.01984.x. [DOI] [PubMed] [Google Scholar]
- 43.Esmon CT. Inflammation and the activated protein C anticoagulant pathway. Semin Thromb Hemost. 2006;32(Suppl 1):49–60. doi: 10.1055/s-2006-939554. [DOI] [PubMed] [Google Scholar]
- 44.Esmon CT. Protein C anticoagulant system--anti-inflammatory effects. Semin Immunopathol. 2012;34:127–132. doi: 10.1007/s00281-011-0284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dahlbäck B. C4b-binding protein: a forgotten factor in thrombosis and hemostasis. Semin Thromb Hemost. 2011;37:355–361. doi: 10.1055/s-0031-1276584. [DOI] [PubMed] [Google Scholar]
- 46.Zöller B, García de Frutos P, Dahlbäck B. Evaluation of the relationship between protein S and C4b-binding protein isoforms in hereditary protein S deficiency demonstrating type I and type III deficiencies to be phenotypic variants of the same genetic disease. Blood. 1995;85:3524–3531. [PubMed] [Google Scholar]
- 47.García de Frutos P, Alim RI, Härdig Y, Zöller B, Dahlbäck B. Differential regulation of alpha and beta chains of C4b-binding protein during acute-phase response resulting in stable plasma levels of free anticoagulant protein S. Blood. 1994;84:815–822. [PubMed] [Google Scholar]
- 48.Ginsberg JS, Demers C, Brill-Edwards P, Bona R, Johnston M, Wong A, Denburg JA. Acquired free protein S deficiency is associated with antiphospholipid antibodies and increased thrombin generation in patients with systemic lupus erythematosus. Am J Med. 1995;98:379–383. doi: 10.1016/S0002-9343(99)80317-9. [DOI] [PubMed] [Google Scholar]
- 49.Tomás JF, Alberca I, Tabernero MD, Cordero M, Del Pino-Montes J, Vicente V. Natural anticoagulant proteins and antiphospholipid antibodies in systemic lupus erythematosus. J Rheumatol. 1998;25:57–62. [PubMed] [Google Scholar]
- 50.Song KS, Park YS, Kim HK. Prevalence of anti-protein S antibodies in patients with systemic lupus erythematosus. Arthritis Rheum. 2000;43:557–560. doi: 10.1002/1529-0131(200003)43:3<557::AID-ANR11>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 51.D’Angelo A, Della Valle P, Crippa L, Pattarini E, Grimaldi LM, Viganò D’Angelo S. Brief report: autoimmune protein S deficiency in a boy with severe thromboembolic disease. N Engl J Med. 1993;328:1753–1757. doi: 10.1056/NEJM199306173282405. [DOI] [PubMed] [Google Scholar]
- 52.Rask O, Hillarp A, Berntorp E, Ljung R. Anti-prothrombin antibodies are associated with thrombosis in children. Thromb Res. 2010;125:19–24. doi: 10.1016/j.thromres.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 53.Ruiz-Irastorza G, Crowther M, Branch W, Khamashta MA. Antiphospholipid syndrome. Lancet. 2010;376:1498–1509. doi: 10.1016/S0140-6736(10)60709-X. [DOI] [PubMed] [Google Scholar]
- 54.Espinosa G, Cervera R. Antiphospholipid syndrome: frequency, main causes and risk factors of mortality. Nat Rev Rheumatol. 2010;6:296–300. doi: 10.1038/nrrheum.2010.47. [DOI] [PubMed] [Google Scholar]
- 55.Meroni PL, Borghi MO, Raschi E, Tedesco F. Pathogenesis of antiphospholipid syndrome: understanding the antibodies. Nat Rev Rheumatol. 2011;7:330–339. doi: 10.1038/nrrheum.2011.52. [DOI] [PubMed] [Google Scholar]
- 56.Lim W, Crowther MA, Eikelboom JW. Management of antiphospholipid antibody syndrome: a systematic review. JAMA. 2006;295:1050–1057. doi: 10.1001/jama.295.9.1050. [DOI] [PubMed] [Google Scholar]
- 57.Erkan D, Lockshin MD. What is antiphospholipid syndrome? Curr Rheumatol Rep. 2004;6:451–457. doi: 10.1007/s11926-004-0024-1. [DOI] [PubMed] [Google Scholar]
- 58.de Groot PG, Lutters B, Derksen RH, Lisman T, Meijers JC, Rosendaal FR. Lupus anticoagulants and the risk of a first episode of deep venous thrombosis. J Thromb Haemost. 2005;3:1993–1997. doi: 10.1111/j.1538-7836.2005.01485.x. [DOI] [PubMed] [Google Scholar]
- 59.Roldan V, Lecumberri R, Munoz-Torrero JF, Vicente V, Rocha E, Brenner B, Monreal M RIETE Investigators. Thrombophilia testing in patients with venous thromboembolism. Findings from the RIETE registry. Thromb Res. 2009;124:174–177. doi: 10.1016/j.thromres.2008.11.003. [DOI] [PubMed] [Google Scholar]
- 60.Devreese K, Hoylaerts MF. Challenges in the diagnosis of the antiphospholipid syndrome. Clin Chem. 2010;56:930–940. doi: 10.1373/clinchem.2009.133678. [DOI] [PubMed] [Google Scholar]
- 61.Bengtsson A, Zöller B, de Frutos PG, Dahlbäck B, Sturfelt G. Factor V:Q506 mutation and anticardiolipin antibodies in systemic lupus erythematosus. Lupus. 1996;5:598–601. doi: 10.1177/096120339600500607. [DOI] [PubMed] [Google Scholar]
- 62.Rees JD, Lança S, Marques PV, Gómez-Puerta JA, Moco R, Oliveri C, Khamashta MA, Hughes GR, D’Cruz DP. Prevalence of the antiphospholipid syndrome in primary systemic vasculitis. Ann Rheum Dis. 2006;65:109–111. doi: 10.1136/ard.2004.034231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pierrot-Deseilligny Despujol C, Michel M, Khellaf M, Gouault M, Intrator L, Bierling P, Godeau B. Antiphospholipid antibodies in adults with immune thrombocytopenic purpura. Br J Haematol. 2008;142:638–643. doi: 10.1111/j.1365-2141.2008.07228.x. [DOI] [PubMed] [Google Scholar]
- 64.de Carvalho JF, Caleiro MT. Primary antiphospholipid syndrome and thyroid involvement. J Clin Rheumatol. 2010;16:164–167. doi: 10.1097/RHU.0b013e3181df5592. [DOI] [PubMed] [Google Scholar]
- 65.Rottem M, Krause I, Fraser A, Stojanovich L, Rovensky J, Shoenfeld Y. Autoimmune hemolytic anaemia in the antiphospholipid syndrome. Lupus. 2006;15:473–477. doi: 10.1191/0961203306lu2336oa. [DOI] [PubMed] [Google Scholar]
- 66.Pahor A, Hojs R, Holc I, Ambrozic A, Cucnik S, Kveder T, Rozman B. Antiphospholipid antibodies as a possible risk factor for atherosclerosis in patients with rheumatoid arthritis. Immunobiology. 2006;211:689–694. doi: 10.1016/j.imbio.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 67.Gaubitz M. Epidemiology of connective tissue disorders. Rheumatology (Oxford) 2006;45(Suppl 3):iii3–iii4. doi: 10.1093/rheumatology/kel282. [DOI] [PubMed] [Google Scholar]
- 68.Palatinus A, Adams M. Thrombosis in systemic lupus erythematosus. Semin Thromb Hemost. 2009;35:621–629. doi: 10.1055/s-0029-1242716. [DOI] [PubMed] [Google Scholar]
- 69.Burgos PI, Alarcón GS. Thrombosis in systemic lupus erythematosus: risk and protection. Expert Rev Cardiovasc Ther. 2009;7:1541–1549. doi: 10.1586/erc.09.137. [DOI] [PubMed] [Google Scholar]
- 70.Somers E, Magder LS, Petri M. Antiphospholipid antibodies and incidence of venous thrombosis in a cohort of patients with systemic lupus erythematosus. J Rheumatol. 2002;29:2531–2536. [PubMed] [Google Scholar]
- 71.Brouwer JL, Bijl M, Veeger NJ, Kluin-Nelemans HC, van der Meer J. The contribution of inherited and acquired thrombophilic defects, alone or combined with antiphospholipid antibodies, to venous and arterial thromboembolism in patients with systemic lupus erythematosus. Blood. 2004;104:143–148. doi: 10.1182/blood-2003-11-4085. [DOI] [PubMed] [Google Scholar]
- 72.Sarabi ZS, Chang E, Bobba R, Ibanez D, Gladman D, Urowitz M, Fortin PR. Incidence rates of arterial and venous thrombosis after diagnosis of systemic lupus erythematosus. Arthritis Rheum. 2005;53:609–612. doi: 10.1002/art.21314. [DOI] [PubMed] [Google Scholar]
- 73.Calvo-Alén J, Toloza SM, Fernández M, Bastian HM, Fessler BJ, Roseman JM, McGwin G Jr, Vilá LM, Reveille JD, Alarcón GS LUMINA Study Group. Systemic lupus erythematosus in a multiethnic US cohort (LUMINA). XXV. Smoking, older age, disease activity, lupus anticoagulant, and glucocorticoid dose as risk factors for the occurrence of venous thrombosis in lupus patients. Arthritis Rheum. 2005;52:2060–2068. doi: 10.1002/art.21149. [DOI] [PubMed] [Google Scholar]
- 74.Chang ER, Pineau CA, Bernatsky S, Neville C, Clarke AE, Fortin PR. Risk for incident arterial or venous vascular events varies over the course of systemic lupus erythematosus. J Rheumatol. 2006;33:1780–1784. [PubMed] [Google Scholar]
- 75.Romero-Díaz J, García-Sosa I, Sánchez-Guerrero J. Thrombosis in systemic lupus erythematosus and other autoimmune diseases of recent onset. Romero-Díaz J, García-Sosa I, Sánchez-Guerrero J. J Rheumatol. 2009;36:68–75. doi: 10.3899/jrheum.071244. [DOI] [PubMed] [Google Scholar]
- 76.Bergen JA, Barker NW. Extensive arterial and venous thrombosis complicating chronic ulcerative colitis. Arch Intern Med. 1936;58:17–31. [Google Scholar]
- 77.Talbot RW, Heppell J, Dozois RR, Beart RW Jr. Vascular complications of inflammatory bowel disease. Mayo Clin Proc. 1986;61:140–145. doi: 10.1016/s0025-6196(12)65200-8. [DOI] [PubMed] [Google Scholar]
- 78.Markowitz RL, Ment LR, Gryboski JD. Cerebral thromboembolic disease in pediatric and adult inflammatory bowel disease: case report and review of the literature. Pediatr Gastroenterol Nutr. 1989;8:413–420. doi: 10.1097/00005176-198904000-00028. [DOI] [PubMed] [Google Scholar]
- 79.Paradis K, Bernstein ML, Adelson JW. Thrombosis as a complication of inflammatory bowel disease in children: a report of four cases. J Pediatr Gastroenterol Nutr. 1985;4:659–662. doi: 10.1097/00005176-198508000-00029. [DOI] [PubMed] [Google Scholar]
- 80.Bernstein CN, Blanchard JF, Houston DS, Wajda A. The incidence of deep venous thrombosis and pulmonary embolism among patients with inflammatory bowel disease: a population-based cohort study. Thromb Haemost. 2001;85:430–434. [PubMed] [Google Scholar]
- 81.Miehsler W, Reinisch W, Valic E, Osterode W, Tillinger W, Feichtenschlager T, Grisar J, Machold K, Scholz S, Vogelsang H, Novacek G. Is inflammatory bowel disease an independent and disease specific risk factor for thromboembolism? Gut. 2004;53:542–548. doi: 10.1136/gut.2003.025411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bernstein CN, Nabalamba A. Hospitalization-based major comorbidity of inflammatory bowel disease in Canada. Can J Gastroenterol. 2007;21:507–511. doi: 10.1155/2007/924257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nguyen GC, Sam J. Rising prevalence of venous thromboembolism and its impact on mortality among hospitalized inflammatory bowel disease patients. Am J Gastroenterol. 2008;103:2272–2280. doi: 10.1111/j.1572-0241.2008.02052.x. [DOI] [PubMed] [Google Scholar]
- 84.Wang JY, Terdiman JP, Vittinghoff E, Minichiello T, Varma MG. Hospitalized ulcerative colitis patients have an elevated risk of thromboembolic events. World J Gastroenterol. 2009;15:927–935. doi: 10.3748/wjg.15.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grainge MJ, West J, Card TR. Venous thromboembolism during active disease and remission in inflammatory bowel disease: a cohort study. Lancet. 2010;375:657–663. doi: 10.1016/S0140-6736(09)61963-2. [DOI] [PubMed] [Google Scholar]
- 86.Kappelman MD, Horvath-Puho E, Sandler RS, Rubin DT, Ullman TA, Pedersen L, Baron JA, Sørensen HT. Thromboembolic risk among Danish children and adults with inflammatory bowel diseases: a population-based nationwide study. Gut. 2011;60:937–943. doi: 10.1136/gut.2010.228585. [DOI] [PubMed] [Google Scholar]
- 87.Jess T, Gamborg M, Munkholm P, Sørensen TI. Overall and cause-specific mortality in ulcerative colitis: meta-analysis of population-based inception cohort studies. Am J Gastroenterol. 2007;102:609–617. doi: 10.1111/j.1572-0241.2006.01000.x. [DOI] [PubMed] [Google Scholar]
- 88.Grip O, Svensson PJ, Lindgren S. Inflammatory bowel disease promotes venous thrombosis earlier in life. Scand J Gastroenterol. 2000;35:619–623. doi: 10.1080/003655200750023589. [DOI] [PubMed] [Google Scholar]
- 89.Murthy SK, Nguyen GC. Venous thromboembolism in inflammatory bowel disease: an epidemiological review. Am J Gastroenterol. 2011;106:713–718. doi: 10.1038/ajg.2011.53. [DOI] [PubMed] [Google Scholar]
- 90.Scaldaferri F, Sans M, Vetrano S, Graziani C, De Cristofaro R, Gerlitz B, Repici A, Arena V, Malesci A, Panes J, Grinnell BW, Danese S. Crucial role of the protein C pathway in governing microvascular inflammation in inflammatory bowel disease. J Clin Invest. 2007;117:1951–1960. doi: 10.1172/JCI31027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yoshida H, Russell J, Stokes KY, Yilmaz CE, Esmon CT, Granger DN. Role of the protein C pathway in the extraintestinal thrombosis associated with murine colitis. Gastroenterology. 2008;135:882–888. doi: 10.1053/j.gastro.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Niki Y, Matsumoto H, Hakozaki A, Mochizuki T, Momohara S. Rheumatoid arthritis: a risk factor for deep venous thrombosis after total knee arthroplasty? Comparative study with osteoarthritis. J Orthop Sci. 2010;15:57–63. doi: 10.1007/s00776-009-1410-3. [DOI] [PubMed] [Google Scholar]
- 93.Matta F, Singala R, Yaekoub AY, Najjar R, Stein PD. Risk of venous thromboembolism with rheumatoid arthritis. Thromb Haemost. 2009;101:134–138. [PubMed] [Google Scholar]
- 94.Johannesdottir SA, Schmidt M, Horváth-Puhó E, Sørensen HT. Autoimmune skin and connective tissue diseases and risk of venous thromboembolism: a population-based case-control study. J Thromb Haemost. 2012;10:815–821. doi: 10.1111/j.1538-7836.2012.04666.x. [DOI] [PubMed] [Google Scholar]
- 95.Gomes MP, Deitcher SR. Risk of venous thromboembolic disease associated with hormonal contraceptives and hormone replacement therapy: a clinical review. Arch Intern Med. 2004;164:1965–1976. doi: 10.1001/archinte.164.18.1965. [DOI] [PubMed] [Google Scholar]
- 96.Agnelli G, Becattini C. Venous thromboembolism and atherosclerosis: common denominators or different diseases? J Thromb Haemost. 2006;4:1886–1890. doi: 10.1111/j.1538-7836.2006.02138.x. [DOI] [PubMed] [Google Scholar]
- 97.Prandoni P, Bilora F, Marchiori A, Bernardi E, Petrobelli F, Lensing AW, Prins MH, Girolami A. An association between atherosclerosis and venous thrombosis. N Engl J Med. 2003;348:1435–1441. doi: 10.1056/NEJMoa022157. [DOI] [PubMed] [Google Scholar]
- 98.Schulman S, Lindmarker P, Holmström M, Lärfars G, Carlsson A, Nicol P, Svensson E, Ljungberg B, Viering S, Nordlander S, Leijd B, Jahed K, Hjorth M, Linder O, Beckman M. Post-thrombotic syndrome, recurrence, and death 10 years after the first episode of venous thromboembolism treated with warfarin for 6 weeks or 6 months. J Thromb Haemost. 2006;4:734–742. doi: 10.1111/j.1538-7836.2006.01795.x. [DOI] [PubMed] [Google Scholar]
- 99.Becattini C, Agnelli G, Prandoni P, Silingardi M, Salvi R, Taliani MR, Poggio R, Imberti D, Ageno W, Pogliani E, Porro F, Casazza F. A prospective study on cardiovascular events after acute pulmonary embolism. Eur Heart J. 2005;26:77–83. doi: 10.1093/eurheartj/ehi018. [DOI] [PubMed] [Google Scholar]
- 100.Sørensen HT, Horvath-Puho E, Pedersen L, Baron JA, Prandoni P. Venous thromboembolism and subsequent hospitalisation due to acute arterial cardiovascular events: a 20-year cohort study. Lancet. 2007;370:1773–1779. doi: 10.1016/S0140-6736(07)61745-0. [DOI] [PubMed] [Google Scholar]
- 101.Dahlbäck B, Carlsson M, Svensson PJ. Familial thrombophilia due to a previously unrecognized mechanism characterized by poor anticoagulant response to activated protein C: prediction of a cofactor to activated protein C. Proc Natl Acad Sci USA. 1993;90:1004–1008. doi: 10.1073/pnas.90.3.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bertina RM, Koeleman BP, Koster T, Rosendaal FR, Dirven RJ, de Ronde H, van der Velden PA, Reitsma PH. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369:64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 103.Zöller B, Dahlbäck B. Linkage between inherited resistance to activated protein C and factor V gene mutation in venous thrombosis. Lancet. 1994;343:1536–1538. doi: 10.1016/s0140-6736(94)92940-8. [DOI] [PubMed] [Google Scholar]
- 104.Zöller B, Svensson PJ, He X, Dahlbäck B. Identification of the same factor V gene mutation in 47 out of 50 thrombosis-prone families with inherited resistance to activated protein C. J Clin Invest. 1994;94:2521–2524. doi: 10.1172/JCI117623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Holm J, Zöller B, Svensson PJ, Berntorp E, Erhardt L, Dahlbäck B. Myocardial infarction associated with homozygous resistance to activated protein C. Lancet. 1994;344:952–953. doi: 10.1016/s0140-6736(94)92302-7. [DOI] [PubMed] [Google Scholar]
- 106.Ye Z, Liu EH, Higgins JP, Keavney BD, Lowe GD, Collins R, Danesh J. Seven haemostatic gene polymorphisms in coronary disease: meta-analysis of 66,155 cases and 91,307 controls. Lancet. 2006;367:651–658. doi: 10.1016/S0140-6736(06)68263-9. [DOI] [PubMed] [Google Scholar]
- 107.Zöller B, Li X, Sundquist J, Sundquist K. Venous thromboembolism does not share strong familial susceptibility with coronary heart disease: a nationwide family study in Sweden. Eur Heart J. 2011;32:2800–2805. doi: 10.1093/eurheartj/ehr223. [DOI] [PubMed] [Google Scholar]
- 108.Zöller B, Li X, Ohlsson H, Sundquist J, Sundquist K. Venous thromboembolism does not share strong familial susceptibility with ischemic stroke: a nationwide family study in Sweden. Circ Cardiovasc Genet. 2011;4:484–490. doi: 10.1161/CIRCGENETICS.111.959882. [DOI] [PubMed] [Google Scholar]
- 109.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–874. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 110.Zöller B, Li X, Sundquist J, Sundquist K. Risk of subsequent coronary heart disease in patients hospitalized for immune-mediated diseases: a nationwide follow-up study from Sweden. PLoS One. 2012;7:e33442. doi: 10.1371/journal.pone.0033442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Poredos P, Jezovnik MK. The role of inflammation in venous thromboembolism and the link between arterial and venous thrombosis. Int Angiol. 2007;26:306–311. [PubMed] [Google Scholar]
- 112.Davies R, Galloway JB, Watson KD, Lunt M, Symmons DP, Hyrich KL BSRBR Control Centre Consortium, British Society for Rheumatology Biologics Register. Venous thrombotic events are not increased in patients with rheumatoid arthritis treated with anti-TNF therapy: results from the British Society for Rheumatology Biologics Register. Ann Rheum Dis. 2011;70:1831–1834. doi: 10.1136/ard.2011.153536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jilma B, Cvitko T, Winter-Fabry A, Petroczi K, Quehenberger P, Blann AD. High dose dexamethasone increases circulating P-selectin and von Willebrand factor levels in healthy men. Thromb Haemost. 2005;94:797–801. doi: 10.1160/TH04-10-0652. [DOI] [PubMed] [Google Scholar]
- 114.van Zaane B, Nur E, Squizzato A, Gerdes VE, Büller HR, Dekkers OM, Brandjes DP. Systematic review on the effect of glucocorticoid use on procoagulant, anti-coagulant and fibrinolytic factors. J Thromb Haemost. 2010;8:2483–2493. doi: 10.1111/j.1538-7836.2010.04034.x. [DOI] [PubMed] [Google Scholar]
- 115.Samama MM, Cohen AT, Darmon JY, Desjardins L, Eldor A, Janbon C, Leizorovicz A, Nguyen H, Olsson CG, Turpie AG, Weisslinger N. A comparison of enoxaparin with placebo for the prevention of venous thromboembolism in acutely ill medical patients. Prophylaxis in Medical Patients with Enoxaparin Study Group. N Engl J Med. 1999;34:793–800. doi: 10.1056/NEJM199909093411103. [DOI] [PubMed] [Google Scholar]
- 116.Leizorovicz A, Cohen AT, Turpie AG, Olsson CG, Vaitkus PT, Goldhaber SZ PREVENT Medical Thromboprophylaxis Study Group. Randomized, placebo-controlled trial of dalteparin for the prevention of venous thromboembolism in acutely ill medical patients. Circulation. 2004;110:874–879. doi: 10.1161/01.CIR.0000138928.83266.24. [DOI] [PubMed] [Google Scholar]
- 117.Cohen AT, Davidson BL, Gallus AS, Lassen MR, Prins MH, Tomkowski W, Turpie AG, Egberts JF, Lensing AW ARTEMIS Investigators. Efficacy and safety of fondaparinux for the prevention of venous thromboembolism in older acute medical patients: randomised placebo controlled trial. BMJ. 2006;332:325–329. doi: 10.1136/bmj.38733.466748.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kakkar AK, Cimminiello C, Goldhaber SZ, Parakh R, Wang C, Bergmann JF LIFENOX Investigators. Low-molecular-weight heparin and mortality in acutely ill medical patients. N Engl J Med. 2011;365:2463–2472. doi: 10.1056/NEJMoa1111288. [DOI] [PubMed] [Google Scholar]
- 119.Lederle FA, Zylla D, MacDonald R, Wilt TJ. Venous thromboembolism prophylaxis in hospitalized medical patients and those with stroke: a background review for an American College of Physicians Clinical Practice Guideline. Ann Intern Med. 2011;155:602–615. doi: 10.7326/0003-4819-155-9-201111010-00008. [DOI] [PubMed] [Google Scholar]
- 120.Kahn SR, Lim W, Dunn AS, Cushman M, Dentali F, Akl EA, Cook DJ, Balekian AA, Klein RC, Le H, Schulman S, Murad MH American College of Chest Physicians. Prevention of VTE in nonsurgical patients: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest. 2012;141(2 Suppl):e195S–e226S. doi: 10.1378/chest.11-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]