

Abstract
Common variable immune deficiency (CVID) B cells have impaired responses to TLR7 and TLR9 agonists including poor cell proliferation, loss of cytokine production, and failure to produce IgG or IgA. We show that TLR7- or 9-activated B cells from CVID subjects with >0.5% peripheral isotype-switched CD27+ B cells (group 2) have increased mature Cγ1 and Cγ2 heavy-chain mRNA transcripts compared to subjects who have <0.5% isotype-switched cells (group 1). While TLR-stimulated CVID plasmacytoid dendritic cells for all subjects had impaired IFN-α production, TLR7 or TLR9 stimulation in the presence IFN-α normalized isotype-switched CD27+ B cells, enhanced activation-induced cytidine deaminase mRNA, and significantly improved IgG production only for group 2 subjects. IFN-α also upregulated TLR7 and TLR9 mRNA expression comparable to normal levels in B cells of group 2 subjects, indicating that the loss of IFN-α could be a significant component of the B-cell defect for these subjects.
Keywords: Toll-like receptor, B cell, IFN-α, common variable immune deficiency
Introduction
Common variable immune deficiency (CVID) is a primary immune defect characterized by reduced levels of serum immunoglobulins (Ig) due to lack of normal B cell differentiation [1, 2]. Since specific exogenous signals are required to differentiate naive B cells into antibody secreting cells, many investigators have examined in vitro Ig synthesis in CVID to dissect the nature of this collection of defects. These studies show that B cells of some CVID subjects retained a capacity for Ig synthesis in vitro while B cells of others did not. While most patients have normal numbers of B cells, modestly reduced numbers of CD27+ (memory) B cells, especially very low numbers of CD27+IgD− (isotype-switched memory) B cells, are correlated to both poorer in vitro and in vivo antibody production [3–6] and selected clinical complications [7, 8].
Differentiation of human CD27− naive B cells into CD27+ memory B and plasma cells generally occurs within the germinal centers in secondary lymphoid organs where antigen-activated naive B cells undergo proliferation, somatic hypermutation of Ig variable (V)-region genes, isotype switching, and ultimately differentiation [9, 10]. In response to antigen re-challenge, memory B cells undergo somatic hypermutation of IgV-region genes and are capable of generating Ig of all isotypes. The loss of isotype-switched memory B cells in CVID correlates well with the restricted variable heavy-chain (VH) gene families usage and reduced somatic hypermutation of VH genes [11, 12]. In fact, it has been shown that the CD27+IgD+ B cells, which undergo somatic hypermutation in normal subjects, are less likely to do so in CVID subjects [4]. As plasma cell differentiation occurs predominantly from CD27+ B cells [13], one predictable outcome is the lack of plasma cells in tissues in CVID [6, 14].
One of the most potent stimulators for B-cell activation and maturation are the endosomal Toll-like receptors (TLRs), whose agonists are single-stranded RNA or various synthetic agonists (TLR7) and unmethylated CpG motifs in microbial DNA (CpG-DNA) (TLR9), respectively. TLR9 binding by CpG-DNA has been shown to activate normal B cells, upregulate the expression of co-stimulatory molecules, trigger the secretion of IL-6 and IL-10, and mediate T-independent isotype switch and specific antibody production independently of B-cell receptor (BCR) ligation [15–19]. Naïve B cells express low levels of TLRs while memory B cells constitutively express TLR7, TLR8, and TLR9 at higher levels [19–22]. Ligation of TLR9 on memory B cells further upregulates its own expression, increasing cell sensitivity independently of the stimulating antigen while concurrently reinforcing specific antibody responses [23, 24]. The ligands for TLR7 also can activate naïve human B cells, leading to both cellular differentiation and Ig production [25, 26]. Interestingly for TLR7-mediated stimulation, removal of plasmacytoid dendritic cells (pDCs) was shown to reduce Ig production, demonstrating that either direct or indirect interaction with pDCs was required. Since the addition of IFN-α restored Ig secretion, this cytokine appears essential for TLR-activated antibody production in normal B-cell cultures [25].
We have previously shown that TLR9-stimulated CVID B cells failed to upregulate CD86 or produce IL-6 or IL-10, and in the presence of TLR7, TLR8, and TLR9 ligands, B cells of many CVID subjects proliferated poorly, retained an immature CD27+IgM+IgD+ phenotype, and did not produce IgG or IgA [27, 28]. These defects were not limited to B cells as pDCs cultured with TLR7 and TLR9 ligands had markedly impaired the production of IFN-α. However, we now demonstrate here that IFN-α restores TLR7- and TLR9-triggered functional responses in B cells of CVID subjects who could be identified based on their peripheral B-cell phenotype.
Materials and Methods
Patients and Controls
Peripheral blood samples were obtained from 35 CVID subjects, ages 18–71, using an IRB-approved protocol and written informed consent. Diagnostic criteria for CVID included reduced serum IgG, IgA, and/or IgM two or more confidence intervals below the normal ranges for age and verified specific antibody deficiency. CVID subjects were divided into groups, based on previous studies [5, 7], into those with isotype-switched CD27+IgM−IgD− memory B cells (group 1) less than 0.55% of peripheral blood lymphocytes and those with greater than 0.55% (group 2). For group 1, isotype-switched memory B cells ranged from 0% to 0.41% (mean 0.13%); for group 2 subjects, the range was 0.58% to 23.3% (mean 4.8%). All subjects were healthy and receiving regular Ig replacement; blood studies were done prior to these treatments. Healthy adult volunteers and normal blood bank donors served as controls.
Cell Isolation and Culture
Peripheral blood mononuclear cells (PBMCs) were isolated from peripheral blood of healthy human volunteers or CVID patients by Ficoll-Hypaque (Pharmacia, Uppsala, Sweden). CD19+ B cells were positively selected by immunomagnetic bead isolation (Miltenyi, Auburn, CA, USA). To ensure no contamination with CD3+ cells, the CD19+ fraction was then incubated with anti-CD3 immunomagnetic beads and passed over a magnetic column. CD27+ memory B cells were then isolated using positive microbead selection (Miltenyi Biotec, Auburn, CA, USA) as previously described [28]. Cell viability was assessed by trypan blue exclusion. Cells were cultured at a density of 2– 4×105 cells/ml in 24-well plates.
For TLR stimulation, cells were incubated for 6 days with optimal amounts of the TLR7 agonist, the guanosine analog loxoribine (500 μM), or the TLR9 agonist, CpG-oligodeoxynucleotide ODN2006 (0.6 μg/ml), (InvivoGen, San Diego, CA, USA) in the presence or absence of increasing amounts of IFN-α (125, 250, 500, 1,000, or 2,000 U/ml) (Schering, Kenilworth, NJ, USA). To examine cell proliferation, B-cell fractions were suspended in pre-warmed PBS with 0.5% BSA and labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE) (Invitrogen, Carlsbad, CA, USA) for 5 min at room temperature, washed with 0.5% BSA/PBS, and cultured with TLR ligands with or without IFN-α. Flow cytometric analysis was performed by gating on viable B cells to enumerate CD27− (naïve), CD27+IgD+ (memory), and CD27+IgD− (isotype-switched memory) B cells and to examine B-cell proliferation in cell cultures using FACSCalibur (BD Biosciences). All cultures were performed in RPMI 1640 medium (Cellgro, Herndon, VA, USA) with L-glutamine, 10% heat-inactivated fetal bovine serum (Sigma, St Louis, MO, USA), 15 μg/ml gentamicin, and 100 U/ml penicillin–streptomycin.
Germline and Mature Cγ1 and Cγ2 and Activation-Induced Cytidine Deaminase Expression
To examine TLR-mediated upregulation of activation-induced cytidine deaminase (AID) and germline and mature Cγ1 and Cγ2 mRNA transcripts, 1×106 CD27+ B cells isolated from patients in CVID groups 1 or 2 and normal controls were cultured with optimum concentrations of loxoribine (500 μM) or ODN2006 (0.6 μg/ml) for 24, 48, or 72 h. After stimulation, mRNA was isolated (RNeasy Mini Kit, Qiagen, Valencia, CA, USA) and reverse transcribed (SuperScript III First-Strand cDNA synthesis kit, Invitrogen, Carlsbad, CA, USA). Stabilized RNAs were frozen at −20°C. The cDNA was then amplified using the following oligonucleotide primers: germline Cγ1 (forward, 5′-ACGAGGAACATGACTG GATGC-3′; reverse, 5′-TGTGAGTTTTGTCACAA GATTTGGG-3′) [29], germline Cγ2 (forward, 5′-TCTCAGCCAGGACCAAGGAC-3′; reverse, 5′-ACTCGA CACAACATTTGCG-3′) [29], mature Cγ1 (forward, 5′-CCTGGTCACCGTCTCCTCA-3′; reverse, 5′-TGTGAGTTTTGTCACAAGATTTGGG-3′) [29], mature Cγ2 (forward, 5′-CCTGGTCACCGTCTCCTCA-3′; reverse, 5′-ACTCGACACAACATTTGCG-3′) [29], AID (forward, 5′-TGCTCTTCCTCGGCTACATCTC-3′; reverse, 5′-AACCTCATACAGGGGCAAAAGG-3′) [30], and control β-actin (forward, 5′-CCCCCTGAACCCCAAGGC CAACCGCGAGAA-3′; reverse, 5′-TAGCCGCGCTCGGT GAGGATCTTCATGAGG-3′) [31]. The amplified products were analyzed on a 1% agarose gel containing ethidium bromide and visualized by UV light illumination. Quantitative real-time PCR was conducted using a LightCycler SYBR Green I Detection System (Roche Diagnostics). For this, 2 μl cDNA in a total volume of 20 μl were run in duplicate samples in glass capillary reaction tubes. Real-time PCR products were quantified by copy number per microgram of RNA relative to β-actin.
TLR7 and TLR9 mRNA Expression
To compare the expression of TLR7 and TLR9 mRNA at baseline and in stimulated CVID and control isolated CD27+ B cells, cells were cultured as above with or without 0.6 μg/ml ODN2006 or 500 μM loxoribine in the presence or absence of 1,000 U/ml IFN-α. After 6 days, mRNA expression was determined by quantitative real-time PCR using the following primers: TLR9 (forward, 5′-CTGCCACATGACCATCGAG-3′; reverse, 5′-GGA CAGGGATATGAGGGATTTGG-3′) [32] and TLR7 (forward, 5′-TGTTTCCAATGTGGACACTGAA-3′; reverse, 5′-TGTTCGTGGGAATACCTCCAG-3′) [32]. Real-time PCR products were expressed as copy number per microgram of RNA normalized to β-actin.
TLR-Induced IgG and IgA Production
PBMCs (5×106 cells/ml) from CVID patients categorized as groups 1 or 2 as above or normal controls were cultured with the optimum amount of loxoribine (500 μM) in the presence or absence of or 250, 500, or 1,000 U/ml IFN-α for 13 days. The IgG and IgA contents in the cell supernatants were determined by ELISAwith a lower detection limit of 7.8 ng/ml (Bethyl Laboratories, Montgomery, TX, USA).
IFN-α Production by pDCs
BDCA-4/neuropilin-1-conjugated magnetic beads (Miltenyi Biotec, Auburn, CA, USA) were used to isolate the pDCs as previously described from the peripheral blood of control and CVID group 1 and 2 subjects [27]. Isolated pDCs were stimulated for 48 h with either 100, 500, or 1,000 μM loxoribine, and IFN-α levels in harvested supernatants were assessed by ELISA (Bender Medsystems, Burlingame, CA, USA).
Statistical Analysis
Statistical analyses were performed using GraphPad Prism v.4.03 (GraphPad Software Inc, San Diego, CA, USA). Data were expressed as mean values and standard deviations, ranges, and, as needed, 10th percentiles and 25th (interquartile) percentiles. Mann–Whitney test was used to compare B-cell populations, PCR mRNA results, and IgG and IgA production between CVID and control subjects. A p<0.05 was considered as statistically significant.
Results
TLR-Mediated Induction of Germline and Mature Cγ1 and Cγ2
TLR ligands such as CpG-DNA (ODN2006) or loxoribine are strong B-cell activators, especially for CD27+ B cells. Binding of these ligands leads to class switch recombination, in which the Ig heavy-chain constant region (CH) is rearranged to alter the antibody isotype. Germline mRNA transcription precedes Ig synthesis; mature mRNA is expressed after class switching. A comparison of the TLR9 activation profile in control and CVID CD27+ B cells (Fig. 1a) reveals that isolated CD27+ B cells from normal controls contain both germline (A) and mature (B) Cγ1 and Cγ2 transcripts in both unstimulated and stimulated cells. The CD27+ B cells of the two CVID subjects, each selected to represent group 1 or group 2 based on their peripheral blood memory B-cell phenotypes (Fig. 1a), had detectable levels of germline Cγ1 and Cγ2 transcripts but had reduced mature transcripts before and after TLR activation. Examination in CVID subjects segregated based on their peripheral blood memory B-cell phenotypes (Fig. 1b) shows that, while TLR9 activation upregulated both germline and mature Cγ1 and Cγ2 transcripts in control CD27+ B cells and to some extent in CD27+ B cells of CVID group 2 subjects, induction of mature transcripts was most impaired in the CD27+ B cells of group 1 subjects with the lowest numbers of isotype-switched B cells.
Fig. 1.

TLR9 activation of germline and mature heavychain transcripts. Isolated CD27+ B cells (1×106 cells/ml) from a representative CVID group 1 (CVID 1), group 2 (CVID 2) subject, and two normal controls (NL) were cultured in medium (−) or with 0.6 μg/ml ODN2006 (+) for an optimal time (72 h) (a). Germline and mature Cγ1 and Cγ2 mRNA expression was assessed by RT-PCR. b Quantitative real time PCR results in CD27+ B cells from controls, ten CVID group 1, and eight CVID group 2 subjects in medium alone (−) or stimulated with 0.6 μg/ml ODN2006 (+). The data are expressed as copy number relative to β-actin and shown as the mean±standard error of the mean (SEM)
Examining the effects of TLR7 activation, CD27+ B cells of normal donors again had substantial levels of mature Cγ1 and Cγ2 transcripts in unstimulated and TLR7-stimulated cultures as opposed to the representative CVID group 1 and group 2 subject (Fig. 2a). While CVID group 1 subjects had little or no baseline expression or induction of mature Cγ transcripts, B cells of group 2 subjects demonstrated somewhat greater enhancement, especially of mature Cγ1 and Cγ2 transcripts (Fig. 2b). We conclude that, although CD27+ B cells were present in all the cultures, the markers of isotype switch, loss of IgM and IgD, are more specific indicators of TLR7 or TLR9 responsiveness.
Fig. 2.

TLR7 activation of germline and mature heavy chain transcripts. Isolated CD27+ B cells (1×106 cells/ml) from a representative CVID group 1 (CVID 1), group 2 (CVID 2) subject, and two normal controls (NL) were cultured in medium (−) or with 500 μM loxoribine (+) for 72 h (a). Germline and mature Cγ1 and Cγ2 mRNA expression was assessed by RT-PCR. b Realtime PCR results in CD27+ B cells from controls, ten CVID group 1, and eight CVID group 2 subjects in medium alone (−) or with 500 μM loxoribine (+). The data are expressed as mean copy number±SEM relative to β-actin
IFN-α Augments TLR-Mediated B-Cell Proliferation and Class Switch in Select CVID Subjects
We previously showed that TLR7- and TLR9-induced pDC production of IFN-α was impaired in CVID [27, 28]. However, adding exogenous IFN-α to TLR7-stimulated B-cell cultures augments cell proliferation (as shown here for one representative CVID subject; Fig. 3a). More interestingly, TLR7 and IFN-α co-stimulation induces a striking increase in the number of isotype-switched memory B cells for group 2 CVID subjects, which was significantly greater than in B cells of group 1 subjects (p=0.008) (Fig. 3c). The B cells of group 2 subjects had responses indistinguishable from normal control B cells (Fig. 3b, c), suggesting that the loss of IFN-α production could contribute to the persistence of an immature B-cell phenotype in these subjects.
Fig. 3.


a TLR7 and IFN-α induce B cell proliferation and isotype switching. The effects of 1000 U/ml IFN-α, 500 μM loxoribine, or both IFN-α and loxoribine on B cell proliferation after 6 days of culture, are demonstrated for a CVID subject (top panel) and a control. CFSE dye dilution (x-axis) tracked cell division of CD19+ B cells (y-axis). b The number of CD27+IgD− class-switched memory B cells in a control subject, were compared to B cells of a CVID Group 1 and 2 subject. Shown here, IFN-α in the presence of loxoribine increased the number of CD27+IgD− cells for both the control and the group 2 subject but not for the group 1 subject. c Using a larger group of 10 controls, 15 group 1 subjects, and 15 group 2 subjects, the B cells of controls and CVID group 2 subjects again responded similarly to stimulation with loxoribine, particularly loxoribine with IFN-α, with significantly increased numbers of CD27+IgD− class-switched memory B cells (**p=NS). Compared to Group 2 subjects, B cells of Group 1 subjects were significantly impaired (*p=0.008). The boxes and whiskers indicate mean and 25th and 75th percentiles
IFN-α Can Enhance TLR-Triggered Upregulation of AID mRNA
Upregulation of AID mRNA normally occurs with isotype switch; thus, we examined AID expression in isolated CD27+ B cells from CVID group 1 and group 2 subjects and controls stimulated with ODN2006 or loxoribine, in the presence or absence of IFN-α, using quantitative real-time PCR (Fig. 4a, b). Compared to baseline expression, either IFN-α, ODN2006, or loxoribine alone increased AID mRNA expression in control CD27+ B cells, while the combination of IFN-α in conjunction with either ODN2006 or loxoribine further enhanced AID mRNA expression. For B cells of CVID group 1 subjects, ODN2006 or IFN-α alone slightly induced AID mRNA (Fig. 4a), and the combination of these two increased AID mRNA only marginally. For CVID group 2 B cells, adding IFN-α alone increased AID mRNA more than the TLR9 agonist alone, but, similar to the controls, the combination of ODN2006 and IFN-α was most effective. For TLR7 activation, stimulation with loxoribine, IFN-α, or their combination enhanced AID mRNA for control cells but, again, only had a slight effect for B cells of CVID group 1 subjects. However, B cells of group 2 subjects were as responsive to these activators as control B cells, with significantly more AID mRNA in loxoribine- and IFN-α-activated cultures than B cells of group 1 subjects (p=0.005) (Fig. 4b).
Fig. 4.

IFN-α augments TLR mediated AID mRNA expression. Isolated B cells from CVID group 1 and group 2 and control subjects were cultured with or without 0.6 μg/ml ODN2006 or 500 μM loxoribine for 72 h in the presence or absence of 1000 U/ml IFN-α. AID mRNA expression was assessed by quantitative real time PCR and expressed as mean copy number ± SEM relative to β-actin. Stimulation with IFN-α alone or either ODN2006 or loxoribine alone increased AID mRNA expression in all subjects, however, mRNA was most increased in B cells cultured with TLR agonist and IFN-α. For ODN-activated cultures, results for group 1 and 2 subjects were not statistically significant (Fig. 4a). However, in the presence of loxoribine, and especially IFN-α and loxoribine, AID mRNA was significantly upregulated for B cells of both controls and CVID group 2 subjects (**p=NS) but not for group 1 subjects (*p=0.005) (Fig. 4b)
IFN-α Promotes TLR7-Induced IgG and IgA Production
To determine what effect IFN-α might exert on in vitro, IgG or IgA production PBMCs from CVID group 1 and 2 subjects were cultured with loxoribine, with or without varying concentrations of IFN-α (Fig. 5). Although the IFN-α added did not increase IgG secretion for TLR7-stimulated CVID group 1 B cells, the dual-stimulated B cells of group 2 subjects demonstrated a significant increase in IgG production (p<0.0006) which was not significantly different from the normal control B cells. As with IgG, IFN-α also induced higher IgA production by TLR7-triggered B cells of normal controls. In contrast, B cells of CVID subjects in either group did not upregulate IgA even with dual stimulation.
Fig. 5.

IFN-α enhances TLR7-mediated IgG and IgA production. PBMCs from CVID group 1 subjects (n=10), CVID group 2 subjects (n=10), or control subjects (n=10) were stimulated with 500 μM loxoribine (Lox) in the presence of IFN-α (250, 500, or 1,000 U/ml) for 13 days. IgG and IgA levels were quantitated by ELISA. TLR7-mediated IgG secretion by control B cells and CVID group 2 subjects correlated with increasing concentrations of IFN-α above that produced in the presence of loxoribine alone (single asterisk, p=NS). In contrast, the addition of IFN-α did not reverse the poor response to loxoribine stimulation in CVID group 1 subjects. However, both CVID group 1 and 2 subjects produced significantly less, if any, IgA as compared to normal controls (two asterisks, p< 0.004) in the presence of both loxoribine and IFN-α. Data are expressed as mean±SEM
IFN-α Increases TLR7 and TLR9 mRNA Expression
To explain the differences in TLR responses between B cells of group 1 and 2 subjects, we then examined the expression of TLR7 or TLR9 in CD27+ B cells of these subjects in comparison to control CD27+ B cells. In fact, unstimulated CD27+ B cells from both CVID group 1 (p= 0.04) and group 2 (p=0.0002) subjects had less TLR9 mRNA expression compared to normal B cells. However, activation with ODN2006, especially ODN2006 in conjunction with IFN-α, increased TLR9 mRNA expression in B cells of group 2 subjects to a level indistinguishable from control B cells (p=NS), while TLR9 expression remained low in TLR-stimulated B cells of group 1 subjects (p= 0.016) (Fig. 6a). Similarly, TLR7 mRNA expression was also significantly reduced in unstimulated group 1 CD27+ B cells (p=0.002), while B cells of group 2 subjects expressed amounts of TLR7 mRNA similar to control B cells (p=NS) (Fig. 6b). As with TLR9, activation of these cells with loxoribine and IFN-α upregulated TLR7 mRNA in B cells of both normal and CVID group 2 subjects (p=NS), while TLR7 expression in CVID group 1 B cells remained significantly impaired (p=0.002). These data suggest that both TLR7 and TLR9 mRNA expression are deficient in CD27+ B cells of CVID subjects, particularly for group 1 subjects. However, activation by either TLR7 or TLR9, especially in concert with IFN-α, can normalize the mRNA expression of these TLRs in B cells of CVID group 2 subjects, potentially explaining the enhanced responses of the cells from these subjects.
Fig. 6.

IFN-α promotes TLR7 and 9 mRNA expression in CD27+ B cells. Isolated CD27+ B cells from controls (n=10), CVID Group 1 (n=10), and CVID Group 2 (n=12) subjects were cultured with either 0.6 μg/ml ODN2006 or 500 μM loxoribine for 24 h with or without 1000 U/ml IFN-α. TLR7 and 9 mRNA expression was examined by quantitative real time PCR, expressed as copy number relative to β-actin. Boxes and whiskers represent mean and 25th and 75th percentiles. Baseline TLR9 expression in control B cells was significantly higher than CVID Group 1 (p=0.04) or Group 2 (p= 0.0002) B cells. While ODN2006 stimulation in the presence or absence of IFN-α did not affect TLR9 expression for CVID Group 1 B cells, IFN-α enhanced TLR9 mRNA expression levels in TLR9-stimulated CVID Group 2 B cells, comparable to control B cells (*p= NS) (Fig. 6a). CVID Group 1 B cells (p=0.0023), but not CVID Group 2 (Fig. 6b), had less TLR7 mRNA at baseline than control B cells. For B cells of Group 2, stimulation with loxoribine and IFN-α upregulated TLR7 expression to levels comparable to control B cells (*p=NS) but this did not occur for B cells of group 1 subjects
TLR7-Mediated IFN-α Production is Similar in CVID Groups 1 and 2 pDCs
We previously showed that CVID pDCs overall had substantially reduced IFN-α production upon TLR7 activation compared to normal controls [28]. To determine if group 1 subjects might, in fact, have more impaired production of this cytokine, TLR7-induced pDC production of IFN-α was compared between CVID group 1 and group 2 subjects over a range of concentrations of loxoribine (Fig. 7). IFN-α production was similarly deficient at 100 and 500 μM loxoribine for subjects in both CVID group 1 (n=16) and group 2 (n=13). Although pDCs from CVID group 2 subjects secreted somewhat higher levels of IFN-α than group 1 subjects at the highest concentration of loxoribine tested (1,000 μM) (p=0.0454), these amounts were still more than tenfold less than normal controls.
Fig. 7.
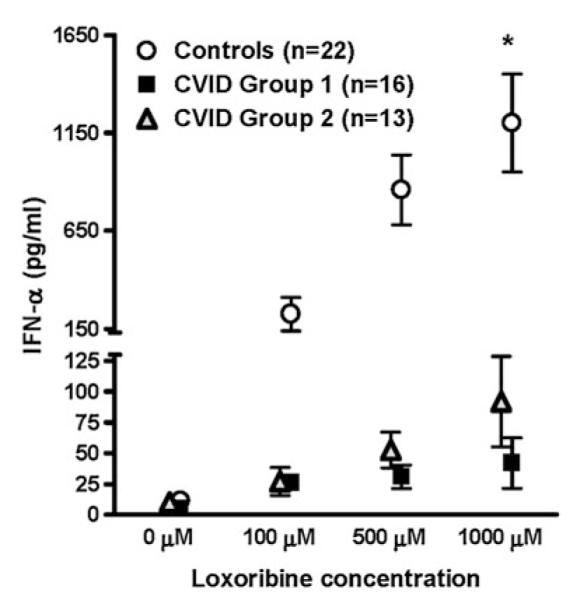
CVID group 1 and group 2 pDCs have similarly impaired IFN-α production. Isolated pDCs from CVID group 1 and group 2 subjects were stimulated with loxoribine for 48 h, and IFN-α levels in the supernatants were measured by ELISA. Normal pDCs served as controls. There was no difference in IFN-α production between CVID group 1 and group 2 subjects at 100 and 500 μM loxoribine although group 2 subjects produced more IFN-α than group 1 subjects at 1,000 μM (p=0.0454)
Discussion
We previously demonstrated that activation through the endosomal TLRs, TLR7, TLR8, and TLR9 resulted in defective B-cell proliferation, lack of IL-6 and IL-10 production, defective upregulation of AID mRNA, blunted isotype switch, and impaired IgG and IgA production in both CD27− naïve and CD27+ memory CVID B cells [27, 28]. Although a panel of TLR ligands led to comparatively normal levels of inflammatory cytokines (TNF-α, IL-6, and IL-12), CVID pDC failed to produce IFN-α when triggered by TLR7, TLR7/TLR8, or TLR9 ligands [28]. In these studies, we show that adding IFN-α to TLR7- and TLR9-activated B-cell cultures improves outcomes by restoring proliferation, promoting isotype switching, upregulation of AID mRNA, and, to some extent, Ig secretion.
While CVID is primarily considered an intrinsic B-cell immunodeficiency [2], other cellular abnormalities such as T-cell activation and proliferative defects, loss of cytokine production, and monocytoid DC defects have been described [33, 34]. Having shown that pDC dysfunction with loss of TLR-mediated IFN-α production is integral to CVID, we now show that impaired production of IFN-α could also impact B-cell maturation and function. The mechanisms by which IFN-α and other type 1 IFNs augment normal B-cell development are complex and exerted at a number of levels. IFN-α is known to induce B-cell activation, amplify the BCR signal, increase B-cell proliferation, and enhance survival [35]. Type I IFNs can also trigger myeloid DCs to upregulate the expression of potent B-cell activators, such as B-cell-activating factor and a proliferation-inducing ligand [36], promoting specific antibody production. When pDCs as the main producers of IFN-α are removed from influenza-stimulated cultures of human PBMCs, the B cells in these cultures lose the capacity to secrete specific antibody to the virus and fail to mature into plasma cells [37]. In contrast, the enhancing, but potentially deleterious, effects of type 1 IFNs on humoral immunity are well known in autoimmune disease, where excess IFN-α appears to play a pathogenic role, leading to the activation and proliferation of auto-reactive B cells [38–40]. In this regard, we do not know if the addition of IFN-α to CVID B-cell cultures might lead to the expansion and isotype switching of B cells which recognize environmental antigens or of self-reactive B cells. Interestingly, although the B cells of CVID group 2 subjects were much more responsive to the added IFN-α in our studies, these subjects are actually less likely to have autoimmune complications [7]. This difference could possibly be attributed to a higher proportion of mature isotype-switched B cells, fewer aberrant auto-reactive B cells at baseline, or another autoimmune regulatory mechanism in the group 2 sub-jects. B cells with auto-reactivity have certainly been observed in some abundance in the peripheral blood of CVID subjects, especially in those with expanded numbers of complement receptor 2/CD21 B cells [41]. However, from a clinical point of view, both subjects with an expansion of CD21 B cells and those with very few isotype-switched memory B cells (group 1) are much more likely to have overt autoimmune disease [5, 7, 8].
The biologic role that TLR ligands and associated adaptors play in B-cell responses overall is unclear. While both TLR7 and TLR9 are potent B-cell activators, MyD88−/− mice mount TLR-independent antibody responses [42] and antibody impairment has not been demonstrated in subjects with MyD88 or IRAK-4 mutations [43, 44], suggesting that TLR signaling might not be essential for optimum B-cell responses. On the other hand, B cell switching from IgM to IgG isotypes requires the simultaneous presence of multiple signals: either direct TLR or CD40 stimulation on antigen-activated B cells or either one of these signals in conjunction with IFN-α [45]. One view is that TLR signaling may enhance T-dependent IgM antibody responses, particularly those arising from marginal zone B cells [46, 47].
Previous work has shown that classification of CVID patients by peripheral blood B-cell phenotypes provides a biomarker related to development of autoimmunity, granulomatous disease, lymphoid infiltrations and splenomegaly, severity of lung disease, and responses to vaccine challenge [5, 7, 8, 48–51]. We show here that while baseline TLR7 and TLR9 mRNA were impaired in CD27+ B cells of all CVID subjects, TLR activation of B cells from group 2 subjects only, in the presence of IFN-α, upregulated the expression of TLR mRNA to the same extent as that for normal controls [25]. As recently demonstrated, B-cell responses to TLR7 ligands in type 1 IFN receptor-deficient mice depend on an IFN feedback loop, as revealed by a selective defect in TLR7 expression in these IFNR−/− B cells [39]. Thus, B cells of CVID group 1 subjects, deprived of TLR-triggered IFN-α, might remain TLR unresponsive, lacking this reinforcement. In conclusion, the data presented here suggest that the numbers of circulating isotype-switched memory B cells in CVID subjects provide a useful marker, not only for the clinical phenotypes which have been described but also to predict inherent differences in TLR responses by both CVID pDCs and B cells.
Acknowledgments
This study was supported by the National Institutes of Health AI-101093, AI-467320, AI-48693 and NIH Contract 03–22, and the David S Gottesman Immunology Chair.
Footnotes
Conflicts of interest CCR has received research support from the National Institutes of Health/National Institute of Allergy and Infectious Diseases and has served on the medical advisory boards of Talecris Corporation and has received research support from Baxter Therapeutics. The rest of the authors have declared that they have no conflicts of interest.
Present Address: J. E. Yu Department of Pediatrics, Weill Cornell Medical College, New York, NY 10021, USA
Contributor Information
Joyce E. Yu, Department of Medicine, Mount Sinai Medical Center, 1425 Madison Avenue, New York 10029 NY, USA; Department of Pediatrics, Mount Sinai Medical Center, New York, NY, USA
Li Zhang, The Immunology Institute, Mount Sinai School of Medicine, New York, NY, USA.
Lin Radigan, The Immunology Institute, Mount Sinai School of Medicine, New York, NY, USA.
Silvia Sanchez-Ramon, The Immunology Institute, Mount Sinai School of Medicine, New York, NY, USA.
Charlotte Cunningham-Rundles, Department of Medicine, Mount Sinai Medical Center, 1425 Madison Avenue, New York 10029 NY, USA; Department of Pediatrics, Mount Sinai Medical Center, New York, NY, USA.
References
- 1.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–86. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 2.Notarangelo LD, Fischer A, Geha RS, et al. Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009;124:1161–78. doi: 10.1016/j.jaci.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brouet JC, Chedeville A, Fermand JP, Royer B. Study of the B cell memory compartment in common variable immunodeficiency. Eur J Immunol. 2000;30:2516–20. doi: 10.1002/1521-4141(200009)30:9<2516::AID-IMMU2516>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 4.Agematsu K, Futatani T, Hokibara S, et al. Absence of memory B cells in patients with common variable immunodeficiency. Clin Immunol. 2002;103:34–42. doi: 10.1006/clim.2001.5197. [DOI] [PubMed] [Google Scholar]
- 5.Warnatz K, Denz A, Drager R, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(−)IgD(−)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99:1544–51. doi: 10.1182/blood.v99.5.1544. [DOI] [PubMed] [Google Scholar]
- 6.Taubenheim N, von Hornung M, Durandy A, et al. Defined blocks in terminal plasma cell differentiation of common variable immunodeficiency patients. J Immunol. 2005;175:5498–503. doi: 10.4049/jimmunol.175.8.5498. [DOI] [PubMed] [Google Scholar]
- 7.Sanchez-Ramon S, Radigan L, Yu JE, Bard S, Cunningham-Rundles C. Memory B cells in common variable immunodeficiency: clinical associations and sex differences. Clin Immunol. 2008;128:314–21. doi: 10.1016/j.clim.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 9.Klein U, Rajewsky K, Kuppers R. Human immunoglobulin (Ig)M+ IgD+peripheral blood B cells expressing the CD27 cell surface antigen carry somatically mutated variable region genes: CD27 as a general marker for somatically mutated (memory) B cells. J Exp Med. 1998;188:1679–89. doi: 10.1084/jem.188.9.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maruyama M, Lam KP, Rajewsky K. Memory B-cell persistence is independent of persisting immunizing antigen. Nature. 2000;407:636–42. doi: 10.1038/35036600. [DOI] [PubMed] [Google Scholar]
- 11.Levy Y, Gupta N, Le Deist F, et al. Defect in IgV gene somatic hypermutation in common variable immuno-deficiency syndrome. Proc Natl Acad Sci U S A. 1998;95:13135–40. doi: 10.1073/pnas.95.22.13135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonhomme D, Hammarstrom L, Webster D, et al. Impaired antibody affinity maturation process characterizes a subset of patients with common variable immunodeficiency. J Immunol. 2000;165:4725–30. doi: 10.4049/jimmunol.165.8.4725. [DOI] [PubMed] [Google Scholar]
- 13.Agematsu K, Hokibara S, Nagumo H, Shinozaki K, Yamada S, Komiyama A. Plasma cell generation from B-lymphocytes via CD27/CD70 interaction. Leuk Lymphoma. 1999;35:219–25. doi: 10.3109/10428199909145724. [DOI] [PubMed] [Google Scholar]
- 14.Ochtrop ML, Goldacker S, May AM, et al. T and B lymphocyte abnormalities in bone marrow biopsies of common variable immunodeficiency. Blood. 118:309–318. doi: 10.1182/blood-2010-11-321695. [DOI] [PubMed] [Google Scholar]
- 15.Krieg AM, Yi AK, Matson S, et al. CpG motifs in bacterial DNA trigger direct B-cell activation. Nature. 1995;374:546–9. doi: 10.1038/374546a0. [DOI] [PubMed] [Google Scholar]
- 16.Gantner F, Hermann P, Nakashima K, Matsukawa S, Sakai K, Bacon KB. CD40-dependent and -independent activation of human tonsil B cells by CpG oligodeoxynucleotides. Eur J Immunol. 2003;33:1576–85. doi: 10.1002/eji.200323444. [DOI] [PubMed] [Google Scholar]
- 17.He B, Qiao X, Cerutti A. CpG DNA induces IgG class switch DNA recombination by activating human B cells through an innate pathway that requires TLR9 and cooperates with IL-10. J Immunol. 2004;173:4479–91. doi: 10.4049/jimmunol.173.7.4479. [DOI] [PubMed] [Google Scholar]
- 18.Poeck H, Wagner M, Battiany J, et al. Plasmacytoid dendritic cells, antigen, and CpG-C license human B cells for plasma cell differentiation and immunoglobulin production in the absence of T-cell help. Blood. 2004;103:3058–64. doi: 10.1182/blood-2003-08-2972. [DOI] [PubMed] [Google Scholar]
- 19.Ruprecht CR, Lanzavecchia A. Toll-like receptor stimulation as a third signal required for activation of human naive B cells. Eur J Immunol. 2006;36:810–6. doi: 10.1002/eji.200535744. [DOI] [PubMed] [Google Scholar]
- 20.Hornung V, Rothenfusser S, Britsch S, et al. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol. 2002;168:4531–7. doi: 10.4049/jimmunol.168.9.4531. [DOI] [PubMed] [Google Scholar]
- 21.Bernasconi NL, Onai N, Lanzavecchia A. A role for toll-like receptors in acquired immunity: up-regulation of TLR9 by BCR triggering in naive B cells and constitutive expression in memory B cells. Blood. 2003;101:4500–4. doi: 10.1182/blood-2002-11-3569. [DOI] [PubMed] [Google Scholar]
- 22.Fillatreau S, Manz RA. Tolls for B cells. Eur J Immunol. 2006;36:798–801. doi: 10.1002/eji.200636040. [DOI] [PubMed] [Google Scholar]
- 23.Bourke E, Bosisio D, Golay J, Polentarutti N, Mantovani A. The toll-like receptor repertoire of human B lymphocytes: inducible and selective expression of TLR9 and TLR10 in normal and transformed cells. Blood. 2003;102:956–63. doi: 10.1182/blood-2002-11-3355. [DOI] [PubMed] [Google Scholar]
- 24.Traggiai E, Puzone R, Lanzavecchia A. Antigen dependent and independent mechanisms that sustain serum antibody levels. Vaccine. 2003;21(Suppl 2):S35–7. doi: 10.1016/s0264-410x(03)00198-1. [DOI] [PubMed] [Google Scholar]
- 25.Bekeredjian-Ding IB, Wagner M, Hornung V, et al. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174:4043–50. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 26.Glaum MC, Narula S, Song D, et al. Toll-like receptor 7-induced naive human B-cell differentiation and immunoglobulin production. J Allergy Clin Immunol. 2009;123:224–230. doi: 10.1016/j.jaci.2008.09.018. [DOI] [PubMed] [Google Scholar]
- 27.Cunningham-Rundles C, Radigan L, Knight AK, Zhang L, Bauer L, Nakazawa A. TLR9 activation is defective in common variable immune deficiency. J Immunol. 2006;176:1978–87. doi: 10.4049/jimmunol.176.3.1978. [DOI] [PubMed] [Google Scholar]
- 28.Yu JE, Knight AK, Radigan L, et al. Toll-like receptor 7 and 9 defects in common variable immunodeficiency. J Allergy Clin Immunol. 2009;124:349–356. e341–343. doi: 10.1016/j.jaci.2009.05.019. 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Y, Agematsu K, Ochs HD, Sugane K. Functional analysis of human memory B-cell subpopulations: IgD+CD27+ B cells are crucial in secondary immune response by producing high affinity IgM. Clin Immunol. 2003;108:128–37. doi: 10.1016/s1521-6616(03)00092-5. [DOI] [PubMed] [Google Scholar]
- 30.Litinskiy MB, Nardelli B, Hilbert DM, et al. DCs induce CD40-independent immunoglobulin class switching through BLyS and APRIL. Nat Immunol. 2002;3:822–9. doi: 10.1038/ni829. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.Greeve J, Philipsen A, Krause K, et al. Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood. 2003;101:3574–80. doi: 10.1182/blood-2002-08-2424. [DOI] [PubMed] [Google Scholar]
- 32.Martin HJ, Lee JM, Walls D, Hayward SD. Manipulation of the toll-like receptor 7 signaling pathway by Epstein–Barr virus. J Virol. 2007;81:9748–58. doi: 10.1128/JVI.01122-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cunningham-Rundles C, Radigan L. Deficient IL-12 and dendritic cell function in common variable immune deficiency. Clin Immunol. 2005;115:147–53. doi: 10.1016/j.clim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 34.Scott-Taylor TH, Green MR, Raeiszadeh M, Workman S, Webster AD. Defective maturation of dendritic cells in common variable immunodeficiency. Clin Exp Immunol. 2006;145:420–7. doi: 10.1111/j.1365-2249.2006.03152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braun D, Caramalho I, Demengeot J. IFN-alpha/beta enhances BCR-dependent B cell responses. Int Immunol. 2002;14:411–9. doi: 10.1093/intimm/14.4.411. [DOI] [PubMed] [Google Scholar]
- 36.Cerutti A, Qiao X, He B. Plasmacytoid dendritic cells and the regulation of immunoglobulin heavy chain class switching. Immunol Cell Biol. 2005;83:554–62. doi: 10.1111/j.1440-1711.2005.01389.x. [DOI] [PubMed] [Google Scholar]
- 37.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, Banchereau J. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity. 2003;19:225–34. doi: 10.1016/s1074-7613(03)00208-5. [DOI] [PubMed] [Google Scholar]
- 38.Uccellini MB, Busconi L, Green NM, et al. Autoreactive B cells discriminate CpG-rich and CpG-poor DNA and this response is modulated by IFN-alpha. J Immunol. 2008;181:5875–84. doi: 10.4049/jimmunol.181.9.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Green NM, Laws A, Kiefer K, et al. Murine B cell response to TLR7 ligands depends on an IFN-beta feedback loop. J Immunol. 2009;183:1569–76. doi: 10.4049/jimmunol.0803899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hall JC, Rosen A. Type I interferons: crucial participants in disease amplification in autoimmunity. Nat Rev Rheumatol. 6:40–49. doi: 10.1038/nrrheum.2009.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Isnardi I, Ng YS, Menard L, et al. Complement receptor 2/CD21-human naive B cells contain mostly autoreactive unresponsive clones. Blood. 115:5026–5036. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nemazee D, Gavin A, Hoebe K, Beutler B. Immunology: toll-like receptors and antibody responses. Nature. 2006;441:E4. doi: 10.1038/nature04875. discussion E4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ku CL, von Bernuth H, Picard C, et al. Selective predisposition to bacterial infections in IRAK-4-deficient children: IRAK-4-dependent TLRs are otherwise redundant in protective immunity. J Exp Med. 2007;204:2407–22. doi: 10.1084/jem.20070628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Picard C, von Bernuth H, Ghandil P, et al. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89:403–425. doi: 10.1097/MD.0b013e3181fd8ec3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heer AK, Shamshiev A, Donda A, et al. TLR signaling fine-tunes anti-influenza B cell responses without regulating effector T cell responses. J Immunol. 2007;178:2182–91. doi: 10.4049/jimmunol.178.4.2182. [DOI] [PubMed] [Google Scholar]
- 46.Meyer-Bahlburg A, Rawlings DJ. B cell autonomous TLR signaling and autoimmunity. Autoimmun Rev. 2008;7:313–6. doi: 10.1016/j.autrev.2007.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rubtsov AV, Swanson CL, Troy S, Strauch P, Pelanda R, Torres RM. TLR agonists promote marginal zone B cell activation and facilitate T-dependent IgM responses. J Immunol. 2008;180:3882–8. doi: 10.4049/jimmunol.180.6.3882. [DOI] [PubMed] [Google Scholar]
- 48.Piqueras B, Lavenu-Bombled C, Galicier L, et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol. 2003;23:385–400. doi: 10.1023/a:1025373601374. [DOI] [PubMed] [Google Scholar]
- 49.Ko J, Radigan L, Cunningham-Rundles C. Immune competence and switched memory B cells in common variable immunodeficiency. Clin Immunol. 2005;116:37–41. doi: 10.1016/j.clim.2005.03.019. [DOI] [PubMed] [Google Scholar]
- 50.Carsetti R, Rosado MM, Donnanno S, et al. The loss of IgM memory B cells correlates with clinical disease in common variable immunodeficiency. J Allergy Clin Immunol. 2005;115:412–7. doi: 10.1016/j.jaci.2004.10.048. [DOI] [PubMed] [Google Scholar]
- 51.Alachkar H, Taubenheim N, Haeney MR, Durandy A, Arkwright PD. Memory switched B cell percentage and not serum immunoglobulin concentration is associated with clinical complications in children and adults with specific antibody deficiency and common variable immunodeficiency. Clin Immunol. 2006;120:310–8. doi: 10.1016/j.clim.2006.05.003. [DOI] [PubMed] [Google Scholar]