

Summary of recent advances
Stress induced activation or denudation of the endothelium elicits arrest and activation of platelets with attendant triggering of coagulation, culminating in a physical barrier to limit blood loss. Recently, coagulation-activated osteopontin, chemerin, and protease activated receptor signaling, as well as platelet-derived molecules including platelet factor 4, serotonin, P-selectin, and CD154 (CD40L) have been revealed as new links between hemostasis and adaptive immunity. The initiation of hemostasis establishes a local state of inflammation that serves as an adjuvant system for antigen presentation, consequently influencing the onset and functional characteristics of an evolving adaptive immune response. In this context, the hemostatic system and its associated signaling pathways warrant further study as novel therapeutic targets that may enhance, abrogate, or otherwise selectively direct the adaptive immune response.
Introduction
Hemostasis is an exquisite orchestration of physical and biochemical forces to arrest bleeding. Trauma initiates local vasoconstriction and enhanced extravasation of blood to the surrounding tissue. The endothelium lining the luminal surface of blood vessels can be “activated” or physically denuded by injury to generate a highly prothrombotic interface. As a consequence, circulating platelets bind and activate on the injured site, while coagulation is triggered by exposed tissue factor (TF). These two systems are independent but highly complementary. Platelet phospholipid membrane is an optimal substrate to amplify the coagulation cascade, which generates thrombin that enables fibrin crosslinking to firmly bind platelet clusters culminating in hemostatic plug formation. Thrombin, in turn, is a powerful physiological activator of platelets, leading to the release of molecular mediators that promote hemostatic reactions and initiate an inflammatory response.
Tantamount to pathogen recognition, “danger” signals elicited by inflammation are obligatory for activation of antigen presenting cells (APCs) to direct the maturation of naïve precursor T and B lymphocytes. In the absence of danger cues, quiescent “semi-mature” APCs maintain peripheral tolerance to self antigens (see review [1]). A growing body of evidence suggests that hemostasis, in this regard, tips the balance towards the onset of adaptive immunity. Thrombin's ability to activate inflammatory mediators, particularly protease activated receptors (PAR), as well as osteopontin and chemerin, has emerged as a new molecular mechanism contributing to the pathogenesis of a number of immune disorders. In collaboration, platelets express and secrete a complex set of pro-inflammatory mediators such as serotonin, platelet factor 4, P-selectin, as well as CD154 (CD40 ligand), an indispensible co-stimulatory molecule for activation of naïve lymphocytes. A number of recent observations in vivo implicate platelet-derived CD154 in enhancing germinal center formation, protecting against infection, and inducing allograft rejection. This review will summarize and interpret recently accumulated evidence that substantiates a previously underappreciated role by hemostatic components as adjuvants in the initiation and development of adaptive immunity.
Platelets and coagulation in the pas de deux of hemostasis
Current understanding of hemostatic mechanisms is largely based on in vitro studies, and their functional significance in vivo remains a topic of ongoing investigation [2,3]. The physiologically important soluble activators of platelets include thromboxane A2 (TxA2), adenosine diphosphate (ADP), and thrombin. Alternatively, exposed collagen at sites of vascular injury facilitates site-specific platelet adhesion and activation. Physiological initiation of the coagulation cascade occurs when sub-endothelial TF binds factor VIIa, the enzymatically active form of factor VII. The VIIa/TF “tenase” complex converts factor X to Xa, which assembles with cofactor Va and Ca2+ on the platelet phospholipid membrane to form a “prothrombinase” complex that cleaves prothrombin into active thrombin. Secondary to VIIa/TF-induced onset of coagulation, thrombin production is sustained by an alternate tenase, the complex of VIIIa and IXa, and amplified through positive feedback by thrombin's activation of VIIIa and Va, as well as XIa required for IX activation. Ultimately, thrombin cleaves fibrinogen and activates factor XIII to enable fibrin crosslinking. Activated platelets further augment hemostasis by secreting their granular cargo consisting of TxA2, ADP, thrombin, fibrinogen, as well as factor V. As a result, small amounts of thrombin can trigger coagulation and platelet activation, which rapidly intensify in magnitude to generate a hemostatic plug (Figure 1).
Figure 1.
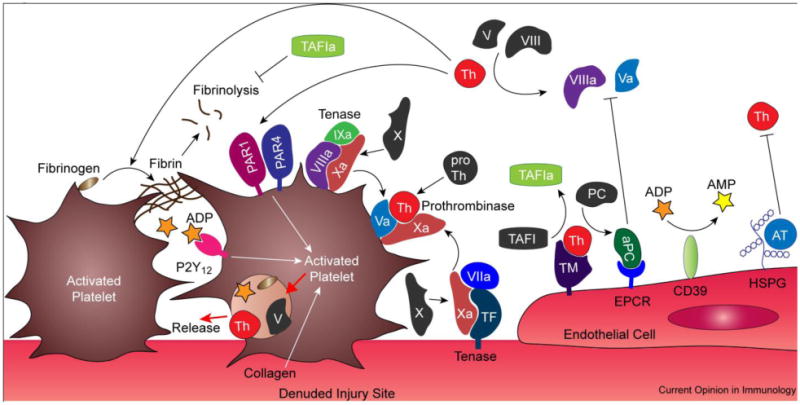
Physiological elaboration of coagulation and platelet activation is quarantined by quiescent endothelial cells. Generation of thrombin (Th) from the coagulation cascade facilitates fibrin crosslinking to form a hemostatic plug, and is intimately linked to activation of platelets through PAR1/PAR4 heterodimers. Self-reinforcing feedback systems amplify production of Th through its activation of factor VIII and V, as well as platelet activation which release agonists such as Th, factor V, and ADP. Endogenous regulators of coagulation in the endothelium include heparan sulfate proteoglycans (HSPG) that catalyze the inactivation of Th by antithrombin III (AT), as well as thrombomodulin (TM), a cofactor for Th that reverses its prothrombotic activity to accelerate generation of activated protein C (aPC) which disrupts formation of tenase and prothrombinase complexes. The TM-Th complex also catalyzes formation of thrombin-activatable fibrinolysis inhibitor (TAFI) which stabilizes crosslinked fibrin. In concert, platelet activation is abrogated by endothelial regulators, such as CD39 that hydrolyze ADP to AMP. Mediators of hemostasis are increasingly associated with establishing a state of inflammation that is linked to the initiation and development of adaptive immunity. Abbreviations: TF, tissue factor.
Multiple regulatory mechanisms are present in the vascular endothelium to establish an activation threshold for hemostasis. The endothelium synthesizes prostacyclin (prostaglandin I2, PGI2) which limits platelet response to TxA2, nitric oxide (NO) that decreases intracellular Ca2+ to suppress αIIbβ3 binding with fibrinogen, and expresses ecto-diphosphohydrolase (CD39) to hydrolyze ADP (reviewed in [3]). Notably, PGI2 and NO are vasodilators, which enhance the convective efflux of hemostatic mediators, thus limiting their participation in surface reactions. Circulating antithrombin III irreversibly inactivates thrombin in a process catalyzed by heparan sulfate proteoglycans, a constituent of the endothelium. Thrombomodulin (TM), expressed in quiescent endothelial cells, forms a 1:1 complex with thrombin, reversing its procoagulant activity to accelerate production of activated protein C (aPC) that suppresses coagulation through inactivation of cofactors Va and VIIIa. In this role, aPC bound to endothelial protein C receptors (EPCR) appears more important than soluble aPC for factor Va inactivation. Furthermore, factor Va associated with prothrombinase complex on platelets is significantly more resistant to aPC inactivation than factor Va bound to the endothelium [4]. The TM-thrombin complex also catalyzes activation of thrombin-activatable fibrinolysis inhibitor (TAFI) that stabilizes crosslinked fibrin. Hence, rather than competitively inhibiting hemostasis after its onset, regulatory mechanisms within the endothelium “cordon off” injury sites where hemostasis is elaborated, thereby limiting deleterious systemic thrombosis.
Protease activated receptors at the nexus between coagulation and adaptive immunity
Coagulation serine proteases engage cell surface PARs, an emerging signaling paradigm affecting the onset of adaptive immunity. Of the four known PARs, PAR1 can be activated by the transient VIIa/TF/Xa complex, thrombin, as well as EPCR-bound aPC (see review [4,5]). Thrombin's potent capacity to activate platelets via PAR1 underscores the intricate ties between the cellular and plasma compartments of hemostasis (reviewed in [6]). Both plasmacytoid and myeloid DCs express PAR1, and can be stimulated by thrombin to produce monocyte chemotactic protein-1 (MCP-1), IL-10, and IL-12 [7]. Thrombin activation of PAR1 in mature DCs can also induce expression of CCL18, a potent chemoattractant for immature DCs and T lymphocytes [8]. In a xenotransplantation model, proinflammatory MCP-1 cytokine production by activated donor endothelial cells required PAR1 engagement [9•]. These studies suggest that, immediately following tissue injury, the thrombin/PAR1 signaling pathway aids in homing and activation of APCs to maximize antigen presentation. In support of this view, deletion of PAR1 protected mice against carbon tetrachloride induced liver fibrosis, concomitant with a reduction in T cell infiltration [10]. The VIIa/TF/Xa complex can additionally activate PAR2. DC activation and migration to the lymph nodes, as well as T cell activation were dependent on the engagement of PAR2 in mice [11]. PAR2 and toll-like receptor 4 (TLR4) have synergistic effects in NFκB induction, a possible mechanism for augmenting DC response to “danger” signals [12]. However, it does not appear that human DCs express PAR2 [8].
Activation of platelet PAR1 by thrombin and consequent activation of sphingosine kinase generates a local gradient of sphingosine 1-phosphate (S1P), an essential regulator of vascular permeability, as well as immune cell trafficking and differentiation (reviewed in [13,14]). Notably, S1P appears to shift the phenotype of effector T cells towards Th2 and Th17 while hampering Th1 type responses [13]. S1P's signaling capacity is partially transmitted through a family of five S1P receptors (S1PR), which exhibit differential constitutive and inducible tissue expression. The S1P receptor 1 (S1PR1) is abundantly expressed in endothelial cells, and both S1PR1 and S1PR3 are highly expressed in the lymphoid organs. S1PR1 signaling was recently shown to be critical in maintaining basal vascular integrity and blunting lethal responses to leak-inducing agents in mice [15•]. Immature DCs preferentially express S1PR1, but upregulate S1PR3 upon maturation. Consistent with this observation, thrombin stimulation of DCs induced S1P production, which activates S1PR3, leading to an autocrine amplification of coagulation and inflammation through increased tissue factor and IL-1β production [16••]. Engagement of lymphocyte S1PR1 was instrumental in promoting T cell retention in peripheral tissues in response to inflammation, which increase S1P levels [17•]. S1PR1 also appears responsible for suppressing regulatory T cell development and function through activation of the Akt-mTOR pathway [18].
Downstream PAR1 signaling pathways from activation by thrombin and aPC, two opposing forces in the coagulation cascade, exert opposite effects on inflammation. Thrombin-PAR1 signaling increased inflammation-induced vascular leakage that is dependent on activation of S1PR3, while aPC/EPCR-PAR1 signaling and transactivation of S1PR1 produced the opposite effect in LPS challenged mice [19••]. While aPC could mediate this effect in vivo by blocking thrombin generation, it was demonstrated that aPC's PAR signaling activity was more significant than its anticoagulant function in reducing mortality in this model [20]. In addition to the divergent downstream S1P receptor signaling pathways of PAR1, the barrier-protective effects of aPC/EPCR also requires the trans-activation of endothelial PAR2 [21•]. This is consistent with other studies that demonstrate PAR3 heterodimerization with PAR1 allosterically modulates thrombin-mediated PAR1 activation in endothelial cells [22•]. Likewise, the PAR4 heterodimerizes with PAR1 on platelets and enables their activation by thrombin [23•]. It is increasingly clear that both heterodimerization of PARs and divergent S1PR engagement influence the signaling outcome of PAR1 activation.
Coagulation-activated inflammatory mediators and adaptive immune disorders
Osteopontin (Opn), a phosphorylated glycoprotein, is traditionally tied to its physiological roles in bone and tissue remodeling. Proteolytic cleavage of Opn by thrombin exposes hidden α4β1 integrin (also known as VLA4) and CD44 binding domains to enable migration of antigen-specific T cells to sites of inflammation. Conversely, TAFI renders thrombin-cleaved Opn inactive to suppress inflammation [24]. Considerable evidence were compiled recently using several disease models, which reveal Opn's role in augmenting Th1 and Th17 mediated immune responses [25]. Antigen-specific CD4+ and CD8+ memory T cells secreted Opn upon activation and steered effector T cells towards a Th1 phenotype by down-modulating their IL-4 production in murine models of chronic allergic contact dermatitis (ACD) [26]. Likewise, during the sensitization stage of ACD, autocrine Opn production was a vital component for DC migration to the skin-draining lymph nodes [27]. In experimental autoimmune encephalomyelitis (EAE), Opn was abundantly expressed in mouse DCs, which induced IL-17 and IFN-γ production by T cells in a process mediated by integrins, while diminishing IL-10 production through CD44 engagement [28]. Further, engagement of type I interferon receptors on DCs suppressed production of Opn and inhibited IL-17 secretion [29••]. In agreement with these observations, interferon β, a standard treatment for relapsing forms of multiple sclerosis (MS), decreased Opn and IL-17 production, as well as T cell migration [30]. Opn induced NF-κB activation, inhibited pro-apoptotic FOXO3A activity, and altered Bim, Bak, and Bax expression, which collectively prolonged survival of stimulated CD4+ and CD8+ T cells to exacerbate EAE relapse and progression [31••]. In light of these discoveries, Opn is increasingly recognized as a central mediator of relapsing MS (reviewed in [32]).
Highlighting the growing importance of bioinformatics in the field of immunology, a proteomic analysis of human MS lesions identified tissue factor and protein C inhibitor unique to chronic acute plaques. Respective inhibition of the physiological activity of these target molecules with hirudin and aPC significantly ameliorated EAE in mice. aPC exerted these effects by suppressing IL-17 production and NF-κB signaling by activated T cells [33••]. A potential route by which aPC mediated this effect is through attenuating thrombin-mediated activation of Opn. These results also raise the possibility that aPC can directly modulate the functional behavior of immune cells, though the extent of this influence remains unexplored. Indeed, it was demonstrated that on leukocytes where EPCR expression is low, the integrin CD11b/CD18 could play a bigger role in mediating aPC-PAR1 signaling [34]. Our understanding of aPC as an anti-inflammatory mediator is far from complete, and their binding partners and signaling pathways in cellular compartment of immunity should present exciting new areas of research.
Chemerin is a recently characterized chemoattractant that is increasingly studied for its role in regulating inflammation. Similar to Opn, chemerin circulates as an inactive precursor that can be activated by serine proteases involved in coagulation and fibrinolysis, the most potent of which are factor XIIa and plasmin, and to a lesser extent factor VIIa. Additionally, thrombin-stimulated platelets appear to release partially cleaved chemerin [35]. Chemerin signaling via its cognate receptor ChemR23 (CMKLR1) may be involved in the accumulation of DC in lesions from patients with psoriasis [36] as well as oral lichen planus [37]. Emerging evidence also implicate chemerin in the development of EAE [38]. Sequential cleavage of chemerin by plasmin and TAFI in vitro synergistically enhanced the activity of chemerin to affect plasmacytoid DC migration through ChemR23 [35]. The significance of this observation in vivo is unclear, as TAFI inhibits plasmin generation. Surprisingly, chemerin can reduce the production of proinflammatory mediators and macrophage activation through ChemR23 but requires additional C-terminal processing by cysteine proteases [39••]. This suggests an intriguing link with the secretion of cysteine proteases by Staphylococcus aureus that could inhibit effective host defense to clear infections [40]. In view of these observations, chemerin exhibits incredible plasticity to transform into ligands that exert opposite effects on inflammation through the same receptor, a process that deserves further study.
Platelet activation strengthens antigen presentation
Platelet activation is increasingly implicated in a number of chronic adaptive immune diseases, including inflammatory bowel disease (IBD), atherosclerosis, infections, and transplant rejection. In a mouse model of IBD, depletion of platelets by thrombocytopenia commensurately reduced leukocyte adhesion in the inflamed colonic venules [41]. Antibody blockade of TF function significantly blunted colonic inflammation induced by dextran sodium sulfate (DSS) in mice. Remarkably, the treatment nearly abolished both leukocyte and platelet recruitment to the venules of inflamed colons [42•]. Genetic deletion of CD39 significantly exacerbated IBD in mice, and lower CD39 expression in humans was correlated with increased susceptibility to Crohn's disease [43]. Activated platelets, which express P-selectin (CD62P), appear to directly interact with DCs. Following initial contact between CD62P and PSGL-1 expressed on DCs, junctional adhesion molecule C (JAM-C) and Mac-1 mediated firm DC adherence to platelets in vitro [44]. JAM-C deficient mice showed decreased persistence of specific circulating IgG titers and impaired germinal center formation [45]. Elevated soluble CD62P levels, a marker for platelet activation, is associated with an increased risk for atherosclerosis in ApoE−/− mice [46].
A large repertoire of inflammatory mediators can be released or expressed by platelets upon activation. Among these, platelet factor 4 (PF4, or CXCL4) was recently found to induce migration of activated T lymphocytes in a process mediated by the native chemokine receptor CXCR3 in addition to the previously known interactions with the alternatively spliced variant CXCR3b [47]. A flow-cytometry based evaluation revealed constitutive CXCL4 expression on DCs, B cells, and some T cells. Upon in vitro bacterial infection, DCs reduced their expression of CXCL4 [48]. Both observations were unexpected and deserve further study. Heterodimerization of CXCL4 and RANTES (CCL5) released by activated platelets was critical for in vivo monocyte recruitment and atherogenesis [49]. Elimination of CXCL4 from platelets reduced the development of atherosclerosis in both wild type and ApoE−/− mice [50].
Despite their considerable importance as a neurotransmitter, serotonin (5-HT) is increasingly known for its role as an inflammatory mediator. Platelets sequester 5-HT in their dense granules and release them upon activation. Human monocytes treated with 5-HT exhibited enhanced capacity to stimulate allogenic T cells in vitro and reduced susceptibility to Fas-FasL mediated apoptosis [51]. Exogenous 5-HT induced activation of ERK1/2 and NFκB as well as endogenous production of 5-HT in naïve T cells, contributing to their activation and proliferation [52]. Interestingly, in a murine model of noncytopathic lymphocytic choriomeningitis viral infection, platelet-derived 5-HT delayed activated virus-specific CD8+ T cell infiltration but prolonged their persistence in the liver primarily through altering the sinusoidal microcirculation [53]. Hence, 5-HT appears to exert proinflammatory effects through physical and biochemical enhancement of T cell activation and survival.
Platelet-derived CD154 appears to be an important component in directing the process of antigen presentation so as to optimize the maturation of naïve adaptive immune precursors. A number of recent observations in vivo implicate platelet-derived CD154 in enhancing germinal center formation, protecting against infection, and inducing allograft rejection. TLR-4 engagement on platelets induced release of soluble CD154, implicating platelet activation in immune response to infections [54]. DCs adherent to platelets were able to stimulate lymphocyte proliferation only in the presence of CD154 [44]. Platelet-derived membrane vesicles were capable of delivering CD154 to sites distant from location of activation to stimulate antigen-specific IgG production and to enhance CD4+ T cell mediated germinal center formation [55••]. Platelet CD154 secretion correlated with B lymphocyte activation and IgG production [56], and augmented cytotoxic T cell response to Listeria challenge [57]. Activation of platelets was required for virus-specific cytotoxic T lymphocyte response, a process that depended on CD154 [58••]. Soluble CD154 induced rejection 30 days following allogeneic cardiac transplantation in mice, while platelet infusion had no effect [59•]. Thus, in vivo activation of platelets from surgical trauma at the time of transplantation could serve as the sole exogenous source of CD154 to instigate allograft rejection. Both CD40−/− and CD154−/− mice were protected from DSS induced colitis, concomitant with a reduction in inflammatory angiogenesis [60]. Hyperinsulinemia and hyperglycemia induced greater circulating levels of CD154+ platelets, as well as tissue factor procoagulant activity, leading to a prothrombotic state that could be linked to the greater incidence of atherosclerosis in patients with type 2 diabetes [61,62].
Conclusions and perspectives
The link between adaptive immunity and hemostasis has long been suspected through observations by transplant surgeons associating acute allograft rejection with significant thrombosis. Despite recent progress, therapeutic targeting of the hemostatic system and its related signaling pathways to treat disorders of adaptive immunity has not been well defined. Clinical trials in the last decade have revealed benefits of anticoagulants, such as aPC (Xigris®) to treat severe sepsis. Undesirable bleeding risks associated with these formulations may be overcome by engineering newer aPC variants with impaired anticoagulant activity but intact PAR signaling capacity [4]. Alternatively, thrombomodulin exhibits thrombin-dependent, “on-demand” generation of aPC and TAFI, whose anti-inflammatory downstream pathways are only beginning to be uncovered. Our laboratory showed exogenous TM infusion enhanced long-term engraftment in an allogeneic murine model of intraportal islet transplantation, substantially suppressing the hallmark endogenous “danger” signals IL-1β and TNF-α in the liver [63•]. Risks of bleeding and dosage requirement can be further reduced through recombinant TM fusion proteins with targeting moieties for TF [64], as well as techniques to site-specifically tether TM to tissue surfaces [65].
Two decades after their initial discovery, the PAR and downstream S1P signaling pathway has emerged as a new paradigm linking inflammation and a plethora of adaptive immune disorders. Phase III clinical trials showed fingolimod (Gilenia®), a synthetic S1P analogue, significantly reduced relapse rates in MS patients compared with placebo and IFN β-1a therapy [66,67]. Fingolimod is an agonist of four of the five S1P receptors and influences lymphocyte trafficking, though its immune-modulatory mechanisms are currently not fully characterized. In 2010, a US Food and Drug Administration advisory panel unanimously recognized the safety and effectiveness of fingolimod, and overwhelmingly recommended its approval as a first-line treatment for relapsing MS. Therapeutic intervention with a specific inhibitor of sphingosine kinase-1 has also been demonstrated to inhibit production of proinflammatory cytokines in a mouse model of sepsis [68]. These encouraging developments have demarcated the lipid metabolic and signaling pathways as a new frontier in the quest to create a unified model of inflammation onset and resolution.
In conclusion, recent research substantiates an elegant system whereby activation of coagulation and platelets in response to trauma contributes to a state of inflammation that is central to the development of an effective host defense response as well as numerous adaptive immune disorders (Figure 2). We anticipate that the evolution of bioinformatics, as well as in vitro and in vivo models of immune dysfunction will further expand our understanding of the molecular networks through which hemostatic reactions affect the onset of adaptive immunity, as well as our repertoire of available drug targets that provide new opportunities to modulate this process.
Figure 2.
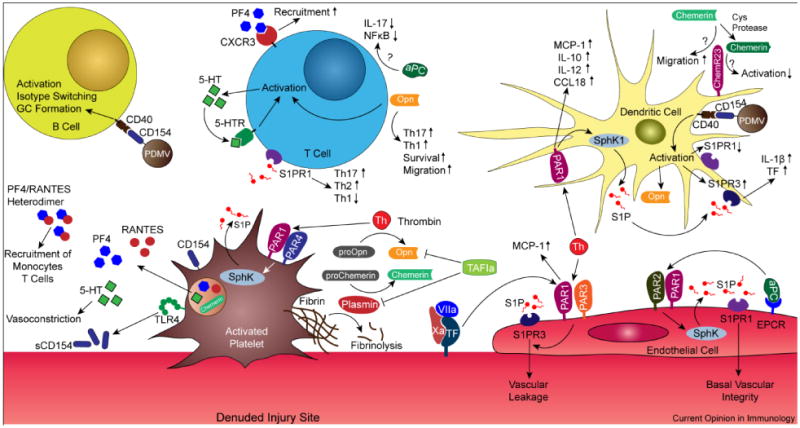
Interface between hemostasis and adaptive immunity. Thrombin activation of PARs and pro-osteopontin (proOpn), in concert with platelet-derived CD154, serotonin (5-HT), platelet factor 4 (PF4), and RANTES, have now been discovered to optimize the process of antigen presentation to initiate and steer the phenotype of subsequent adaptive immune responses. Platelets express CD154 which can be secreted by TLR4 induction or remotely delivered by platelet-derived microvesicles (PDMV) to augment both dendritic and B cell activation. In a similar capacity, 5-HT can activate T cells and induce autocrine production of 5-HT to amplify this process. PF4 directly interacts with CXCR3 to increase T cell recruitment, or forms dimers with RANTES that recruits monocytes and T cells. Activation of PAR1 on dendritic cells (DC) enhanced the production of proinflammatory and prothrombotic mediators, a potential feedback system to amplify antigen presentation. A critical signaling pathway downstream of PAR engagement is activation of sphingosine kinase (SphK) to generate sphingosine-1 phosphate (S1P) that act on S1P receptors (S1PR). Extravasation of inflammatory cells through the endothelium is enhanced by thrombin (Th) activation of the S1PR3 pathway, while activated protein C (aPC) maintains the endothelial barrier through S1PR1. Differential engagement of PAR heterodimers by aPC and Th is also responsible for this divergent effect. Engagement of S1PR1 steers T helper cells away from Th1 phenotype towards a Th2-type response. aPC attenuates Th17 responses and downregulate T cell activation. Osteopontin (Opn) has been implicated in prolonging T cell survival and driving a Th17 response. Chemerin depends on precise protease processing to either enhance or attenuate inflammation through the same receptor ChemR23. Activated thrombin-activatable fibrinolysis inhibitor (TAFIa) deactivates Opn and inhibits chemerin cleavage by plasmin that yields a proinflammatory ligand for ChemR23. Substantial crosstalk between the hemostatic and adaptive immune compartments is therefore an indispensible component in the onset of effective host defense as well as immune dysfunction. Abbreviations: MCP-1, monocyte chemotactic protein-1; sCD154, soluble CD154; TF, tissue factor.
Acknowledgments
This work was supported by grants from the National Institutes of Health and Juvenile Diabetes Research Foundation.
Footnotes
Conflicts of interest: The authors hereby declare no conflicts of interest that relate to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Recommended Reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7:610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 2.Furie B, Furie BC. Mechanisms of disease: Mechanisms of thrombus formation. N Engl J Med. 2008;359:938–949. doi: 10.1056/NEJMra0801082. [DOI] [PubMed] [Google Scholar]
- 3.Davi G, Patrono C. Platelet activation and atherothrombosis. N Engl J Med. 2007;357:2482–2494. doi: 10.1056/NEJMra071014. [DOI] [PubMed] [Google Scholar]
- 4.Weiler H. Regulation of inflammation by the protein C system. Crit Care Med. 2010;38:S18–25. doi: 10.1097/CCM.0b013e3181c9cbb5. [DOI] [PubMed] [Google Scholar]
- 5.Shpacovitch V, Feld M, Hollenberg MD, Luger TA, Steinhoff M. Role of protease-activated receptors in inflammatory responses, innate and adaptive immunity. J Leukoc Biol. 2008;83:1309–1322. doi: 10.1189/jlb.0108001. [DOI] [PubMed] [Google Scholar]
- 6.Jennings LK. Mechanisms of platelet activation: need for new strategies to protect against platelet-mediated atherothrombosis. Thromb Haemost. 2009;102:248–257. doi: 10.1160/TH09-03-0192. [DOI] [PubMed] [Google Scholar]
- 7.Yanagita M, Kobayashi R, Kashiwagi Y, Shimabukuro Y, Murakami S. Thrombin regulates the function of human blood dendritic cells. Biochem Biophys Res Commun. 2007;364:318–324. doi: 10.1016/j.bbrc.2007.10.002. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Syrovets T, Paskas S, Laumonnier Y, Simmet T. Mature dendritic cells express functional thrombin receptors triggering chemotaxis and CCL18/pulmonary and activation-regulated chemokine induction. J Immunol. 2008;181:1215–1223. doi: 10.4049/jimmunol.181.2.1215. [DOI] [PubMed] [Google Scholar]
- •9.Chen DX, Carpenter A, Abrahams J, Chambers RC, Lechler RI, McVey JH, Dorling A. Protease-activated receptor 1 activation is necessary for monocyte chemoattractant protein 1-dependent leukocyte recruitment in vivo. J Exp Med 2008. 205:1739–1746. doi: 10.1084/jem.20071427. In a xenogenic mouse heart to rat transplant model, donor PAR1 activation was necessary for recruitment of inflammatory cells, establishing the communication link between thrombosis and acute organ rejection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rullier A, Gillibert-Duplantier J, Costet P, Cubel G, Haurie V, Petibois C, Taras D, Dugot-Senant N, Deleris G, Bioulac-Sage P, et al. Protease-activated receptor 1 knockout reduces experimentally induced liver fibrosis. Am J Physiol Gastrointest Liver Physiol. 2008;294:G226–235. doi: 10.1152/ajpgi.00444.2007. [DOI] [PubMed] [Google Scholar]
- 11.Ramelli G, Fuertes S, Narayan S, Busso N, Acha-Orbea H, So A. Protease-activated receptor 2 signalling promotes dendritic cell antigen transport and T-cell activation in vivo. Immunology. 2010;129:20–27. doi: 10.1111/j.1365-2567.2009.03144.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rallabhandi P, Nhu QM, Toshchakov VY, Piao W, Medvedev AE, Hollenberg MD, Fasano A, Vogel SN. Analysis of proteinase-activated receptor 2 and TLR4 signal transduction - A novel paradigm for receptor cooperativity. J Biol Chem. 2008;283:24314–24325. doi: 10.1074/jbc.M804800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rivera J, Proia RL, Olivera A. The alliance of sphingosine-1-phosphate and its receptors in immunity. Nat Rev Immunol. 2008;8:753–763. doi: 10.1038/nri2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- •15.Camerer E, Regard JB, Cornelissen I, Srinivasan Y, Duong DN, Palmer D, Pham TH, Wong JS, Pappu R, Coughlin SR. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119:1871–1879. doi: 10.1172/JCI38575. This work showed that the S1PR1 signaling pathway was critical for maintaining basal vascular integrity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••16.Niessen F, Schaffner F, Furlan-Freguia C, Pawlinski R, Bhattacharjee G, Chun J, Derian CK, Andrade-Gordon P, Rosen H, Ruf W. Dendritic cell PAR1-S1P3 signalling couples coagulation and inflammation. Nature. 2008;452:654–658. doi: 10.1038/nature06663. This work implicates the PAR1-S1PR3 signaling pathway in the activation of dendritic cells, suggesting a potential adjuvant effect by thrombin signaling on antigen presentation. [DOI] [PubMed] [Google Scholar]
- •17.Ledgerwood LG, Lal G, Zhang N, Garin A, Esses SJ, Ginhoux F, Merad M, Peche H, Lira SA, Ding Y, et al. The sphingosine 1-phosphate receptor 1 causes tissue retention by inhibiting the entry of peripheral tissue T lymphocytes into afferent lymphatics. Nat Immunol. 2008;9:42–53. doi: 10.1038/ni1534. Links S1PR1 signaling pathway with increased retention of T cells in inflamed tissue, which contain elevated levels of S1P. [DOI] [PubMed] [Google Scholar]
- 18.Liu GW, Burns S, Huang GH, Boyd K, Proia RL, Flavell RA, Chi HB. The receptor S1P(1) overrides regulatory T cell-mediated immune suppression through Akt-mTOR. Nat Immunol. 2009;10:769–U132. doi: 10.1038/ni.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••19.Niessen F, Furlan-Freguia C, Fernandez JA, Mosnier LO, Castellino FJ, Weiler H, Rosen H, Griffin JH, Ruf W. Endogenous EPCR/aPC-PAR1 signaling prevents inflammation-induced vascular leakage and lethality. Blood. 2009;113:2859–2866. doi: 10.1182/blood-2008-12-192385. This work creates a working model by which thrombin and aPC exert opposite effects on inflammation via PAR1, by activation of the thrombin/S1PR3 and aPC/EPCR/S1PR1 downstream signaling pathways. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Mosnier LO, Zampolli A, Kerschen EJ, Schuepbach RA, Banerjee Y, Fernandez JA, Yang XV, Riewald M, Weiler H, Ruggeri ZM, et al. Hyperantithrombotic, noncytoprotective Glu149Ala-activated protein C mutant. Blood. 2009;113:5970–5978. doi: 10.1182/blood-2008-10-183327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •21.Kaneider NC, Leger AJ, Agarwal A, Nguyen N, Perides G, Derian C, Covic L, Kuliopulos A. ‘Role reversal’ for the receptor PAR1 in sepsis-induced vascular damage. Nat Immunol. 2007;8:1303–1312. doi: 10.1038/ni1525. PAR1 signaling in endothelial cells is a central modulator of vascular permeability. While PAR1 signaling is responsible for vascular disruption during sepsis, transactivation of PAR2 converted PAR1 to preserve the vascular barrier. Consequently, the downstream signaling pathways of PAR1 may depend on crossactivation of alternate PARs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •22.McLaughlin JN, Patterson MM, Malik AB. Protease-activated receptor-3 (PAR3) regulates PAR1 signaling by receptor dimerization. Proc Natl Acad Sci U S A. 2007;104:5662–5667. doi: 10.1073/pnas.0700763104. Demonstrates heterodimerization of PAR1 and PAR3 on endothelial cells is crucial in potentiating the responsiveness of PAR1 to thrombin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •23.Leger AJ, Jacques SL, Badar J, Kaneider NC, Derian CK, Andrade-Gordon P, Covic L, Kuliopulos A. Blocking the protease-activated receptor 1-4 heterodimer in platelet-mediated thrombosis. Circulation. 2006;113:1244–1254. doi: 10.1161/CIRCULATIONAHA.105.587758. Demonstrated thrombin is a bivalent agonist for PAR1 and PAR4, which form a stable heterodimer on platelets. [DOI] [PubMed] [Google Scholar]
- 24.Leung LLK, Myles T, Nishimura T, Song JJ, Robinson WH. Regulation of tissue inflammation by thrombi n-activatable carboxypeptidase B (or TAFI) Mol Immunol. 2008;45:4080–4083. doi: 10.1016/j.molimm.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Scatena M, Liaw L, Giachelli CM. Osteopontin: a multifunctional molecule regulating chronic inflammation and vascular disease. Arterioscler Thromb Vasc Biol. 2007;27:2302–2309. doi: 10.1161/ATVBAHA.107.144824. [DOI] [PubMed] [Google Scholar]
- 26.Seier AM, Renkl AC, Schulz G, Uebele T, Sindrilaru A, Iben S, Liaw L, Kon S, Uede T, Weiss JM. Antigen-Specific Induction of Osteopontin Contributes to the Chronification of Allergic Contact Dermatitis. Am J Pathol. 2010;176:246–258. doi: 10.2353/ajpath.2010.090488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schulz G, Renkl AC, Seier A, Liaw L, Weiss JM. Regulated osteopontin expression by dendritic cells decisively affects their migratory capacity. J Invest Dermatol. 2008;128:2541–2544. doi: 10.1038/jid.2008.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murugaiyan G, Mittal A, Weiner HL. Increased osteopontin expression in dendritic cells amplifies IL-17 production by CD4+ T cells in experimental autoimmune encephalomyelitis and in multiple sclerosis. J Immunol. 2008;181:7480–7488. doi: 10.4049/jimmunol.181.11.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••29.Shinohara ML, Kim JH, Garcia VA, Cantor H. Engagement of the type I interferon receptor on dendritic cells inhibits T helper 17 cell development: Role of intracellular osteopontin. Immunity. 2008;29:68–78. doi: 10.1016/j.immuni.2008.05.008. Inhibition of intracellular Opn by type I IFN receptor engagement on dendritic cells supressed Th17 cell development, providing further insight into the modulatory effects of Opn on T cell phenotype as well as the therapeutic mechanism behind type I IFN therapy in MS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen MY, Chen GJ, Nie H, Zhang X, Niu XY, Zang YCQ, Skinner SM, Zhang JZW, Killian JM, Hong J. Regulatory effects of IFN-beta on production of osteopontin and IL-17 by CD4(+) T Cells in MS. Eur J Immunol. 2009;39:2525–2536. doi: 10.1002/eji.200838879. [DOI] [PubMed] [Google Scholar]
- ••31.Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol. 2007;8:74–83. doi: 10.1038/ni1415. This work reveals the molecular pathways behind observations that osteopontin (Opn) worsened outcomes in mouse models of multiple sclerosis. Opn promotes survival of myelin-reactive T cells through activation of transcription factor NFkB and down-modulation of proapoptotic proteins Bim, Bak, and Bax. [DOI] [PubMed] [Google Scholar]
- 32.Steinman L. A molecular trio in relapse and remission in multiple sclerosis. Nat Rev Immunol. 2009;9:440–447. doi: 10.1038/nri2548. [DOI] [PubMed] [Google Scholar]
- ••33.Han MH, Hwang SI, Roy DB, Lundgren DH, Price JV, Ousman SS, Fernald GH, Gerlitz B, Robinson WH, Baranzini SE, et al. Proteomic analysis of active multiple sclerosis lesions reveals therapeutic targets. Nature. 2008;451:1076–1081. doi: 10.1038/nature06559. An important study utilizing proteomics to identify molecular targets in human multiple sclerosis, many of which possess currently unknown physiological function. Revealed protein C inhibitor and tissue factor as unique therapeutic targets in chronic active plaques using aPC and the thrombin inhibitor hirudin treatment in a mice EAE model. [DOI] [PubMed] [Google Scholar]
- 34.Cao C, Gao Y, Li Y, Antalis TM, Castellino FJ, Zhang L. The efficacy of activated protein C in murine endotoxemia is dependent on integrin CD11b. J Clin Invest. 2010 doi: 10.1172/JCI40380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Du XY, Zabel BA, Myles T, Allen SJ, Handel TM, Lee PP, Butcher EC, Leung LL. Regulation of chemerin bioactivity by plasma carboxypeptidase N, carboxypeptidase B (activated thrombin-activable fibrinolysis inhibitor), and platelets. J Biol Chem. 2009;284:751–758. doi: 10.1074/jbc.M805000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albanesi C, Scarponi C, Pallotta S, Daniele R, Bosisio D, Madonna S, Fortugno P, Gonzalvo-Feo S, Franssen JD, Parmentier M, et al. Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J Exp Med. 2009;206:249–258. doi: 10.1084/jem.20080129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parolini S, Santoro A, Marcenaro E, Luini W, Massardi L, Facchetti F, Communi D, Parmentier M, Majorana A, Sironi M, et al. The role of chemerin in the colocalization of NK and dendritic cell subsets into inflamed tissues. Blood. 2007;109:3625–3632. doi: 10.1182/blood-2006-08-038844. [DOI] [PubMed] [Google Scholar]
- 38.Graham KL, Zabel BA, Loghavi S, Zuniga LA, Ho PP, Sobel RA, Butcher EC. Chemokine-like receptor-1 expression by central nervous system-infiltrating leukocytes and involvement in a model of autoimmune demyelinating disease. J Immunol. 2009;183:6717–6723. doi: 10.4049/jimmunol.0803435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••39.Cash JL, Hart R, Russ A, Dixon JP, Colledge WH, Doran J, Hendrick AG, Carlton MB, Greaves DR. Synthetic chemerin-derived peptides suppress inflammation through ChemR23. J Exp Med. 2008;205:767–775. doi: 10.1084/jem.20071601. By constructing a series of chemerin peptides, the authors demonstrated cysteine protease processing was required to generate a ligand that surprisingly inhibited inflammation through its cognate receptor ChemR23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kulig P, Zabel BA, Dubin G, Allen SJ, Ohyama T, Potempa J, Handel TM, Butcher EC, Cichy J. Staphylococcus aureus-derived staphopain B, a potent cysteine protease activator of plasma chemerin. J Immunol. 2007;178:3713–3720. doi: 10.4049/jimmunol.178.6.3713. [DOI] [PubMed] [Google Scholar]
- 41.Vowinkel T, Wood KC, Stokes KY, Russell J, Tailor A, Anthoni C, Senninger N, Krieglstein CF, Granger DN. Mechanisms of platelet and leukocyte recruitment in experimental colitis. Am J Physiol Gastrointest Liver Physiol. 2007;293:G1054–1060. doi: 10.1152/ajpgi.00350.2007. [DOI] [PubMed] [Google Scholar]
- •42.Anthoni C, Russell J, Wood KC, Stokes KY, Vowinkel T, Kirchhofer D, Granger DN. Tissue factor: a mediator of inflammatory cell recruitment, tissue injury, and thrombus formation in experimental colitis. J Exp Med. 2007;204:1595–1601. doi: 10.1084/jem.20062354. Clinically, inflammatory bowel disease is associated with a heighten state of coagulation activation. This study provides evidence that tissue factor mediates IBD pathogenesis through recruitment of inflammatory cells and platelets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Friedman DJ, Kunzli BM, YI AR, Sevigny J, Berberat PO, Enjyoji K, Csizmadia E, Friess H, Robson SC. From the Cover: CD39 deletion exacerbates experimental murine colitis and human polymorphisms increase susceptibility to inflammatory bowel disease. Proc Natl Acad Sci U S A. 2009;106:16788–16793. doi: 10.1073/pnas.0902869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langer HF, Daub K, Braun G, Schonberger T, May AE, Schaller M, Stein GM, Stellos K, Bueltmann A, Siegel-Axel D, et al. Platelets recruit human dendritic cells via Mac-1/JAM-C interaction and modulate dendritic cell function in vitro. Arterioscler Thromb Vasc Biol. 2007;27:1463–1470. doi: 10.1161/ATVBAHA.107.141515. [DOI] [PubMed] [Google Scholar]
- 45.Zimmerli C, Lee BPL, Palmer G, Gabay C, Adams R, Aurrand-Lions M, Imhof BA. Adaptive Immune Response in JAM-C-Deficient Mice: Normal Initiation but Reduced IgG Memory. J Immunol. 2009;182:4728–4736. doi: 10.4049/jimmunol.0803892. [DOI] [PubMed] [Google Scholar]
- 46.Kisucka J, Chauhan AK, Zhao BQ, Patten IS, Yesilaltay A, Krieger M, Wagner DD. Elevated levels of soluble P-selectin in mice alter blood-brain barrier function, exacerbate stroke, and promote atherosclerosis. Blood. 2009;113:6015–6022. doi: 10.1182/blood-2008-10-186650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mueller A, Meiser A, McDonagh EM, Fox JM, Petit SJ, Xanthou G, Williams TJ, Pease JE. CXCL4-induced migration of activated T lymphocytes is mediated by the chemokine receptor CXCR3. J Leukoc Biol. 2008;83:875–882. doi: 10.1189/jlb.1006645. [DOI] [PubMed] [Google Scholar]
- 48.Eberlein J, Nguyen TT, Victorino F, Golden-Mason L, Rosen HR, Homann D. Comprehensive assessment of chemokine expression profiles by flow cytometry. J Clin Invest. 2010;120:907–923. doi: 10.1172/JCI40645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, Kramp BK, Piccinini AM, Paludan SR, Kowalska MA, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. 2009;15:97–103. doi: 10.1038/nm.1898. [DOI] [PubMed] [Google Scholar]
- 50.Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb Haemost. 2007;98:1108–1113. [PubMed] [Google Scholar]
- 51.Soga F, Katoh N, Inoue T, Kishimoto S. Serotonin activates human monocytes and prevents apoptosis. J Invest Dermatol. 2007;127:1947–1955. doi: 10.1038/sj.jid.5700824. [DOI] [PubMed] [Google Scholar]
- 52.Leon-Ponte M, Ahern GP, O'Connell PJ. Serotonin provides an accessory signal to enhance T-cell activation by signaling through the 5-HT7 receptor. Blood. 2007;109:3139–3146. doi: 10.1182/blood-2006-10-052787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lang PA, Contaldo C, Georgiev P, El-Badry AM, Recher M, Kurrer M, Cervantes-Barragan L, Ludewig B, Calzascia T, Bolinger B, et al. Aggravation of viral hepatitis by platelet-derived serotonin. Nat Med. 2008;14:756–761. doi: 10.1038/nm1780. [DOI] [PubMed] [Google Scholar]
- 54.Cognasse F, Hamzeh-Cognasse H, Lafarge S, Delezay O, Pozzetto B, McNicol A, Garraud O. Toll-like receptor 4 ligand can differentially modulate the release of cytokines by human platelets. Br J Haematol. 2008;141:84–91. doi: 10.1111/j.1365-2141.2008.06999.x. [DOI] [PubMed] [Google Scholar]
- ••55.Sprague DL, Elzey BD, Crist SA, Waldschmidt TJ, Jensen RJ, Ratliff TL. Platelet-mediated modulation of adaptive immunity: unique delivery of CD154 signal by platelet-derived membrane vesicles. Blood. 2008;111:5028–5036. doi: 10.1182/blood-2007-06-097410. This work demonstrates the capacity for platelet-derived membrane vesicles to transport CD154 and stimulate antigen-specific B cell maturation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cognasse F, Hamzeh-Cognasse H, Lafarge S, Chavarin P, Cogne M, Richard Y, Garraud O. Human platelets can activate peripheral blood B cells and increase production of immunoglobulins. Exp Hematol. 2007;35:1376–1387. doi: 10.1016/j.exphem.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 57.Elzey BD, Schmidt NW, Crist SA, Kresowik TP, Harty JT, Nieswandt B, Ratliff TL. Platelet-derived CD154 enables T-cell priming and protection against Listeria monocytogenes challenge. Blood. 2008;111:3684–3691. doi: 10.1182/blood-2007-05-091728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••58.Iannacone M, Sitia G, Isogawa M, Whitmire JK, Marchese P, Chisari FV, Ruggeri ZM, Guidotti LG. Platelets prevent IFN-alpha/beta-induced lethal hemorrhage promoting CTL-dependent clearance of lymphocytic choriomeningitis virus. Proc Natl Acad Sci U S A. 2008;105:629–634. doi: 10.1073/pnas.0711200105. Illustrates the critical role of platelet-derived CD154 (CD40L) in eliciting effective cytotoxic T cell response to resolve viral infections. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •59.Xu H, Zhang X, Mannon RB, Kirk AD. Platelet-derived or soluble CD154 induces vascularized allograft rejection independent of cell-bound CD154. J Clin Invest. 2006;116:769–774. doi: 10.1172/JCI27155. Allograft rejection may be instigated through CD154 derived from trauma-induced activation of platelets. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Danese S, Scaldaferri F, Vetrano S, Stefanelli T, Graziani C, Repici A, Ricci R, Straface G, Sgambato A, Malesci A, et al. Critical role of the CD40 CD40-ligand pathway in regulating mucosal inflammation-driven angiogenesis in inflammatory bowel disease. Gut. 2007;56:1248–1256. doi: 10.1136/gut.2006.111989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vaidyula VR, Rao AK, Mozzoli M, Homko C, Cheung P, Boden G. Effects of hyperglycemia and hyperinsulinemia on circulating tissue factor procoagulant activity and platelet CD40 ligand. Diabetes. 2006;55:202–208. [PubMed] [Google Scholar]
- 62.Boden G, Vaidyula VR, Homko C, Cheung P, Rao AK. Circulating tissue factor procoagulant activity and thrombin generation in patients with type 2 diabetes: effects of insulin and glucose. J Clin Endocrinol Metab. 2007;92:4352–4358. doi: 10.1210/jc.2007-0933. [DOI] [PubMed] [Google Scholar]
- •63.Cui W, Wilson JT, Wen J, Angsana J, Qu Z, Haller CA, Chaikof EL. Thrombomodulin improves early outcomes after intraportal islet transplantation. Am J Transplant. 2009;9:1308–1316. doi: 10.1111/j.1600-6143.2009.02652.x. Demonstrates therapeutic benefits of thrombomodulin in an murine allograft model of intraportal islet transplantation, possibly through downmodulation of danger signals TNF-a and IL-1b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Light DB, Mclean K. Methods of using novel tissue factor targeted thrombomodulin fusion proteins as anticoagulants. US Patent. 2009:7622122.
- 65.Stabler CL, Sun XL, Cui W, Wilson JT, Haller CA, Chaikof EL. Surface re-engineering of pancreatic islets with recombinant azido-thrombomodulin. Bioconjug Chem. 2007;18:1713–1715. doi: 10.1021/bc7002814. [DOI] [PubMed] [Google Scholar]
- 66.Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, Selmaj K, Agoropoulou C, Leyk M, Zhang-Auberson L, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- 67.Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, Pelletier J, Capra R, Gallo P, Izquierdo G, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- 68.Puneet P, Yap CT, Wong L, Lam Y, Koh DR, Moochhala S, Pfeilschifter J, Huwiler A, Melendez AJ. SphK1 regulates proinflammatory responses associated with endotoxin and polymicrobial sepsis. Science. 2010;328:1290–1294. doi: 10.1126/science.1188635. [DOI] [PubMed] [Google Scholar]