

Abstract
Objective
To describe the development of progressive multifocal leukoencephalopathy (PML) in patients with rheumatoid arthritis (RA) treated with rituximab.
Design
Case study.
Setting
Clinical care for patients with rheumatologic diseases. Most were referred to academic centers for care after diagnosis (Washington University, St Louis, Missouri; Karolinska Insitute, Stockholm, Sweden; and Royal Melbourne Hospital, Melbourne, Australia) while one was cared for in a neurology practice in Dallas, Texas, with consultation by an academic neurovirologist from the University of Colorado in Denver.
Patients
Four patients developing PML in the setting of rituximab therapy for RA.
Intervention
Rituximab therapy.
Main Outcome Measures
Clinical and pathological observations.
Results
Four patients from an estimated population of 129 000 exposed to rituximab therapy for RA are reported in whom PML developed after administration of this drug. All were women older than 50 years, commonly with Sjögren syndrome and a history of treatment for joint disease ranging from 3 to 14 years. One case had no prior biologic and minimal immunosuppressive therapy. Progressive multifocal leukoencephalopathy presented as a progressive neurological disorder, with diagnosis confirmed by detection of JC virus DNA in the cerebrospinal fluid or brain biopsy specimen. Two patients died in less than 1 year from PML diagnosis, while 2 remain alive after treatment withdrawal. Magnetic resonance scans and tissue evaluation confirmed the frequent development of inflammatory PML during the course of the disease.
Conclusion
These cases suggest an increased risk, about 1 case per 25 000 individuals, of PML in patients with RA being treated with rituximab. Inflammatory PML may occur in this setting even while CD20 counts remain low.
Progressive multifocal leukoencephalopathy (PML) is a serious demyelinating infection of the brain caused by the JC virus.1,2 Serosurveys suggest that primary infection usually occurs during childhood, with more than 50% of adults typically being seropositive.3 Following primary infection, the virus becomes latent in kidney epithelial cells, lymphoid tissues, bone marrow, and potentially the brain. Periodic reactivation is common and cross-sectional studies have found JC virus DNA in the urine of about 25% of healthy individuals and more rarely in the blood (<1%).4 In immunocompromised individuals, reactivation can result in the development of PML. A dramatic increase in the prevalence of PML was associated with human immunodeficiency virus (HIV) infection, and AIDS remains the disease with the highest associated risk of PML.5 Prior to the HIV pandemic, PML had most commonly been associated with hematologic malignancies, organ transplant, and, occasionally, inflammatory diseases.6
More recently, renewed focus on PML has occurred as an unanticipated complication of natalizumab treatment for multiple sclerosis and inflammatory bowel disease.7–11 Since PML had never been previously associated with multiple sclerosis, it was immediately clear that this monoclonal antibody developed to block α-4 integrin in some way substantially augmented the risk for PML. While the mechanism remains unknown, the emergence of PML cases with several other monoclonal antibody therapies, most notably efalizumab used for the treatment of chronic plaque psoriasis, heightened attention to these medications with regard to PML.12
Rituximab, a chimeric monoclonal anti-CD20, is one of the most widely used monoclonal antibody drugs.13 It is widely used in the treatment of lymphoproliferative diseases such as chronic lymphocytic leukemia and CD20+ non-Hodgkins lymphoma. A recent report found 57 cases of PML associated with rituximab use in HIV-negative patients. Unlike the situation with multiple sclerosis or psoriasis, many of the underlying diseases for which rituximab therapy was used had previously been associated with PML, with more than 90% of cases with complicating lymphoproliferative conditions.14 Determining any increased risk of PML attributable to rituximab is confounded by the uncertainties about the number of exposed patients, the use by most patients of multiple immunosuppressive drugs, and the lack of reliable incidence data for PML in lymphoproliferative and rheumatological diseases in the absence of rituximab therapy. A Food and Drug Administration alert concerning 2 cases of PML associated with rituximab use in systemic lupus erythematosus drew further attention to this problem (http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafety-InformationforPatientsandProviders/ucm126519.htm). Reviews by Calabrese and Molloy15–17 have focused on PML risk in rheumatic disease, finding a somewhat higher risk in systemic lupus erythematosus than other settings such as rheumatoid arthritis (RA).
Until very recently, treatment of RA with rituximab had not been associated with development of PML. However, recently, a patient with RA who had been treated with rituximab and developed PML was reported.18 In this previously reported case, attribution of risk for PML was complicated by a history of malignancy that had been treated with chemotherapy and irradiation a short time before PML onset. Herein, we report the clinical and pathological results from 4 additional cases of PML that developed in the context of treatment for RA using rituximab. Our results suggest that exposure to rituximab leads to an increased risk of PML.
METHODS
Patients presented with clinical signs of possible PML that were further worked up at each of the contributing institutions. Each of the cases was reported to Genentech, the company that markets rituximab. Clinical data were compiled with the assistance of treating physicians.
RESULTS
Table 1 provides a summary of demographics of the 4 new cases of rituximab-associated PML in HIV-negative patients with underlying RA that we have collected, as well as the previously reported information from the original case.18 In each, symptomatic PML was confirmed by either biopsy or cerebral spinal fluid detection of JC virus subsequent to treatment with rituximab. All of the patients were women, with the median age of patients being 67 years (range, 51–73 years). All patients had been diagnosed with moderate to severe RA with disease duration of at least 3 years. Currently, rituximab is not considered a first line of therapy for patients with RA, so treatment with this drug was started in each patient only after failure of other interventions, including methotrexate, other biologics, or corticosteroids. In each case, the rituximab was administered at the recommended dose for refractory RA of two 1000-mg infusions at a 2-week interval for each course. Two patients received only 1 course. The maximal number of courses was 5 (case 4), where the final dose was given between clinical presentation and diagnosis or PML. All patients were negative for HIV. Two may have had enhanced risk due to cancer, 1 having breast cancer treated with surgery and chemotherapy, while another (previously reported18) developed superficial squamous cell carcinoma of the oropharynx with chemotherapy and irradiation after rituximab therapy and before development of PML. Lymphopenia was reported in 4 of 5 cases, but comprehensive testing of immune competence was not routinely available.
Table 1.
Demographic and Historical Features of Patients With Rituximab-Associated PML
| Parameter | Case
|
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 (From Fleischmann18) | |
| Age, y/sex | 73/F | 72/F | 67/F | 62/F | 51/F |
| RA duration, y | 3 | 30 | 3 | >10 | 14 |
| Rheumatologic diagnoses and serologic markers | Hypothyroid; Sjögren syndrome; RF−; CCP−; SPEP− normal | Sjögren syndrome; RF level, 20.8 IU/mL; anti-CCP+; ANA titer, 1:640; SSA+; SSB+; thyroperoxidase+; dsDNA titer, 1:40 | Sjögren syndrome; ANA IFL titer, 1:200; SSA−; SSA titer, 52; RF level, 26 IU/mL; CCP titer, 930 U/mL | CCP+; RF+; other antibodies negative | Sjögren syndrome; RA+; ANA titer, 1:40 speckled; SSA+; SSB+ |
| HIV status | Negative | Negative | Negative | Negative | Negative |
| Malignancy history | None | None | Breast cancer 2006; XRT/surgery | None | Superficial papillary squamous cell carcinoma of the oropharynx 700 |
| Baseline absolute lymphocyte count,/μL | 1411 | 100–600 over past 3 y | 600 | 500 | |
| Baseline lymphocyte profile at diagnosis of PML,/μL | 441; CD20 count, 0 | Absolute lymphocyte count, 213; CD4 count, 119; CD19 count, <20; CD8 count, 63 | NA | NA | CD4 count, 398 (N > 400); CD8 count, 87 (N > 200) |
| Possible PML risk-contributing factors | Leflunomide treatment | Chronic lymphopenia; concurrent methotrexate treatment | XRT; lymphopenia | Chronic lymphopenia for at least 6 y; concurrent methotrexate treatment | Prior chemotherapy after XRT and before PML/XRT; low complement (C2 and C4); chronic low CD19+ count, <20/μL |
| Prior RA therapy including biologic therapies | Leflunomide, Nov 2006 to Aug 2009; hydroxychloroquine sulfate, Mar 2008 to Aug 2009; prednisone, 10 mg/d (Oct 2006) tapered to 2.5 mg/d (Apr 2007); rituximab Feb 2009 | Etanercept 2002–2005; adalimumab; methotrexate >10 y use (stopped after onset of symptoms, Sep 2008) | Methotrexate; low-dose oral corticosteroids | Etanercept; adalimumab; anakinra; hydroxychloroquine; leflunomide; gold; sulfasalazine; meloxicam | Infliximab; cetuximab; etodolac; prednisone; hydroxychloroquine; methotrexate, 20 mg/wk after rituximab stopped |
Abbreviations: ANA, antinuclear antibody; CCP, cyclic citrullinated peptide antibody; dsDNA, double-stranded DNA; HIV, human immunodeficiency virus; IFL, immunofluorescence; NA, not available; PML, progressive multifocal leukoencephalopathy; RA, rheumatoid arthritis; RF, rheumatoid factor; SPEP, serum protein electrophoresis; SSA, Sjögren syndrome antibody Ro; SSB, Sjögren syndrome antibody La; XRT, irradiation therapy.
SI conversion factor: To convert lymphocyte count to ×109/L, multiply by 0.001.
The clinical, laboratory, and radiological findings of PML for each case are summarized in Table 2. Onset of symptoms in 3 cases occurred 5 to 7 months subsequent to the last rituximab infusion. Case 3 differed substantially with onset of disease 16 months following the infusion, but this patient had prolonged-duration CD19 suppression that only began to normalize after PML onset, when a clear immune reconstitution inflammatory syndrome (IRIS) was well documented on magnetic resonance (MR) scans. The other delayed presentation was the previously published case where the onset of disease occurred during immune recovery from chemotherapy 18 months following the rituximab exposure. Presenting symptoms consistent with documented MR lesions were typical of PML, with a variety of presentations including dysesthetic symptoms, ataxia, dysphasia, cognitive decline, and focal dystonic tremor and segmental myoclonus. With the exception of case 2, brain MR scans had characteristic T2 hyperintensities without contrast enhancement (Figure 1). The lesion in case 2 was poorly visible early, but a posterior fossa lesion consistent with the clinical presentation of progressive ataxia evolved after rituximab withdrawal in the presence of stable multifocal lesions more consistent with unrelated preexisting vascular disease. In case 1, MR scans with pathologic correlation show the utility of diffusion-weighted imaging, which is bright at the active front of the lesion (Figure 2). This diffusion-weighted imaging finding was seen in case 1 at presentation and was thought to represent ischemic disease in this elderly woman with stroke history. Her scans illustrate the reported presentation of PML with diffusion-weighted imaging bright lesions that may be confused with stroke.19,20 The development of edema and contrast enhancement typical of inflammatory PML was also seen repeatedly, typical of evolution of inflammatory PML in this setting. Figure 3 demonstrates serial MR scan images of case 4’s developing and resolving IRIS after plasma exchange (PLEX) initiated following brain biopsy diagnosis of PML. In this patient, a rituximab dose had been given after onset of symptoms later attributed to PML, so PLEX was tried since this drug has a long half-life. The abrupt augmentation of lesions on MR imaging between the September 2009 and November 2009 scans is consistent with development of inflammation in the lesions following PLEX. Survival in this case is also consistent with an effective inflammatory control of PML after withdrawal of immunosuppression.
Table 2.
Features of Rituximab-Associated PML
| Feature | Case
|
||||
|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 (From Fleischmann18) | |
| Interval from rituximab infusion to PML onset, mo | 5 | 7 | 16 | 5 (after 3rd of 4 cycles) | 18 (CD20 count recovery to 7 mo, then chemotherapy for cancer, then recovery and onset of PML) |
| Presenting symptom/sign | Focal R-hand dysesthesia; ataxia/dysphasia | Right hemiataxia in arm, leg, and trunk, with falls | Cognitive decline, dysphasia | Cortical R-hand dystonic tremor, evolved to segmental myoclonus (unresponsive to valproate sodium and levetiracetam; some improvement with clonazepam) | Inability to walk (in setting of malnutrition, pneumonia) |
| MRI at baseline | Multiple WM lesions, no enhancement, DWI bright at onset | Multiple punctate WM bright T2/FLAIR lesions, atrophy | L gyrus frontalis white matter lesion (Sep 2009) | L precentral gyrus lesion, small T2 hyperintense/T1 hypointense lesion | Multifocal WM lesions, R frontal predominant |
| CSF JC viral load, copies/mL, baseline | Undetectable initially, detected 1 mo late | JC virus DNA detected, 9138 copies at NIH 4 mo after symptoms by history | JC virus DNA not detected Sep 2009; 280 copies/mL JC virus DNA Nov 2009 | Not detected; brain biopsy for diagnosis | NA |
| Therapy used | Mefloquine | Mirtazapine, up to 45 mg/d; mefloquine Dec 2008 onward | Mirtazapine, 30 mg/d; mefloquine; and prednisolone Jan 2010 | Mefloquine; mirtazapine; plasma exchange | None |
| MRI evolution | Lesions increasing in size and number over weeks; late contrast enhancement | New T2/FLAIR lesion in pons/peduncle Dec 2008, no enhancement (4 mo after last infusion), further progression Mar 2009 | Nov 2009 to Feb 2010 expansion of lesions, develop Gd enhancement, new cerebellar lesions | New parietal and occipital lesions, enlargement, Gd enhancement at 5 mo | Progression in weeks, extension in L hemisphere, as well as R hemisphere lesions (no contrast mentioned) |
| CSF JC viral load evolution | Increased to 47 000, then declined to 1859 copies/mL | Declined to 684 | Declined while scan worsened (not detected) | Not detected in CSF (biopsy diagnosis) | NA |
| Evidence of IRIS | Developing Gd contrast on MRI, spasms/seizures?; MRI DWI | None | Worsened scan, developed contrast enhancement, CD19 counts approaching normal range during IRIS | Evolved enlargement and contrast enhancement 2 mo after diagnosis, mass effect developed, mass and contrast improved after 10 mo | Inflammatory changes on biopsy specimen; rapid progression to death in weeks with no steroid treatment |
| Course/outcome | Spasms, progression to death | Progression, brainstem/cerebellar findings, progressive disability to death 11 mo from first symptoms | Improved, regained walking, improved speech, still cognitively impaired | Alive without progression of neurological symptoms | Progressed to death in 4 wk |
| Histological features | Inflammatory PML; abundant JC virus DNA on immunostain; perivascular inflammatory response; CD8 cells abundant; CD20 cells present in the brain | NA | NA | Biopsy had no CD20 cells at diagnosis, CD4 > CD8 count, patient survived, typical PML histological features with demonstration of virus with histochemical and electron microscopic analysis | Biopsy at diagnosis: inflammatory changes with T/B/plasma cells, macrophages, gliosis |
Abbreviations: CSF, cerebrospinal fluid; DWI, diffusion-weighted imaging; FLAIR, fluid-attenuated inversion recovery; Gd, gadolinium; IRIS, immune reconstitution inflammatory syndrome; L, left; MRI, magnetic resonance imaging; NA, not available; NIH, National Institutes of Health; PML, progressive multifocal leukoencephalopathy; R, right; WM, white matter.
Figure 1.
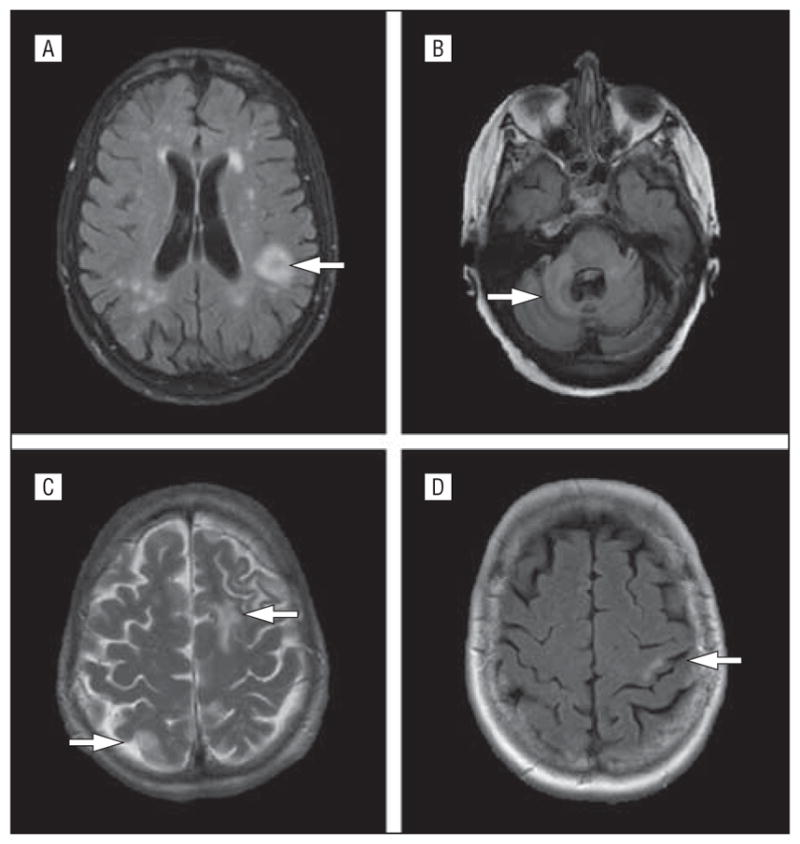
Magnetic resonance imaging of progressive multifocal leukoencephalopathy lesions from cases 1 to 4. A, Case 1 T2 fluid-attenuated inversion recovery (FLAIR) brain image demonstrating predominant left parietal lesion (arrow) in the setting of many presumed vascular lesions that had been seen on a previous scan a year prior to onset of progressive multifocal leukoencephalopathy symptoms and signs. B, T2 FLAIR image from case 2, who developed a right cerebellar lesion (arrow) radiating into the brainstem that was not visible at the onset of disease but was seen here at 8 months after clinical concerns began. C, Case 3 presented with left frontal white matter disease seen on this T2 magnetic resonance image (arrows). D, T2 FLAIR image from case 4. The patient presented with a small left posterior parietal subcortical white matter lesion (arrow) that subsequently progressed markedly (see Figure 3).
Figure 2.
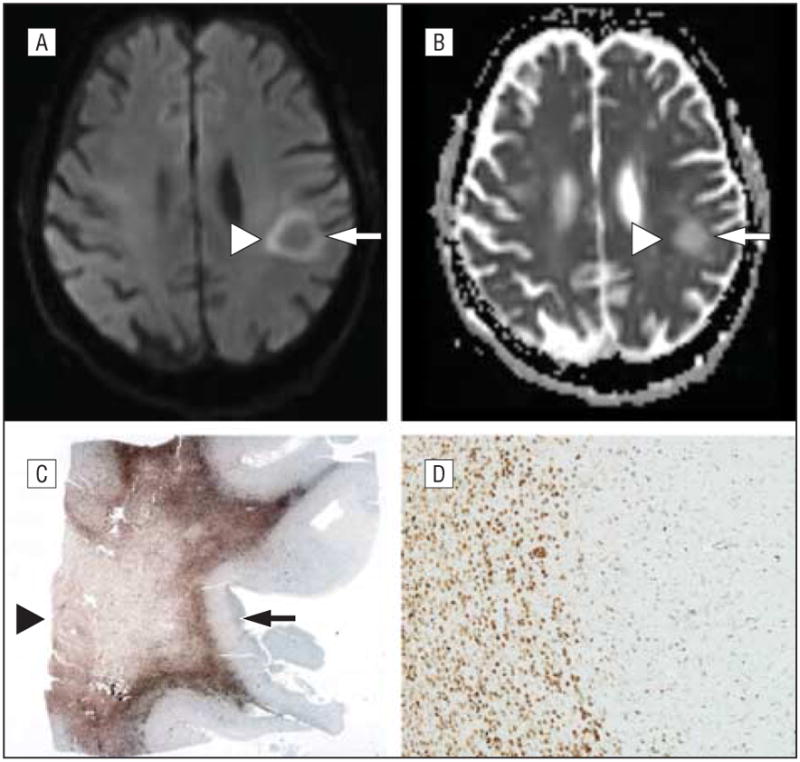
Magnetic resonance characteristics of a progressive multifocal leukoencephalopathy lesion. A-D, Apparent diffusion coefficient (ADC) (A) and diffusion-weighted (B) imaging with the pathologic correlates at post mortem (C and D). Note the inverse relationship of the gliotic lesion where ADC is bright (arrowhead) (A) contrasted with the necrotic core bright on the diffusion-weighted image (arrowhead) (B) and seen at the arrowhead in part C. The active rim of the lesion where viral replication is inducing cytotoxic edema and swelling of oligodendrocytes is highlighted in the ADC image (A). Whole-mount image immunostained for CD68, a marker of macrophages, shows the necrotic core lesion (arrowhead) and the rim of gliosis (arrow) (C) (original magnification ×1). CD68 microscopic image shows many macrophages from the rim of active disease (D) (original magnification ×40).
Figure 3.

Evolution of progressive multifocal leukoencephalopathy in the setting of rituximab therapy. The earliest lesion on fluid-attenuated inversion recovery images is seen in the May 2009 series, with slow evolution particularly in the right occipital lesion over the months from initial symptoms until diagnosis in September 2009 at the time of a brain biopsy. Plasma exchange was performed prior to the November 2009 series and the marked inflammatory response that followed then waned in this surviving patient. Typical of progressive multifocal leukoencephalopathy immune reconstitution inflammatory syndrome, the magnetic resonance images show marked expansion with perilesional edema that then resolved over months following immune control of progressive multifocal leukoencephalopathy. These same lesions demonstrated gadolinium contrast enhancement (not shown).
Diagnosis of PML was confirmed based on clinical presentation with progressive neurological deficits in all cases, laboratory study results including cerebrospinal fluid (CSF) JC viral levels or a positive brain biopsy specimen (case 4), and MR imaging lesions suspicious for PML. JC virus DNA was frequently not detected in the CSF at initial presentation and was found only after repeated lumbar punctures as the disease progressed. In fact, in case 4, results of CSF studies were repeatedly negative, making a brain biopsy necessary to confirm PML. The previously published case (case 5) also required a brain biopsy but CSF JC viral copy levels were never reported. With progression of the disease, an increase in the number and/or size of MR imaging lesions occurred over time in all cases. Three cases developed MR gadolinium enhancement typical of IRIS, suggesting that this is often a complication of rituximab-associated PML. In all cases where CSF JC viral levels were detected, the loads declined over time, consistent with the experience from those with HIV-associated PML who survive.21,22 The outcome of this complication underscores the serious nature of this disease, with 60% dying while surviving patients typically have significant impairment. Three of the 5 cases had brain tissue examined, either post mortem or at biopsy. In 2 of the 3, notable inflammatory PML was documented. Immunohistochemical examination of brain tissue revealed the presence of many lymphocytes. In case 1, some CD20+ cells could be found in the postmortem brain, while no CD20+ cells were identified in the lesion from a diagnostic biopsy specimen from case 4 at an earlier time in the course of the PML. In all 3 cases, JC virus was confirmed in brain tissue (Figures 2, 4, and 5).
Figure 4.

Pathology of rituximab-associated progressive multifocal leukoencephalopathy. A, Case 1 demonstrating a large demyelinated and focally necrotic white matter lesion at autopsy, especially prominent at the arrow. B, Hematoxylineosin–stained section of the brain from case 1 showing marked perivascular inflammation consistent with immune reconstitution inflammatory syndrome in progressive multifocal leukoencephalopathy. The inset is a CD3 immunostain demonstrating the presence of T lymphocytes in both perivascular and parenchymal regions (original magnification ×100; inset, ×200). C, Histological analysis from the brain biopsy in case 4 demonstrating viral inclusions (arrows) and gliotic changes, with some perivascular inflammation (lower right inset), and the immunohistochemical detection of JC virus (upper right inset) (original magnification ×400; upper right inset, ×200; lower right inset, ×100). D, Electron micrograph from the brain biopsy specimen of case 4 demonstrating classic intranuclear papova viral particles, consistent with JC virus (original magnification × 100 000).
Figure 5.

Immunohistochemical staining performed at autopsy of case 1. A, CD3 immunostain marks T cells, here demonstrating increased overall numbers as well as focal clumping around neurons (arrowhead), an atypical feature of progressive multifocal leukoencephalopathy that is more reminiscent of the neuronophagia encountered in cases of viral encephalitis (original magnification ×400). B, CD20 immunostain demonstrates B cells present in small numbers in the brain of the rituximab-treated patient (arrow) (original magnification ×200). C, CD68 immunostain shows numerous macrophages within an area of active demyelination (arrow) (original magnification ×200). D, JC virus immunohistochemical stain identifies numerous infected oligodendrocytes in the postmortem brain (arrow), even though rapidly declining JC viral loads were found in the blood prior to patient death (original magnification ×400).
COMMENT
We document 4 additional cases of rituximab-associated PML occurring in the setting of underlying RA, extending the information available from the prior single published case report.18 Given the rarity of RA-associated PML, these cases support the hypothesis that rituximab increases the risk of PML in this setting. Rheumatoid arthritis has not been commonly associated with PML, although patients living with it commonly take other immunosuppressive drugs that have been associated with this condition.23 Calabrese and Molloy15–17 used the Nationwide Inpatient Sample data to document that the rate of PML in patients with RA is 0.4 per 100 000 discharges, compared with 0.2 for the general population (excluding acknowledged high-risk conditions of HIV, malignancy, and organ transplant) while another incidence study found no cases of RA-associated PML.23 With 5 described cases of rituximab-associated PML in patients with RA and an estimated 129 000 exposed (A. Kelman, MD, Genentech, Inc, written communication, May 20, 2010), incidence is probably at least 1 in 25 000 exposed patients, especially given that not all involved patients may have been accurately diagnosed or reported. While this evidence suggests that risk is increased, it is less than the apparent risk of PML with natalizumab treatment, which is about 1 in 1000 patients exposed for more than 24 months,10,11 or for efalizumab, where risk may have been as high as 1 in 400 exposed patients.24
Risk of PML associated with rituximab has been particularly difficult to characterize. While many cases of PML are reported,14 in most cases they have occurred in individuals with a well-known concomitant risk of PML. This factor, as well as the absence of a clear denominator for exposure, and probable missed cases of PML have made it challenging even for interested professionals collecting all available data to decide if any increased risk exists. To our knowledge, the present series of cases is the most decisive evidence available, documenting a growing number of cases in a setting where PML was very rare, including 1 case (case 1) where there were minimal other significant risks outside the recent exposure to rituximab.
This case series is instructive in an additional way, since PML in this setting differs from that seen in the case of malignancy or untreated HIV by the common occurrence of PML with IRIS. This factor is critical for clinicians, since it impacts diagnosis and management. Detection of JC virus DNA in CSF was insensitive for PML diagnosis early in the disease onset. This may reflect low copy numbers, a phenomenon recently reported in natalizumab-associated PML cases.11,25 If PML presents during IRIS, CSF viral loads are likely to be low or undetectable, making a brain biopsy necessary to confirm the diagnosis. If inflammatory changes are under way, the lesions may transform, accounting for the appearance of gadolinium enhancement, which is atypical for PML in other settings. This inflammatory component may exacerbate neurological signs and symptoms and cause a further decline in the patient’s status. Immune reconstitution inflammatory syndrome associated with PML can be a life-threatening complication requiring therapy such as corticosteroids to optimize survival and best functional outcomes.
The mechanism of increased risk of PML in association with rituximab remains unknown. The immune deficiency experience in AIDS, where the greatest risk for PML occurs, is typified by cellular immune deficiency with progressive loss of CD4 lymphocytes but with relative preservation of humoral immunity. Given the routine presence of antibody to JC virus when PML develops, and the important association of JC virus–specific CD8 cells to the prognosis for survival from PML, the humoral immune system has generally been thought to be of secondary importance in this disorder.26,27 Lymphopenia was chronic in 75% of our cases, providing a prolonged setting in which JC virus may have spread and transformed to enhance neurovirulence. Therapy that targets CD20 cells and the humoral immune system was thought theoretically to carry less risk for PML. Our cases suggest that optimal control of JC virus requires both intact B and T cells. An alternative possibility may be that B-cell precursors, believed to be a site of infection in the marrow, may be critical to activation and spread of the virus with subsequent risk of PML. Depletion and reconstitution of CD20 cells occurring during rituximab therapy may enhance the spread of virus from marrow to the brain. It is interesting that our cases appear to occur during immune reconstitution following rituximab therapy, rather than when CD20 cells are at their nadir.
It is now apparent that a modest increase in the risk of PML should be considered with the use of rituximab, and potentially other agents that target CD20 cells. Given the benefit that this drug provides many patients, it reinforces the need to find means of detecting those at increased risk for this complication and ways to prevent its occurrence. In the case of natalizumab treatment, it has been suggested that patients who are JC virus sero-negative prior to therapy are likely at reduced risk of developing PML, and a similar situation is likely to occur in the setting of rituximab treatment.3 Progressive multifocal leukoencephalopathy is presumed to result from reactivation of latent JC virus rather than from primary exposure. The presence of antibodies is indicative of past exposure to virus and probably identifies a higher-risk population. Conversely, the absence of antibodies likely indicates that primary infection with JC virus has not occurred and that risk of reactivation is therefore theoretically nonexistent.3,27
In the absence of prevention, early diagnosis of PML should be enhanced by clinical vigilance; education of practitioners, patients, and their families; and appropriate diagnostic efforts. Early discontinuation of therapy may allow for earlier immune reconstitution and improved outcomes. Effective direct antiviral treatment for the JC virus has not been demonstrated. An urgent need exists to find active drugs for treating PML. This includes both cytosine arabinoside28 and cidofovir,29 which are still occasionally tried in spite of significant evidence that they are not effective. Mefloquine hydrochloride was used in several of our cases and has in vitro activity against this virus. However, a recent clinical trial was stopped for lack of demonstrable efficacy.30 Similarly, clinicians continue to prescribe mirtazapine on a theoretical basis related to its potential efficacy in blocking serotonin receptors used for viral entry, despite absence for documented clinical efficacy.31
Assuming that rituximab contributes to PML in these cases, reversal of the drug’s effect would be appropriate. Plasma exchange has become a standard practice with natalizumab-associated PML.32 Rituximab is given infrequently in RA since CD20 counts remain depressed for 6 to 9 months after each treatment. While rituximab may remain detectable in plasma for 2 to 3 months, PLEX is unlikely to speed immune recovery after this period. However, if PML is discovered shortly after an infusion of rituximab, PLEX could be considered in this setting, as was performed in case 4 in our series. The apparent brisk increase in inflammatory changes of lesions following PLEX in this case and survival of the patient suggest the possibility this intervention may have been of benefit.
Immune reconstitution inflammatory syndrome, which occurred in our cases, provides an additional potential therapeutic avenue. While controlled trials are not available, the most commonly used treatment for inflammatory PML has been high-dose corticosteroid pulses, often 1 g of intravenous methylprednisolone daily for 5 days, which is repeated if symptoms respond and then recur. Steroid infusions have been reported to stop neurological decline in the setting of IRIS and initiate recovery, without evidence of increased risk.33 Physicians should be aware that this may be required even when the CD20 count suggests ongoing immune compromise on the basis of the prior rituximab therapy. The present cases support the possibility that with early diagnosis and careful management, patients may survive this complication, some with modest deficits. Until more experience is acquired, it is reasonable to encourage support and therapy for these patients, since the dire prognosis of PML acquired when associated with advanced malignancy or AIDS may be exaggerated when considering this disease in other settings. Indeed, current experience with natalizumab suggests survival of as many as 70% to 80% of patients with PML with concentrated efforts for early detection and reversal of immunosuppression (medinfo.biogenidec.com).11
Physicians considering the use of rituximab treatment of rheumatic diseases including RA should be aware that there is a potential, albeit modest risk of developing PML. Because of the morbidity and mortality of PML, however, it is important to consider this in the choice of treatments and to inform patients that this possibility must be considered with therapy including rituximab. In patients treated with rituximab, aggressive evaluation of new and progressing neurological deficits is very important to allow early diagnosis. No further rituximab should be used if a suspicious neurological symptom or sign appears until the diagnosis is successfully excluded.
Acknowledgments
Funding/Support: Dr Clifford has received research support from the National Institute of Mental Health, National Institute of Neurological Disorders and Stroke, National Institute of Allergy and Infectious Diseases, and Fogarty Institutes of the National Institutes of Health.
Footnotes
Author Contributions: Study concept and design: Clifford, Ances, Rosen-Schmidt, Alvarez, and Tyler. Acquisition of data: Clifford, Ances, Costello, Rosen-Schmidt, Andersson, Parks, Perry, Yerra, Schmidt, Alvarez, and Tyler. Analysis and interpretation of data: Clifford, Ances, Andersson, Parks, Perry, Schmidt, Alvarez, and Tyler. Drafting of the manuscript: Clifford, Alvarez, and Tyler. Critical revision of the manuscript for important intellectual content: Clifford, Ances, Costello, Rosen-Schmidt, Andersson, Parks, Perry, Yerra, Schmidt, Alvarez, and Tyler. Administrative, technical, and material support: Clifford, Ances, Andersson, Parks, Perry, and Alvarez. Study supervision: Clifford and Schmidt.
Financial Disclosure: Dr Clifford serves on data safety boards for Millennium, Genzyme, Genentech, and Pfizer. He has been a consultant to Genentech, Wyeth, Bristol-Myers Squibb, Millennium, Biogen Idec, and Pfizer. He has received speaker fees from Biogen Idec, GlaxoSmithKline, and Millennium. He has received travel support from Biogen Idec. He has received research support from Novartis, Biogen Idec, Schering-Plough, Bavarian Nordic, NeurogesX, Tibotec, Pfizer, and Lilly. Dr Tyler has done expert consulting in the area of JC virus and progressive multifocal leukoencephalopathy for Genentech, Biogen Idec, and Pfizer.
Additional Contributions: We are grateful to the patients and families who contributed to this report. Other physicians contributing to the evaluations in these cases included Indrastha Rasaratnam, MD, Meng Tan, MD, Mark Marriott, MD, Michael Gonzales, MD, and Patricia Desmond, MD.
References
- 1.Tan CS, Koralnik IJ. Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol. 2010;9(4):425–437. doi: 10.1016/S1474-4422(10)70040-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koralnik IJ. Progressive multifocal leukoencephalopathy revisited: has the disease outgrown its name? Ann Neurol. 2006;60(2):162–173. doi: 10.1002/ana.20933. [DOI] [PubMed] [Google Scholar]
- 3.Gorelik L, Lerner M, Bixler S, et al. Anti-JC virus antibodies: implications for PML risk stratification. Ann Neurol. 2010;68(3):295–303. doi: 10.1002/ana.22128. [DOI] [PubMed] [Google Scholar]
- 4.Rudick RA, O’Connor PW, Polman CH, et al. Assessment of JC virus DNA in blood and urine from natalizumab-treated patients. Ann Neurol. 2010;68(3):304–310. doi: 10.1002/ana.22107. [DOI] [PubMed] [Google Scholar]
- 5.Berger JR, Pall L, Lanska D, Whiteman M. Progressive multifocal leukoencephalopathy in patients with HIV infection. J Neurovirol. 1998;4(1):59–68. doi: 10.3109/13550289809113482. [DOI] [PubMed] [Google Scholar]
- 6.Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR. Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin Microbiol Rev. 1992;5(1):49–73. doi: 10.1128/cmr.5.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kleinschmidt-DeMasters BK, Tyler KL. Progressive multifocal leukoencephalopathy complicating treatment with natalizumab and interferon beta-1a for multiple sclerosis. N Engl J Med. 2005;353(4):369–374. doi: 10.1056/NEJMoa051782. [DOI] [PubMed] [Google Scholar]
- 8.Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N Engl J Med. 2005;353(4):375–381. doi: 10.1056/NEJMoa051847. [DOI] [PubMed] [Google Scholar]
- 9.Van Assche G, Van Ranst M, Sciot R, et al. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn’s disease. N Engl J Med. 2005;353(4):362–368. doi: 10.1056/NEJMoa051586. [DOI] [PubMed] [Google Scholar]
- 10.Yousry TA, Major EO, Ryschkewitsch C, et al. Evaluation of patients treated with natalizumab for progressive multifocal leukoencephalopathy. N Engl J Med. 2006;354(9):924–933. doi: 10.1056/NEJMoa054693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clifford DB, De Luca A, Simpson DM, Arendt G, Giovannoni G, Nath A. Natalizumab-associated progressive multifocal leukoencephalopathy in patients with multiple sclerosis: lessons from 28 cases [published correction appears in Lancet Neurol. 2010;9(5):463] Lancet Neurol. 2010;9(4):438–446. doi: 10.1016/S1474-4422(10)70028-4. [DOI] [PubMed] [Google Scholar]
- 12.Carson KR, Focosi D, Major EO, et al. Monoclonal antibody-associated progressive multifocal leucoencephalopathy in patients treated with rituximab, natalizumab, and efalizumab: a review from the Research on Adverse Drug Events and Reports (RADAR) Project. Lancet Oncol. 2009;10(8):816–824. doi: 10.1016/S1470-2045(09)70161-5. [DOI] [PubMed] [Google Scholar]
- 13.Carson KR, Bennett CL. Rituximab and progressive multi-focal leukoencephalopathy: the jury is deliberating. Leuk Lymphoma. 2009;50(3):323–324. doi: 10.1080/10428190902779257. [DOI] [PubMed] [Google Scholar]
- 14.Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood. 2009;113 (20):4834–4840. doi: 10.1182/blood-2008-10-186999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calabrese LH, Molloy ES. Progressive multifocal leucoencephalopathy in the rheumatic diseases: assessing the risks of biological immunosuppressive therapies. Ann Rheum Dis. 2008;67(suppl 3):iii64–iii65. doi: 10.1136/ard.2008.097972. [DOI] [PubMed] [Google Scholar]
- 16.Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy in patients with rheumatic diseases: are patients with systemic lupus erythematosus at particular risk? Autoimmun Rev. 2008;8(2):144–146. doi: 10.1016/j.autrev.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 17.Molloy ES, Calabrese LH. Progressive multifocal leukoencephalopathy: a national estimate of frequency in systemic lupus erythematosus and other rheumatic diseases. Arthritis Rheum. 2009;60(12):3761–3765. doi: 10.1002/art.24966. [DOI] [PubMed] [Google Scholar]
- 18.Fleischmann RM. Progressive multifocal leukoencephalopathy following rituximab treatment in a patient with rheumatoid arthritis. Arthritis Rheum. 2009;60(11):3225–3228. doi: 10.1002/art.24906. [DOI] [PubMed] [Google Scholar]
- 19.da Pozzo S, Manara R, Tonello S, Carollo C. Conventional and diffusion-weighted MRI in progressive multifocal leukoencephalopathy: new elements for identification and follow-up. Radiol Med. 2006;111(7):971–977. doi: 10.1007/s11547-006-0095-3. [DOI] [PubMed] [Google Scholar]
- 20.Mader I, Herrlinger U, Klose U, Schmidt F, Küker W. Progressive multifocal leukoencephalopathy: analysis of lesion development with diffusion-weighted MRI. Neuroradiology. 2003;45(10):717–721. doi: 10.1007/s00234-003-0966-4. [DOI] [PubMed] [Google Scholar]
- 21.Giudici B, Vaz B, Bossolasco S, et al. Highly active antiretroviral therapy and progressive multifocal leukoencephalopathy: effects on cerebrospinal fluid markers of JC virus replication and immune response. Clin Infect Dis. 2000;30(1):95–99. doi: 10.1086/313598. [DOI] [PubMed] [Google Scholar]
- 22.Cinque P, Koralnik IJ, Gerevini S, Miro JM, Price RW. Progressive multifocal leukoencephalopathy in HIV-1 infection. Lancet Infect Dis. 2009;9(10):625–636. doi: 10.1016/S1473-3099(09)70226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amend KL, Turnbull B, Foskett N, Napalkov P, Kurth T, Seeger J. Incidence of progressive multifocal leukoencephalopathy in patients without HIV. Neurology. 2010;75(15):1326–1332. doi: 10.1212/WNL.0b013e3181f73600. [DOI] [PubMed] [Google Scholar]
- 24.Korman BD, Tyler KL, Korman NJ. Progressive multifocal leukoencephalopathy, efalizumab, and immunosuppression: a cautionary tale for dermatologists. Arch Dermatol. 2009;145(8):937–942. doi: 10.1001/archdermatol.2009.175. [DOI] [PubMed] [Google Scholar]
- 25.Ryschkewitsch CF, Jensen PN, Monaco MC, Major EO. JC virus persistence following progressive multifocal leukoencephalopathy in multiple sclerosis patients treated with natalizumab. Ann Neurol. 2010;68(3):384–391. doi: 10.1002/ana.22137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lima MA, Marzocchetti A, Autissier P, et al. Frequency and phenotype of JC virus-specific CD8+ T lymphocytes in the peripheral blood of patients with progressive multifocal leukoencephalopathy. J Virol. 2007;81(7):3361–3368. doi: 10.1128/JVI.01809-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tyler KL. Progressive multifocal leukoencephalopathy: can we reduce risk in patients receiving biological immunomodulatory therapies? Ann Neurol. 2010;68(3):271–274. doi: 10.1002/ana.22185. [DOI] [PubMed] [Google Scholar]
- 28.Hall CD, Dafni U, Simpson D, et al. Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection: AIDS Clinical Trials Group 243 Team. N Engl J Med. 1998;338(19):1345–1351. doi: 10.1056/NEJM199805073381903. [DOI] [PubMed] [Google Scholar]
- 29.De Luca A, Ammassari A, Pezzotti P, et al. Gesida 9/99, IRINA, ACTG 363 Study Groups. Cidofovir in addition to antiretroviral treatment is not effective for AIDS-associated progressive multifocal leukoencephalopathy: a multicohort analysis. AIDS. 2008;22(14):1759–1767. doi: 10.1097/QAD.0b013e32830a5043. [DOI] [PubMed] [Google Scholar]
- 30.Clifford D, Nath A, Cinque P, et al. Mefloquine treatment in patients with progressive multifocal leukoencephalopathy. Neurology. 2011;76(9, suppl 4):A28. [Google Scholar]
- 31.Marzocchetti A, Tompkins T, Clifford DB, et al. Determinants of survival in progressive multifocal leukoencephalopathy. Neurology. 2009;73(19):1551–1558. doi: 10.1212/WNL.0b013e3181c0d4a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khatri BO, Man S, Giovannoni G, et al. Effect of plasma exchange in accelerating natalizumab clearance and restoring leukocyte function. Neurology. 2009;72 (5):402–409. doi: 10.1212/01.wnl.0000341766.59028.9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tan K, Roda R, Ostrow L, McArthur J, Nath A. PML-IRIS in patients with HIV infection: clinical manifestations and treatment with steroids. Neurology. 2009;72(17):1458–1464. doi: 10.1212/01.wnl.0000343510.08643.74. [DOI] [PMC free article] [PubMed] [Google Scholar]