

Abstract
Epidermodysplasia verruciformis (EV) is a rare genetic disorder characterized by increased susceptibility to specific human papillomaviruses, the betapapillomaviruses. These EV-HPVs cause warts and increase the risk of skin carcinomas in otherwise healthy individuals. Inactivating mutations in epidermodysplasia verruciformis 1 (EVER1) or EVER2 have been identified in most, but not all, patients with autosomal recessive EV. We found that 2 young adult siblings presenting with T cell deficiency and various infectious diseases, including persistent EV-HPV infections, were homozygous for a mutation creating a stop codon in the ras homolog gene family member H (RHOH) gene. RHOH encodes an atypical Rho GTPase expressed predominantly in hematopoietic cells. Patients’ circulating T cells contained predominantly effector memory T cells, which displayed impaired TCR signaling. Additionally, very few circulating T cells expressed the β7 integrin subunit, which homes T cells to specific tissues. Similarly, Rhoh-null mice exhibited a severe overall T cell defect and abnormally small numbers of circulating β7-positive cells. Expression of the WT, but not of the mutated RHOH, allele in Rhoh–/– hematopoietic stem cells corrected the T cell lymphopenia in mice after bone marrow transplantation. We conclude that RHOH deficiency leads to T cell defects and persistent EV-HPV infections, suggesting that T cells play a role in the pathogenesis of chronic EV-HPV infections.
Introduction
Epidermodysplasia verruciformis (EV) is a rare, lifelong disorder characterized by disseminated and persistent flat warts or pityriasis versicolor–like (PV-like) lesions due to an abnormal and selective susceptibility to a specific group of weakly virulent related keratinocyte-tropic human betapapillomavirus genotypes (EV-HPVs), including the oncogenic human PV type 5 (HPV-5) (OMIM 226400). EV is associated with an increase in the risk of non-melanoma skin carcinomas (NMSC) (1), with no other clinical signs in most patients (2). Cockayne et al. first suggested in 1933 that EV could be inherited as an autosomal recessive (AR) condition (3). However, EV was not considered as a primary immunodeficiency (PID) until the identification of inactivating mutations in epidermodysplasia verruciformis 1 (EVER1) (TMC6) and in EVER2 (TMC8) (4–8). Mutations in these two genes account for approximately 75% of the studied cases (9–16). Both genes are expressed in circulating lymphocytes (refs. 15, 17) and keratinocytes, in which the EVER proteins form a complex with the zinc transporter ZnT1, which regulates intracellular zinc distribution and, consequently, controls the activity of zinc-dependent transcription factors and cell proliferation (18). We investigated 2 siblings, now aged 20 and 31 years, who were born to first-cousin parents and have displayed since childhood a phenotype related to but distinct from EV, with persistent EV-HPV infections and other clinical manifestations, including bronchopulmonary disease and Burkitt lymphoma in one patient. Mutations in EVER1 and EVER2 have been excluded in these patients, and a putative disease-causing locus has been mapped to a broad region of chromosome 2p21–p24 by genome-wide linkage analysis (GWL) and homozygosity mapping with a few hundred polymorphic microsatellites (19). We further investigated the molecular genetic basis of the disease in this kindred by a GWL approach, this time using a high-density SNP map. This approach led to the identification of additional linked regions and, finally, to the discovery of a disease-causing nonsense mutation in the ras homolog gene family member H (RHOH) gene on chromosome 4p13.
Results
Identification of a nonsense mutation in the RHOH gene.
We investigated 2 patients (P1 and P2) with persistent EV-HPV infections and other clinical manifestations (Supplemental Figure 1 and detailed case report in Methods; supplemental material available online with this article; doi: 10.1172/JCI62949DS1) born to consanguineous French parents (Figure 1A). We conducted a new GWL study in this family, by homozygosity mapping with a high-density SNP array (Supplemental Methods). Five regions, including the region originally identified on chromosome 2p21–p24 (19), distributed on 3 chromosomes, with an equal maximum lod score of 2.1, were identified in this analysis (Supplemental Figure 2, A–C). These 5 putative disease-associated regions contained 174 protein-coding genes and 108 RNA genes (Supplemental Tables 1–3 and data not shown). Whole-exome sequencing was carried out for both patients, covering 172 protein-coding genes in the linked regions with a mean of 81.8 and 98.7 reads per gene for P1 and P2, respectively. Only 2 protein-coding genes in the linked regions carried homozygous variants present in both patients and absent from public databases (Ensembl, National Center for Biotechnology Information, 1000 Genomes): FAM179A and RHOH. The best candidate variant was a homozygous nucleotide substitution (C>G) at position 114 in exon 3 of the RHOH gene, which is located on chromosome 4p13, resulting in the replacement of a tyrosine residue with a stop codon (Y38X) (Figure 1B). The RHOH gene encodes an atypical, hematopoietic cell–specific member of the Rho GTPase family. Canonical Rho GTPases function as intracellular switches, transducing signals from various membrane receptors, including T and B cell receptors. Atypical Rho GTPases, including RHOH, lack GTPase activity and therefore remain in the active, GTP-bound conformation (20, 21). The premature termination codon, Y38X, is located upstream from sequences encoding 2 important functional protein domains of RHOH: the immunoreceptor tyrosine-based activation motif–like (ITAM-like) motif not found in any other Rho GTPases and the CAAX box, a lipid modification site common to all Rho GTPases that mediates the localization of the protein to membranes (Figure 1C and ref. 22). The other 2 siblings and the parents were healthy and heterozygous for the nonsense Y38X RHOH allele (Figure 1A). This allele was not found in any of the 1,050 healthy controls from 51 ethnic groups investigated (Human Genome Diversity Project–Centre d’Etude du Polymorphisme Humain [HGDP-CEPH] panel), suggesting that this is a rare pathogenic mutation rather than an irrelevant polymorphism. Moreover, RHOH has no known polymorphic nonsense or frameshift alleles present in public databases. Somatic mutations in RHOH have previously been found in various B cell cancers, including Burkitt lymphoma (23–26). The established link between RHOH and B cell cancers therefore suggests that homozygosity for Y38X may also have contributed to the development of Burkitt lymphoma in P1. Collectively, these data suggest that the Y38X RHOH allele is responsible for the disease in this kindred.
Figure 1. Homozygous RHOH loss-of-expression mutation in 2 patients with persistent EV-HPV infections.

(A) Pedigree of the family with susceptibility to EV-HPV infections and other infectious manifestations. Generations are designated by a Roman numeral (I, II, III, IV, and V). P1 and P2 are represented by black symbols. The proband is indicated by an arrow. The asterisk indicates the individuals genotyped with the Affymetrix Genome-Wide SNP 6.0 array. (B) Automated sequencing profile showing the Y38X RHOH mutation in genomic DNA (gDNA) extracted from EBV-B cells from the patients and comparison with the sequence obtained from a healthy control. (C) Schematic representation of the structure of the RHOH protein adapted from the work of Fueller et al. (17). Y38X is situated between the phosphate-binding loop and the ITAM-like domain. The 4 possible reinitiation sites downstream from the mutation are indicated by small black arrows. (D) RHOH mRNA production, as assessed by qRT-PCR on total RNA isolated from saimiri T cells from the 2 patients, 1 healthy sibling (S1), and the father (F), both heterozygous for the mutant allele, and 2 healthy controls (C1 and C2). Mean + SD for 3 experiments is presented for all samples except for P1 (2 experiments). (E) Immunoblot analyses of 30 μg of total protein extracted from the saimiri T cells of P1, P2, S1, F, C1, and C2, with an antibody directed against RHOH and an antibody against GAPDH as a protein-loading control. (F) Immunoblot analysis of NIH/3T3 cells transfected with WT RHOH, Y38X RHOH, or 139-C RHOH. NT, not transduced.
The Y38X RHOH allele results in a loss-of-function protein.
RHOH mRNA levels were normal, as shown by quantitative RT-PCR (qRT-PCR) analysis of the patients’ herpesvirus saimiri–transformed T cell lines (saimiri T cells), which were compared with those of healthy controls and healthy members of the family (Figure 1D). RHOH protein levels were assessed by immunoblotting lysates of the patients’ saimiri T cells with an antibody recognizing an epitope upstream from the mutated residue. The 21-kDa RHOH protein was absent from the saimiri T cells of both patients, but was present in the cells of 2 healthy controls and 2 healthy family members heterozygous for the mutant allele (Figure 1E). Translation reinitiation is possible, as 4 different ATG start codons are located downstream from the mutation (Figure 1C) and such reinitiation has been documented for upstream nonsense mutations in other genes (27). Thus, both a cDNA corresponding to the N-terminal HA-tagged Y38X RHOH sequence and an HA-tagged cDNA corresponding to the 145–amino acid C terminus that would be translated from the first downstream ATG at nucleotide position 139 of the gene (139-C RHOH) were inserted into a retroviral vector. NIH/3T3 cells were transfected with these vectors, together with an HA-tagged WT RHOH vector used as a positive control. No protein from Y38X RHOH–transfected cells was detected on immunoblots with an anti-HA antibody, whereas a protein smaller than WT-RHOH was detected on immunoblots of cells producing RhoH 139-C (Figure 1F). Thus, the possibility of translation reinitiation cannot be formally excluded. We previously showed that thymocyte selection is defective and T cell maturation is blocked at the DN3 and DP stages in Rhoh–/– mice, which consequently display profound T cell lymphopenia (28, 29). We directly assessed the function of the proteins encoded by the human Y38X RHOH and 139-C RHOH mutant genes in T cell rescue experiments in vivo by transducing bone marrow cells from Rhoh–/– mice with a retroviral vector containing the human Y38X RHOH mutant, the 139-C RHOH truncated cDNA, or the WT RHOH cDNA. The transduced cells were then transplanted into sublethally irradiated Rag2–/– recipient mice. Mice receiving bone marrow cells transduced with the Y38X RHOH or 139-C RHOH cDNA had no more T cells than mice receiving cells transduced with the empty vector 3 months after transplantation. In contrast, as previously reported, mice receiving cells transduced with WT RHOH cDNA had significantly larger numbers of T cells, resulting in the partial rescue of Rhoh–/– T cell lymphopenia (Figure 2A). The mutant Y38X RHOH allele is therefore loss of function.
Figure 2. The Y38X RHOH allele is loss of function in the mouse model and is associated with an impaired phosphorylation of ZAP70 upon TCR stimulation in the patients’ saimiri T cells.

(A) Bone marrow cells from Rhoh–/– mice (KO) of mixed background were transduced with an empty retroviral vector encoding YFP or a retroviral vector coexpressing the YFP gene and WT RHOH, Y38X RHOH, or 139-C RHOH genes and transplanted into sublethally irradiated Rag2–/– recipient mice. Normal bone marrow cells transduced with an empty vector were included as a control. The percentage of CD3+ cells within the YFP+-transduced T cell group was assessed in the blood of recipient mice 3 months after transplantation by flow cytometry (mean ± SEM, n = 4 recipients per construct). *P < 0.05; **P < 0.005. (B) Representative immunoblots of cell lysates extracted from saimiri T cells of a healthy control and the 2 patients (P1 and P2) following OKT3 stimulation, probed with anti-ZAP70 and anti–phospho-ZAP70 antibodies. β-Actin was used as an additional protein-loading control. (C) Representative immunoblot analysis of cell lysates extracted from YPF+ sorted saimiri T cells (control and patient P2) transduced with a vector encoding YFP and the HA-tagged mutant Y38X RHOH (HA-Y38X), YFP and the HA-tagged WT RHOH (HA-WT), or YFP alone (MOCK), subsequently stimulated with OKT3. Lysates were blotted with anti-ZAP70 and anti-phospho-ZAP70 antibodies. β-Actin was used as an additional protein-loading control.
Impact of human RHOH deficiency on T cells.
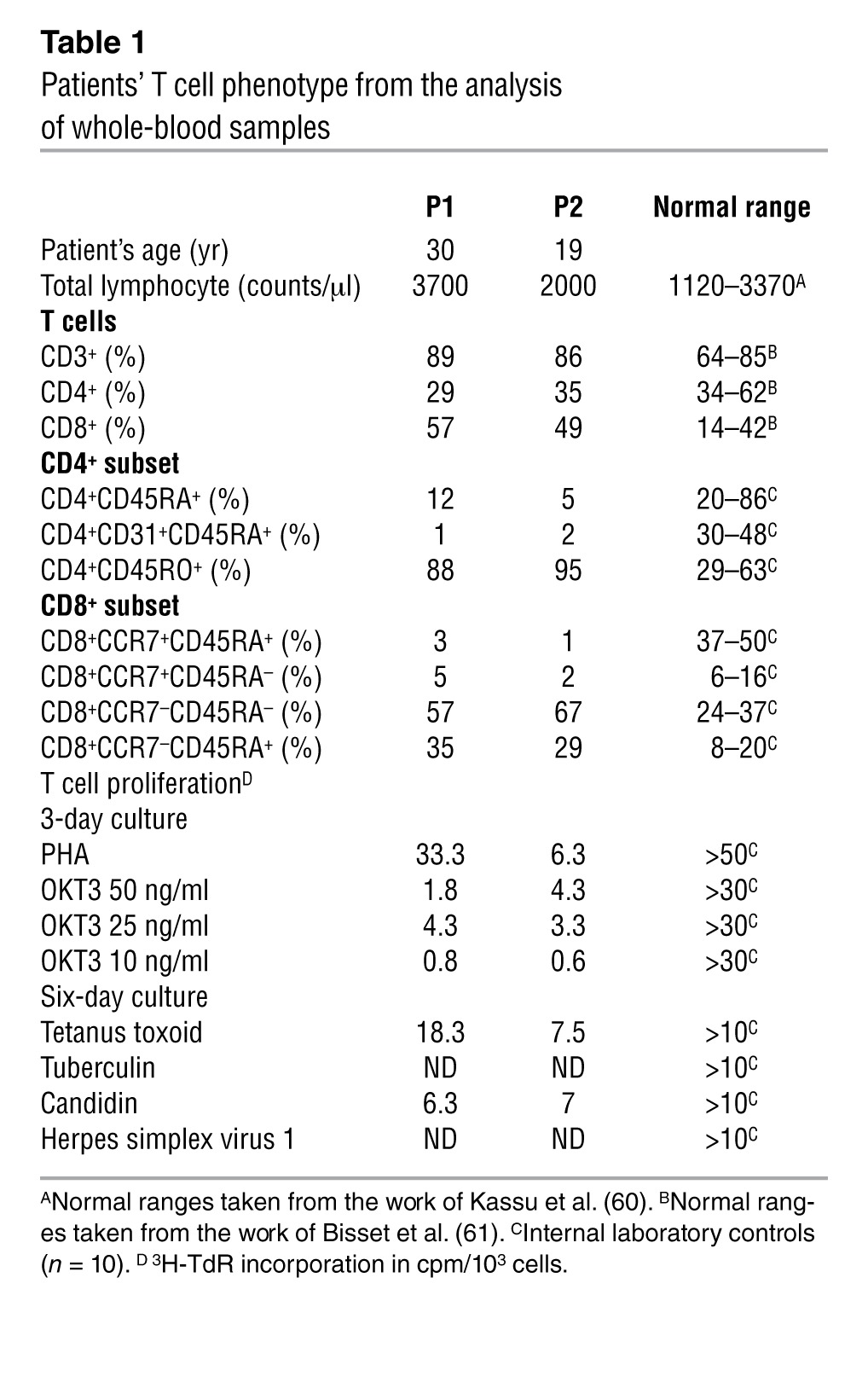
Unlike EVER1 and EVER2, the RHOH gene is expressed only in hematopoietic cells, including myeloid cells, NK cells, B cells, and T cells (20, 30, 31). We carried out general immunophenotyping on whole-blood samples or cryopreserved PBMCs from the patients to assess the various blood lineage subsets and on serum samples from the patients to assess their Ig levels. There were no major abnormalities in the frequencies of B cell subsets, NK cells, NKT cells (Supplemental Figure 3 and Supplemental Table 4), monocytes and polymorphonuclear cells (Supplemental Table 4), and in antibody production (Supplemental Table 6). The patients’ total T cell counts fell within the normal range. However, both patients had slightly lower frequencies of CD4+ T cells than normal, but higher frequencies of CD8+ T cells and normal frequencies of γδ T cells (Table 1 and Supplemental Tables 4 and 5). We then investigated the various naive and memory subsets of the patients’ T cells at different ages (Table 1, Supplemental Table 5, and Supplemental Figure 4). Percentages of naive CD4+CD45RA+, CD4+CCR7+CD45RA+, and CD8+CCR7+CD45RA+ cells and of recent thymic T cell emigrants CD4+CD31+CD45RA+ were consistently very low in both patients. Central memory CD4+CCR7+CD45RA– and CD8+CCR7+CD45RA– cells were reduced to a lesser extent. However, memory CD4+CD45RO+, effector memory (TEM) CD4+CD45RA–CCR7– and CD8+CCR7–CD45RA– and revertant memory (TEMRA) CD8+CCR7–CD45RA+ T cell percentages were above the upper limit of the normal range. An immunoscope analysis of the TCR Vαβ and Vγδ repertoires showed a loss of the normal Gaussian-like polyclonal distribution for the Vα8b, Vα10, Vβ28, Vβ13, Vγ9, and Vδ1 families (Supplemental Figure 5). This suggested restricted TCR usage and the clonal expansion of certain families, consistent with the expansion of the effector memory T cell compartment in the patients. Interestingly, the expanded memory cell populations in both the CD4+ and CD8+ compartments showed signs of exhaustion, with the upregulation of 2B4, CX3CR1, CD57, granzyme B, and perforin and the downregulation of CD127, CD27, and CD62L with respect to healthy controls (Supplemental Figure 6), with some variability of statistical significance between markers and subsets. This exhaustion phenotype and the expansion of memory T cell subsets were previously observed in other cases of chronic infection, including for patients with PIDs (32, 33), suggesting that the increase of memory T cells and their exhaustion phenotype are more likely to be consequences of chronic infection rather than causes of the disease. Thus, RHOH deficiency is associated with a lack of naive T cells in the patients’ peripheral blood, suggesting a developmental defect consistent with the defect in thymic T cell development observed in Rhoh–/– mice (28).
Table 1.
Patients’ T cell phenotype from the analysis of whole-blood samples
Defective TCR signaling in the patients’ T cells.
We then assessed the impact of RHOH deficiency on the function of the patients’ T cells. PBMCs were isolated from the patients at different ages and stimulated with phytohemagglutinin (PHA), an anti-CD3 antibody (OKT3), OKT3 plus anti-CD28 Ab, PMA/ionomycin, or recall antigens (tetanus toxoid, tuberculin, candidin). Their proliferation was assessed by evaluating the incorporation of tritiated thymidine (Table 1, Supplemental Table 5, and data not shown). The T cells of both patients responded to various recall antigens in terms of proliferation, with some antigens eliciting weaker responses than others. In contrast, they failed to proliferate in response to various doses of OKT3, but proliferated in response to OKT3 plus anti-CD28 Ab, PHA, or PMA/ionomycin, although proliferation rates remained below the normal range (Table 1 and Supplemental Table 5). Consistent with the known role of RHOH in TCR signaling (28, 29, 34, 35), these data suggest abnormalities in TCR-mediated activation that are partially compensated by PMA/ionomycin for the stimulation of cells downstream from RHOH or PHA for the stimulation of other membrane-bound receptors, such as CD2 (36). As murine Rhoh–/– T cells have a defect in the localization of ZAP70 to the plasma membrane and its activation upon CD3 stimulation (refs. 28, 29, and Supplemental Table 7), we investigated ZAP70 phosphorylation in response to CD3 stimulation in saimiri T cells from the patients and a healthy control. Basal level of ZAP70 phosphorylation was high in the control cell line and was further increased by CD3 stimulation. In contrast, little or no ZAP70 phosphorylation was observed in the presence or absence of CD3 stimulation in the cells of P1 and P2, confirming the defective TCR signaling pathway observed ex vivo (Figure 2B). We investigated whether the impairment of ZAP70 activation was due to a loss of RHOH function in the patients’ saimiri T cells by transfecting cells from P2 with an empty retroviral control vector (MOCK) or a WT RHOH–expressing retroviral vector and then stimulating the transfected cells with OKT3. In parallel, saimiri T cells from a healthy donor were transduced with an empty vector (MOCK) or a vector expressing the mutant Y38X RHOH allele as controls. Patients’ cells transduced with WT RHOH had higher levels of residual and OKT3-induced ZAP70 phosphorylation than MOCK-transduced cells (Figure 2C), demonstrating the association of the RHOH mutation with the observed phenotype.
Alterations in tissue-homing T cell subsets in the patients.
The occurrence of persistent EV-HPV infections in both patients suggested a focal defect in the immune system. We thus hypothesized that the development of some skin-homing T cells might be affected by the loss of RHOH. The most specific skin-homing markers of human T cells are the cutaneous lymphocyte antigen (CLA) and the chemokine receptors CCR4, CCR6, and CCR10 (37). We assessed the different tissue-homing subsets expressing these markers as percentages of CD3+ cells and counts per million PBMCs (Figure 3, A–C, Supplemental Figure 7, A, B, D, E, and Supplemental Figure 8). We also evaluated the CLA+ subset in the various naive and memory compartments of the CD4+ and CD8+ T cell subsets (Supplemental Figure 9, A and B). The CD4+CLA+ and CD8+CLA+ T cell subsets in the patients’ PBMCs appeared to be smaller than normal. Surprisingly, the CD4+ and CD8+ populations had a much higher proportion of CLA+ naive T cells and a lower proportion of CLA+ central and effector memory T cells than for healthy controls. The CD3+CCR6+ and CD3+CCR10+ populations and the CD3+CLA+CCR4+, CD3+CLA+CCR6+, and CD3+CLA+CCR10+ subsets were also smaller than those in healthy controls. The decrease in the size of the subsets expressing only CLA or both CLA and one of the chemokine receptors was slightly less pronounced in the CD8+ subset than in the CD4+ subset. Integrins are also involved in tissue-homing of T lymphocytes (21). The αEβ7 integrin has been associated with lung, gut, and skin homing, whereas the α4β7 integrin has been associated with gut homing (21). Skin-homing αE+β7+ T cells also express CLA (38). We therefore investigated the expression of the αEβ7 integrin in the whole CD3+ T cell compartment, in the CD3+CLA+ T cell compartment (Figure 3D, Supplemental Figure 7, C and F, and Supplemental Figure 8D), and in the various naive and memory T cell compartments of the patients’ PBMCs (Supplemental Figure 9, C and D). Within the CD3+ T cell population, the proportions and numbers of αE+β7+ and CLA+αE+ cells were normal. Nevertheless, as previously observed for the CLA marker, naive αE+ cells were more abundant, whereas effector memory αE+ cells were slightly less abundant than in healthy controls. We next assessed β7+, α4+, α4+β7+, and α4+β7– cells in the patients’ PBMCs (Figure 3E, Supplemental Figure 7, G–J, and Supplemental Figure 8E). Surprisingly, the β7+ and α4+β7+ subsets were extremely small, whereas the α4+ and α4+β7– subsets were significantly larger than normal in the CD3+, CD3+CD4+, and CD3+CD8+ compartments of the patients’ PBMCs, with some variability of statistical significance between markers and subsets. In Rhoh–/– mice, the proportions of β7+ and α4+ T cells were also affected, with lower and higher than normal percentages of β7+CD3+ and α4+CD3+ T lymphocytes, respectively (Figure 3F). However, in contrast to our findings for patients, the decrease in the size of the β7+ cell population was this time correlated with a decrease in the abundance of αE+β7+ cells, but not in that of α4+β7+ cells. As the Rhoh–/– mice were not infected with papillomaviruses, these data strongly suggest that the decrease in size of the β7+ T cell population in the patients was a consequence of RHOH deficiency rather than of persistent EV-HPV infection or other phenotypes.
Figure 3. Human and mouse RHOH deficiencies lead to an abnormal integrin expression pattern.

(A–E) Tissue-homing T cell subsets were assessed on live CD3+-, CD3+CD4+-, and CD3+CD8+-gated cryopreserved PBMCs from the 2 patients (P1 values indicated by gray squares, P2 values indicated by gray diamonds) and healthy controls (indicated by black circles) by flow cytometry. (A) Skin-homing CLA+ subsets were assessed for both patients and 28 healthy controls. (B) CCR4+, CCR6+, and CCR10+ subsets were assessed for both patients and 12, 17, and 12 healthy controls, respectively. (C) Skin-homing CLA+CCR4+, CLA+CCR6+, and CLA+CCR10+ subsets were assessed for both patients and for 12, 17, and 12 healthy controls, respectively. (D) αE+β7+ cells were assessed for both patients and 14 healthy controls. (E) β7+, α4+, α4+β7+, and α4+β7– subsets were assessed for both patients and 12 healthy controls. The frequencies of the various subsets are expressed in percentages of CD3+ cells. Viability rates were about 95% for all PBMC preparations. Patients’ samples were tested at least twice, except for the chemokine receptors, for which assessments were carried out only once. Mean values are indicated by horizontal bars. The values obtained in all experiments were similar. (F) The frequencies within the CD3+ population of β7+, α4+, αE+β7+, and α4+β7+ cells were assessed by flow cytometry on CD3+-gated peripheral blood cells from Rhoh+/+ (n = 5) and Rhoh–/– mice (mean ± SEM, n = 5 mice of mixed background). *P < 0.05; **P < 0.005; ***P < 0.0005.
Discussion
We report here the discovery of human RHOH deficiency in a kindred with a T cell deficiency and various infectious diseases, including persistent cutaneous lesions caused by EV-HPVs. Unlike EVER1 and EVER2, RHOH is not detectable in keratinocytes, which serve as the first line of defense against EV-HPVs. Moreover, although RHOH is normally expressed in all leukocytes, the T cell phenotype is clearly the strongest phenotype in mice and humans lacking RHOH. Rhoh–/– mice have a mild myeloid phenotype (39, 40); no overt myeloid phenotype was observed in our patients. Moreover, none of the known patients with myeloid PIDs, including disorders affecting mononuclear or polymorphonuclear phagocytes, display persistent EV-HPV infections (41, 42). Finally, a role for human T cells in immunity to cutaneous HPVs has been inferred from histological studies of regressing warts (43). The persistent lesions caused by EV-HPVs in patients with RHOH deficiency are therefore probably associated with the T cell defect. This may not be sufficient, as the contribution of additional factors may account for the expression of the whole EV-HPV genome in keratinocytes. In any case, the patients’ T cell phenotype is complex. The patients present a profound peripheral naive T cell lymphopenia, from at least the age of 26 years for P1 and 15 years for P2, probably due to a developmental defect. The total T cell counts of these patients are, however, normal, due to an increase in memory T cells with an exhaustion phenotype that probably results from chronic viral infection. The patients’ T cells also display an impaired TCR signaling pathway. In the spleen and lymph nodes of Rhoh–/– mice, the frequency of naive T cells is low, whereas that of memory T cells is high (34, 44). In addition, Rhoh-deficient splenocytes and thymocytes show an impaired response to CD3 stimulation (Supplemental Table 7 and refs. 28, 34). RHOH thus seems to play a crucial role following TCR stimulation in both humans and mice.
RHOH deficiency has several immunological features in common with the combined immunodeficiencies (CIDs) involving deficiencies of CD3γ, ZAP70, ITK, STIM1, ORAI1, MAGT1, MST1, and DOCK8 (45–47), in which naive T cells are lost from the CD4+ or CD8+ compartment or both, and the response of peripheral T cells to anti-CD3 mAb stimulation is impaired (Supplemental Table 8). DOCK8 and MST1 deficiencies are also associated with an expansion of the memory cell population with an exhaustion phenotype. However, the immunological and clinical phenotype of the 2 RHOH-deficient patients seems to be less severe than that of patients with these CIDs. Indeed, neither of our patients displays any signs of autoimmunity, and they have a narrower susceptibility to infection than the other patients, despite the bronchopulmonary disease and Burkitt lymphoma observed in P1. These RHOH-deficient patients nonetheless suffer from persistent EV-HPV infections. DOCK8- and most MST1-deficient patients suffer from recurrent cutaneous viral infections, including α-HPV infections, but not from persistent EV-HPV infections (47, 48). Interestingly, we recently found an MST1-deficient patient with persistent EV-HPV infections (A. Crequer, unpublished observations). Nevertheless, the majority of CID patients have never displayed persistent EV-HPV infections. This is unlikely to reflect an ascertainment bias, as the first lesions appeared at the ages of 7 and 4 years in P1 and P2, respectively, whereas patients with other T cell defects did not display EV-HPV–infected lesions at this age. Moreover, EV-HPVs are ubiquitous, and both patients with RHOH deficiency suffer from susceptibility to these viruses. Thus, impaired TCR signaling and a lack of naive T cells, as seen in patients with other T cell defects, may not be sufficient to confer susceptibility to EV-HPVs.
RHOH-deficient patients’ T cells also have an altered expression of tissue-homing markers. The most striking differences between the patients and healthy controls were observed in the β7+ and α4+ subsets, which were significantly smaller and larger in the patients’ PBMCs, respectively. Indeed, the patients had very low counts and frequencies of a specific tissue-homing α4+β7+ T cell subset, despite having a larger than normal α4+ subset, and this may be relevant to this particular issue. It is known that the α4 chain can be associated with either the β7 subunit or the β1 subunit (21). The increased size of the α4+β7– T cell subset therefore reflects an increase of T cells expressing the α4+β1+ integrin, which is known to be strongly expressed on memory cells (49). This increase may therefore be correlated with the excess of memory T cells in the patients’ peripheral blood. The α4+β7+ T cell subset has been shown to home to the gut, although no significant disorder affecting the gut was observed in our patients (50, 51). However, this subset has also been associated with cutaneous homing in situations of immune dysregulation, such as contact hypersensitivity and spongiotic dermatitis (52, 53). Interestingly, Rhoh–/– mice also have a low frequency of β7+ T cells among CD3+ T cells and a high frequency of α4+ T cells (Supplemental Table 7). As Rhoh–/– mice were not challenged with cutaneous EV-HPVs or any other pathogens, their integrin phenotype is unlikely to result from chronic infections. The abnormal integrin profiles of the patients’ T cells are therefore unlikely to be a consequence of persistent EV-HPV infections. Based on the T cell phenotype common to both RHOH-deficient mice and humans, the pathogenesis of susceptibility to EV-HPVs might involve a combination of the various T cell phenotypes documented, including impaired T cell responses, a lack of naive cells, and a smaller than normal proportion of β7+ T cells.
Regardless of the detailed mechanism involved, which remains elusive, these data suggest that T cells may play an important role in protective immunity to skin-tropic EV-HPVs, at least in some individuals. In the literature, there are currently 2 hypotheses, not mutually exclusive, to explain the pathogenesis of EV in EVER deficiency: EVER deficiency alleviates the inhibition of the expression of the whole genome of EV-HPVs in keratinocytes, and EVER deficiency impairs T cell immunity against EV-HPVs (15, 54). The hypothesis of a keratinocyte contribution to EV in these patients is supported by the appearance of EV lesions on the skin of JAK3- and IL2-Rγ–deficient patients after bone marrow transplantation, particularly in cases of successful T cell engraftment (55). Nevertheless, the EVER proteins are also expressed in T cells, and some EV patients seem to have poor cell-mediated immunity (15). Unfortunately, the impact of EVER1 and EVER2 deficiencies has not yet been investigated in T cells. We report here the association of RHOH deficiency with a profound T cell deficiency and persistent EV-HPV infections. The EVER proteins may regulate zinc distribution in T cells, as suggested by Lazarczyk et al. (54). Zinc has been shown to contribute to T cell receptor signaling by increasing ZAP70 phosphorylation (56). It would therefore be interesting to assess T cell function and tissue-homing subsets in EVER-deficient patients to test the hypothesis that T cell immunity is also impaired in patients with EVER deficiency. Conversely, the 2 RHOH-deficient patients may also have elusive keratinocyte abnormalities favoring the development of EV-HPV lesions, which might not solely result from a T cell deficiency. The search for new genetic disorders underlying EV and EV-HPV persistent infections at large will help to clarify the contribution of keratinocytes and T lymphocytes to the pathogenesis of persistent EV-HPV infections in humans.
Methods
Case report: RHOH-deficient patients P1 and P2.
P1 and P2 are French and were born to married first cousins. The pedigree is shown in Figure 1A. P1 was born in 1980. At the age of 5 years, he was diagnosed with Burkitt lymphoma of the abdominal lymph nodes and was treated by chemotherapy and with systemic steroids. At the age of 7 years, disseminated flat wart-like lesions began to appear on his face and on the back of his hands after chemotherapy. In addition, many common warts and several psoriatic-like lesions developed on his arms, forearms, hands, and cheeks. These lesions were persistent in spite of treatment with retinoic acid, cimetidin, and imiquimod, which was relatively effective against the warts. Eruptions of rosacea when the patient reached the ages of 11 and 12 were treated with local metronidazole. HPV genotypes 3, 12, and 20 were identified in DNA preparations obtained from scrapings or biopsy specimens from flat wart-like lesions both by Southern blot hybridization and by the sequencing of amplicons obtained in a nested PCR, as previously described (19). At the age of 7 years, a granulomatous structure was detected in the upper left lung lobe and removed. P1 suffered from emphysema and right pneumothorax at the ages of 11 and 15 years. Attempts to control his chronic bronchial disease included intravenous IgG (polyvalent IVIG) substitution and oral trimethoprim-sulfamethoxazole (cotrimoxazole). P2 was born in 1990. At the age of 4 years, a few psoriatic lesions and flat warts appeared on her scalp and her forehead, respectively. At the age of 9 years, disseminated flat wart-like lesions spread on her face, her neck, and the back of her hands, a papilloma lesion appeared on 1 eyelid, and molluscum lesions appeared on her neck and under her arms. At the age of 14 years, a few PV-like lesions appeared on P2’s chest. All these lesions were treated with imiquimod, to which the flat warts were resistant. P2 also had gingivostomatitis at the age of 2 years. HPV 20 was detected in sections of formaldehyde-fixed, paraffin-embedded flat wart-like lesions. The histological features of the flat-wart-like lesions of both patients are shown in Supplemental Figure 1. Blood samples were collected from 7 individuals of the kindred.
Whole-exome sequencing.
Whole-exome sequencing was carried out as previously described, with the Agilent SureSelect Human All Exon 50 Mb Kit (Agilent Technologies) for exome capture. Paired-end sequencing generated 100-base reads, sequences were aligned with the hg19 build of the human genome, and all calls with a read coverage of less than 2× and a phred-scaled SNP quality of less than 20 were filtered out (57).
Immunoblot analysis.
Immunoblot analysis was carried out as previously described (23). We separated 30 μg of whole saimiri T cell lysates by 12% SDS-PAGE and then analyzed the protein bands obtained by immunoblotting.
TCR stimulation and rescue experiments in saimiri T cells.
Saimiri T cells (1 × 106) were deprived of IL-2 for 3 hours and then stimulated by incubation for 2 minutes with 100 ng/ml monoclonal anti-CD3 antibody OKT3 and crosslinking anti-mouse IgG antibody. The cells were then collected by centrifugation and the cell pellets were immediately frozen at –80°C. For subsequent immunoblot analysis, anti–phospho-ZAP70 (Cell Signaling Technology) and anti-ZAP70 (Upstate) antibodies were used at a dilution of 1:1,000. For in vitro rescue experiments, the patients’ saimiri T cells were then transduced with retroviral vectors coexpressing YFP and HA–WT RHOH. As a control, the patients’ saimiri T cells were transduced with a YFP-expressing retroviral vector, and healthy saimiri cells were transduced with either a YFP-expressing retroviral vector or a retroviral vector coexpressing YFP and HA–Y38X RHOH. YFP+ cells were sorted and used for biochemical analysis. An aliquot of sorted cells was stimulated with anti-CD3 OKT3 antibody, as described above, and immunoblot analysis of phospho- and total ZAP70 was again performed. β-Actin was used as an additional loading control. The production of HA-tagged WT RHOH protein in transduced cells was further confirmed by immunoblotting with an anti-HA antibody (data not shown).
Generation of Rhoh-deficient mice.
The generation of Rhoh–/– mice has been described elsewhere (28).
Mouse bone marrow cell transduction and transplantation.
Mouse bone marrow cells were transduced with retroviruses, as previously described (28). Briefly, the HA-tagged mutant Y38X RHOH cDNA and the HA-tagged cDNA encoding a truncated RHOH protein translated from the downstream ATG (RHOH–139-C), corresponding to the 145 amino acids at the C terminus, were inserted into mouse stem cell virus–based retroviral vectors coexpressing YFP, as previously described for WT RHOH (28). Lin–Sca1+ckit+ (LSK) bone marrow cells from Rhoh–/– mice of mixed genetic background were sorted, stimulated, and infected on fibronectin-coated plates (CH-296; Takara Bio), with a high-titer retrovirus supernatant generated in Phoenix-gp cells. Two days after transduction, YFP+ cells were sorted and transplanted, by intravenous injection, into sublethally irradiated (3 Gy with a 137Cs irradiator) Rag2–/– recipient mice. Three months after transplantation, white blood cell counts were obtained and the percentage of YFP+ T cells in the peripheral blood of the animals was determined by flow cytometry. The expression of HA-WT RHOH and HA-RHOH 139-C and the lack of detectable protein encoded by the HA–Y38X RHOH retrovirus was confirmed by immunoblot analysis.
Flow cytometry experiments.
Murine blood samples were obtained 3 months after transplantation by retroorbital puncture. Before staining, red blood cells were lysed by incubation in BD Pharm Lyse (BD Biosciences) for 10 minutes at room temperature. Cell suspensions were then incubated with the appropriate antibodies for 30 minutes in the dark at 4°C. DAPI staining was carried out to exclude dead cells. Cells were subsequently washed, resuspended in PBS supplemented with 2% FCS, and analyzed with an LSRII cytometer and FACSDiva software (BD Biosciences). Blood samples from the patients and healthy controls were collected into EDTA and stained by incubation for 20 minutes with antibodies in 1% BSA in PBS in the dark at 4°C. Red blood cells were then lysed in FACS lysis solution (0.01 M NaHCO3, 1 mM EDTA, 0.14 M NH4Cl) in 3 successive cycles of incubation for 5 minutes at room temperature in the dark. Samples were washed in 1× PBS (Gibco; Invitrogen) and resuspended in 4% paraformaldehyde in PBS for analysis on a BD FACScanto machine (BD Biosciences). Cryopreserved PBMCs were blocked by incubation for 15 minutes at 4°C with FcR-blocking reagent (MACS) in 0.5 mM EDTA, 1% BSA in PBS, and washed in 1× PBS and subsequently stained by incubation for 30 minutes at 4°C with the various antibodies against tissue-homing markers and the Aqua Live/Dead Marker (Invitrogen). Cells were then washed twice in 1× PBS and resuspended in 100 μl of fixing solution (2% formaldehyde in 1× PBS). Samples were then analyzed on an LSRII machine (BD). Invitrogen mouse beads were used for compensation. At least 10 age-matched healthy controls were tested in addition to the patients for each marker.
Thymidine incorporation assay.
PBMCs were incubated for 3 days alone or with PHA (2.5 μg/ml), the monoclonal soluble anti-CD3 antibody OKT3 (10, 25, 50 ng/ml), or PMA (10–7 or 10–8 M) plus ionomycin (10–5 or 10–6 M) or for 6 days with tetanus toxoid (0.2–0.4 μg/ml), HSV, tuberculin (5 μg/ml), or candidin (50 μg/ml). Cultures were pulsed with tritiated thymidine for the last 18 hours of the incubation period. The radioactivity incorporated was determined with a Matrix 96 beta counter (Canberra Packard). Cell proliferation was assessed by determining the cpm for [3H] thymidine incorporation, as previously described (58, 59).
Statistics.
Two-tailed Student’s t tests were used to compare (a) the percentages of CD3+ cells within the YFP+ reconstituted cell population between the various groups of mice into which cells were transplanted, (b) the percentages of the various naive and memory subsets and counts for the various tissue-homing T cell subsets between patients and healthy controls, and (c) the percentages of the various tissue-homing T cell subsets in WT and Rhoh–/– mice. Differences were considered significant if P values were less than 0.05 for all comparisons.
Study approval.
Written informed consent was obtained from all family members. All human experiments were approved by the IRB of Necker Hospital for Sick Children and the IRB of Rockefeller University. All animal procedures and experiments were approved by the Institutional Animal Care and Use Committee of Children’s Hospital Boston.
Supplementary Material
Acknowledgments
We thank the members of both branches of the Saint Giles Laboratory of Human Genetics of Infectious Diseases, in particular Shen-Ying Zhang for her very helpful suggestions; Lazaro Lorenzo for the preliminary screening of blood samples from the patients; Tatiana Kochetkov for her patience in maintaining saimiri T cells in culture; Corinne Jacques, Chantal Harre, and Stephanie N’Daga for excellent technical assistance; Svetlana Mazel, Gaëlle Breton, Dusan Bogunovic, Vanessa Bryant, and Guillaume Vogt for their suggestions regarding the flow cytometry and expression assays; Tiffany Nivare and Carolina Prando for the recruitment of healthy volunteers; and Yelena Nemirovskaya and Eric Anderson for logistical assistance. We thank Minji Byun, Sophie Cypowyj, Michael Ciancanelli, and Ruben Martínez-Barricarte for critical reading. We thank Laurence Fiette for the histologic pictures. We warmly thank the patients and their family for their participation. This work was supported by grants from INSERM, University Paris Descartes, the Rockefeller University, the Rockefeller University Clinical and Translational Science Awards (CTSA) (5UL1RR024143), the St. Giles Foundation, the National Health and Medical Research Council of Australia, the Deutsche Forschungsgemeinschaft (TR 1005/1-1), the William Lawrence and Blanche Hughes Foundation, the NIH (5R01CA113969-08), and the Akademie der Wissenschaften und der Literatur, Mainz, Germany.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(9):3239–3247. doi:10.1172/JCI62949.
Anja Troeger’s present address is: Clinic for Pediatric Oncology, Hematology and Clinical Immunology, Heinrich Heine University, Düsseldorf, Germany.
References
- 1.Lutzner MA. Epidermodysplasia verruciformis. An autosomal recessive disease characterized by viral warts and skin cancer. A model for viral oncogenesis. Bull Cancer. 1978;65(2):169–182. [PubMed] [Google Scholar]
- 2.Orth G. Host defenses against human papillomaviruses: lessons from epidermodysplasia verruciformis. Curr Top Microbiol Immunol. 2008;321:59–83. doi: 10.1007/978-3-540-75203-5_3. [DOI] [PubMed] [Google Scholar]
- 3.Cockayne E. Inherited Abnormalities Of The Skin And Its Appendages. London, United Kingdom: Oxford University Press; 1933. [Google Scholar]
- 4.Ramoz N, Rueda LA, Bouadjar B, Montoya LS, Orth G, Favre M. Mutations in two adjacent novel genes are associated with epidermodysplasia verruciformis. Nat Genet. 2002;32(4):579–581. doi: 10.1038/ng1044. [DOI] [PubMed] [Google Scholar]
- 5.Alcais A, et al. Life-threatening infectious diseases of childhood: single-gene inborn errors of immunity? Ann N Y Acad Sci. 2010;1214:18–33. doi: 10.1111/j.1749-6632.2010.05834.x. [DOI] [PubMed] [Google Scholar]
- 6.Casanova JL, Abel L. Primary immunodeficiencies: a field in its infancy. Science. 2007;317(5838):617–619. doi: 10.1126/science.1142963. [DOI] [PubMed] [Google Scholar]
- 7.Casanova JL, Abel L. Inborn errors of immunity to infection: the rule rather than the exception. J Exp Med. 2005;202(2):197–201. doi: 10.1084/jem.20050854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alcais A, Abel L, Casanova JL. Human genetics of infections diseases: between proof of principle and paradigm. J Clin Invest. 2009;119(9):2506–2514. doi: 10.1172/JCI38111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aochi S, et al. A novel homozygous mutation of the EVER1/TMC6 gene in a Japanese patient with epidermodysplasia verruciformis. . Br J Dermatol. 2007;157(6):1265–1266. doi: 10.1111/j.1365-2133.2007.08206.x. [DOI] [PubMed] [Google Scholar]
- 10.Berthelot C, et al. Treatment of a patient with epidermodysplasia verruciformis carrying a novel EVER2 mutation with imiquimod. . J Am Acad Dermatol. 2007;56(5):882–886. doi: 10.1016/j.jaad.2007.01.036. [DOI] [PubMed] [Google Scholar]
- 11.Gober MD, Rady PL, He Q, Tucker SB, Tyring SK, Gaspari AA. Novel homozygous frameshift mutation of EVER1 gene in an epidermodysplasia verruciformis patient. . J Invest Dermatol. 2007;127(4):817–820. doi: 10.1038/sj.jid.5700641. [DOI] [PubMed] [Google Scholar]
- 12.Zuo YG, Ma D, Zhang Y, Qiao J, Wang B. Identification of a novel mutation and a genetic polymorphism of EVER1 gene in two families with epidermodysplasia verruciformis. . J Dermatol Sci. 2006;44(3):153–159. doi: 10.1016/j.jdermsci.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 13.Sun XK, Chen JF, Xu AE. A homozygous nonsense mutation in the EVER2 gene leads to epidermodysplasia verruciformis. Clin Exp Dermatol. 2005;30(5):573–574. doi: 10.1111/j.1365-2230.2005.01858.x. [DOI] [PubMed] [Google Scholar]
- 14.Tate G, Suzuki T, Kishimoto K, Mitsuya T. Novel mutations of EVER1/TMC6 gene in a Japanese patient with epidermodysplasia verruciformis. . J Hum Genet. 2004;49(4):223–225. doi: 10.1007/s10038-004-0135-6. [DOI] [PubMed] [Google Scholar]
- 15.Orth G. Genetics of epidermodysplasia verruciformis: Insights into host defense against papillomaviruses. Semin Immunol. 2006;18(6):362–374. doi: 10.1016/j.smim.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 16.Rady PL, et al. Novel homozygous nonsense TMC8 mutation detected in patients with epidermodysplasia verruciformis from a Brazilian family. Br J Dermatol. 2007;157(4):831–833. doi: 10.1111/j.1365-2133.2007.08123.x. [DOI] [PubMed] [Google Scholar]
- 17.Keresztes G, Mutai H, Heller S. TMC and EVER genes belong to a larger novel family, the TM gene family encoding transmembrane proteins. BMC Genomics. 2003;4(1):24. doi: 10.1186/1471-2164-4-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lazarczyk M, Pons C, Mendoza JA, Cassonnet P, Jacob Y, Favre M. Regulation of cellular zinc balance as a potential mechanism of EVER-mediated protection against pathogenesis by cutaneous oncogenic human papillomaviruses. J Exp Med. 2008;205(1):35–42. doi: 10.1084/jem.20071311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramoz N, et al. Evidence for a nonallelic heterogeneity of epidermodysplasia verruciformis with two susceptibility loci mapped to chromosome regions 2p21-p24 and 17q25. J Invest Dermatol. 2000;114(6):1148–1153. doi: 10.1046/j.1523-1747.2000.00996.x. [DOI] [PubMed] [Google Scholar]
- 20.Tybulewicz VL, Henderson RB. Rho family GTPases and their regulators in lymphocytes. Nat Rev Immunol. 2009;9(9):630–644. doi: 10.1038/nri2606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinashi T. Intracellular signalling controlling integrin activation in lymphocytes. Nat Rev Immunol. 2005;5(7):546–559. doi: 10.1038/nri1646. [DOI] [PubMed] [Google Scholar]
- 22.Fueller F, Kubatzky KF. The small GTPase RhoH is an atypical regulator of haematopoietic cells. Cell Commun Signal. 2008;6:6. doi: 10.1186/1478-811X-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Preudhomme C, et al. Nonrandom 4p13 rearrangements of the RhoH/TTF gene, encoding a GTP-binding protein, in non-Hodgkin’s lymphoma and multiple myeloma. Oncogene. 2000;19(16):2023–2032. doi: 10.1038/sj.onc.1203521. [DOI] [PubMed] [Google Scholar]
- 24.Pasqualucci L, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412(6844):341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 25.Hiraga J, et al. Prognostic analysis of aberrant somatic hypermutation of RhoH gene in diffuse large B cell lymphoma. Leukemia. 2007;21(8):1846–1847. doi: 10.1038/sj.leu.2404717. [DOI] [PubMed] [Google Scholar]
- 26.Gaidano G, et al. Aberrant somatic hypermutation in multiple subtypes of AIDS-associated non-Hodgkin lymphoma. Blood. 2003;102(5):1833–1841. doi: 10.1182/blood-2002-11-3606. [DOI] [PubMed] [Google Scholar]
- 27.Gineau L, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest. 2012;122(3):821–832. doi: 10.1172/JCI61014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gu Y, Chae HD, Siefring JE, Jasti AC, Hildeman DA, Williams DA. RhoH GTPase recruits and activates Zap70 required for T cell receptor signaling and thymocyte development. Nat Immunol. 2006;7(11):1182–1190. doi: 10.1038/ni1396. [DOI] [PubMed] [Google Scholar]
- 29.Chae HD, Siefring JE, Hildeman DA, Gu Y, Williams DA. RhoH regulates subcellular localization of ZAP-70 and Lck in T cell receptor signaling. PLoS One. 2010;5(11):e13970. doi: 10.1371/journal.pone.0013970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gu Y, Jasti AC, Jansen M, Siefring JE. RhoH, a hematopoietic-specific Rho GTPase, regulates proliferation, survival, migration, and engraftment of hematopoietic progenitor cells. Blood. 2005;105(4):1467–1475. doi: 10.1182/blood-2004-04-1604. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Bu X, Lu B, Avraham H, Flavell RA, Lim B. The hematopoiesis-specific GTP-binding protein RhoH is GTPase deficient and modulates activities of other Rho GTPases by an inhibitory function. Mol Cell Biol. 2002;22(4):1158–1171. doi: 10.1128/MCB.22.4.1158-1171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Randall KL, et al. DOCK8 deficiency impairs CD8 T cell survival and function in humans and mice. J Exp Med. 2011;208(11):2305–2320. doi: 10.1084/jem.20110345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Appay V, van Lier RA, Sallusto F, Roederer M. Phenotype and function of human T lymphocyte subsets: consensus and issues. Cytometry A. 2008;73(11):975–983. doi: 10.1002/cyto.a.20643. [DOI] [PubMed] [Google Scholar]
- 34.Dorn T, et al. RhoH is important for positive thymocyte selection and T-cell receptor signaling. Blood. 2007;109(6):2346–2355. doi: 10.1182/blood-2006-04-019034. [DOI] [PubMed] [Google Scholar]
- 35.Wang H, Zeng X, Fan Z, Lim B. RhoH modulates pre-TCR and TCR signalling by regulating LCK. Cell Signal. 2011;23(1):249–258. doi: 10.1016/j.cellsig.2010.09.009. [DOI] [PubMed] [Google Scholar]
- 36.O’Flynn K, et al. Different pathways of human T-cell activation revealed by PHA-P and PHA-M. Immunology. 1986;57(1):55–60. [PMC free article] [PubMed] [Google Scholar]
- 37.Kupper TS, Fuhlbrigge RC. Immune surveillance in the skin: mechanisms and clinical consequences. Nat Rev Immunol. 2004;4(3):211–222. doi: 10.1038/nri1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenkinson SE, Whawell SA, Swales BM, Corps EM, Kilshaw PJ, Farthing PM. The alphaE(CD103)beta7 integrin interacts with oral and skin keratinocytes in an E-cadherin-independent manner*. Immunology. 2011;132(2):188–196. doi: 10.1111/j.1365-2567.2010.03352.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Daryadel A, et al. RhoH/TTF negatively regulates leukotriene production in neutrophils. J Immunol. 2009;182(10):6527–6532. doi: 10.4049/jimmunol.0803846. [DOI] [PubMed] [Google Scholar]
- 40.Oda H, et al. RhoH plays critical roles in Fc epsilon RI-dependent signal transduction in mast cells. J Immunol. 2009;182(2):957–962. doi: 10.4049/jimmunol.182.2.957. [DOI] [PubMed] [Google Scholar]
- 41.Klein C. Genetic defects in severe congenital neutropenia: emerging insights into life and death of human neutrophil granulocytes. Annu Rev Immunol. 2011;29:399–413. doi: 10.1146/annurev-immunol-030409-101259. [DOI] [PubMed] [Google Scholar]
- 42.Song E, Jaishankar GB, Saleh H, Jithpratuk W, Sahni R, Krishnaswamy G. Chronic granulomatous disease: a review of the infectious and inflammatory complications. Clin Mol Allergy. 2011;9(1):10. doi: 10.1186/1476-7961-9-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aiba S, Rokugo M, Tagami H. lmmunohistologic analysis of the phenomenon of spontaneous regression of numerous flat warts. Cancer. 1986;58(6):1246–1251. doi: 10.1002/1097-0142(19860915)58:6<1246::AID-CNCR2820580612>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 44.Porubsky S, et al. Rhoh deficiency reduces peripheral T-cell function and attenuates allogenic transplant rejection. Eur J Immunol. 2010;41(1):76–88. doi: 10.1002/eji.201040420. [DOI] [PubMed] [Google Scholar]
- 45.Al-Herz W, et al. Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nehme NT, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T cells survival. Blood. 2012;119(15):3458–3468. doi: 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abdollahpour H, et al. The phenotype of human STK4 deficiency. Blood. 2012;119(15):3450–3457. doi: 10.1182/blood-2011-09-378158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Su HC. Dedicator of cytokinesis 8 (DOCK8) deficiency. Curr Opin Allergy Clin Immunol. 2010;10(6):515–520. doi: 10.1097/ACI.0b013e32833fd718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacheco KA, Tarkowski M, Klemm J, Rosenwasser LJ. CD49d expression and function on allergen-stimulated T cells from blood and airway. Am J Respir Cell Mol Biol. 1998;18(2):286–293. doi: 10.1165/ajrcmb.18.2.2687. [DOI] [PubMed] [Google Scholar]
- 50.Gorfu G, Rivera-Nieves J, Ley K. Role of beta7 integrins in intestinal lymphocyte homing and retention. Curr Mol Med. 2009;9(7):836–850. doi: 10.2174/156652409789105525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salmi M, Jalkanen S. Lymphocyte homing to the gut: attraction, adhesion, and commitment. Immunol Rev. 2005;206:100–113. doi: 10.1111/j.0105-2896.2005.00285.x. [DOI] [PubMed] [Google Scholar]
- 52.Schechner JS, Edelson RL, McNiff JM, Heald PW, Pober JS. Integrins alpha4beta7 and alphaEbeta7 are expressed on epidermotropic T cells in cutaneous T cell lymphoma and spongiotic dermatitis. Lab Invest. 1999;79(5):601–607. [PubMed] [Google Scholar]
- 53.Ohmatsu H, et al. alpha4beta7 Integrin is essential for contact hypersensitivity by regulating migration of T cells to skin. J Allergy Clin Immunol. 2010;126(6):1267–1276. doi: 10.1016/j.jaci.2010.08.048. [DOI] [PubMed] [Google Scholar]
- 54.Lazarczyk M, Cassonnet P, Pons C, Jacob Y, Favre M. The EVER proteins as a natural barrier against papillomaviruses: a new insight into the pathogenesis of human papillomavirus infections. Microbiol Mol Biol Rev. 2009;73(2):348–370. doi: 10.1128/MMBR.00033-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Laffort C, et al. Severe cutaneous papillomavirus disease after haemopoietic stem-cell transplantation in patients with severe combined immune deficiency caused by common gammac cytokine receptor subunit or JAK-3 deficiency. Lancet. 2004;363(9426):2051–2054. doi: 10.1016/S0140-6736(04)16457-X. [DOI] [PubMed] [Google Scholar]
- 56.Yu M, et al. Regulation of T cell receptor signaling by activation-induced zinc influx. J Exp Med. 2011;208(4):775–785. doi: 10.1084/jem.20100031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Byun M, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med. 2010;207(11):2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.de Villartay JP, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. 2005;115(11):3291–3299. doi: 10.1172/JCI25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de Saint Basile G, et al. Severe combined immunodeficiency caused by deficiency in either the delta or the epsilon subunit of CD3. J Clin Invest. 2004;114(10):1512–1517. doi: 10.1172/JCI22588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kassu A, et al. Distribution of lymphocyte subsets in healthy human immunodeficiency virus-negative adult Ethiopians from two geographic locales. Clin Diagn Lab Immunol. 2001;8(6):1171–1176. doi: 10.1128/CDLI.8.6.1171-1176.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bisset LR, Lung TL, Kaelin M, Ludwig E, Dubs RW. Reference values for peripheral blood lymphocyte phenotypes applicable to the healthy adult population in Switzerland. Eur J Haematol. 2004;72(3):203–212. doi: 10.1046/j.0902-4441.2003.00199.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.