

Abstract
Purpose.
This study sought to determine factors involved in nuclear factor erythroid 2–related factor 2 (Nrf2) regulation and their response to oxidative stress in Fuchs endothelial corneal dystrophy (FECD) and normal corneal endothelial cells (CECs).
Methods.
FECD corneal buttons were obtained from transplantations and normal human corneas from tissue banks. Oxidative stress was induced by tert-butyl hydroperoxide (tBHP). Protein and mRNA levels of Nrf2, DJ-1, p53, and Kelch-like ECH-associated protein1 (Keap1) were investigated using Western blotting and real-time PCR. Immunoprecipitation was used to detect levels of oxidized DJ-1 protein and Cullin 3- (Cul3)–regulated degradation of DJ-1 in immortalized FECD (FECDi) and normal CEC (HCECi) cell lines. Nrf2 subcellular localization was assessed by immunocytochemistry.
Results.
Nrf2 protein stabilizer, DJ-1, decreased significantly in FECD CECs compared with normal, whereas Nrf2 protein repressor, Keap1, was unchanged at baseline but increased under oxidative stress. Under oxidative stress, normal CECs upregulated DJ-1 protein synthesis, whereas FECD CECs did not. DJ-1 decline correlated with increased DJ-1 oxidative modification and carbonylation in FECDi as compared with HCECi. Increased labeling of immunoprecipitated DJ-1 protein with anti-Cul3 antibody indicated enhanced DJ-1 degradation in FECDi as compared with HCECi. Following tBHP treatment, Nrf2 translocated from cytoplasm to nuclei in normal CECs, whereas Nrf2 nuclear localization was not observed in FECD.
Conclusions.
Decreased levels of DJ-1 in FECD at baseline and under oxidative stress correlate with impaired Nrf2 nuclear translocation and heightened cell susceptibility to apoptosis. Targeting the DJ-1/Nrf2 axis could yield a mechanism to slow CEC degeneration in FECD.
Decreased levels of DJ-1 in FECD at baseline and under oxidative stress correlate with impaired Nrf2 nuclear translocation and heightened cell susceptibility to apoptosis.
Introduction
Fuchs endothelial corneal dystrophy (FECD) is the most common cause of endogenous corneal endothelial degeneration and can lead to severe impairment of vision from corneal edema.1 In the patients with FECD, the first clinical sign is the formation of corneal guttae and abnormal deposition of extracellular matrix, followed by the decrease of the endothelial cell density observed in the middle of the disease. The only available treatment modality for this disease is corneal transplantation. A better understanding of FECD pathogenesis is key to preventing or arresting the degeneration of corneal endothelial cells (CECs). Recently, different genetic variations have been associated with FECD,2,3 and different pathogenic processes are now being explored—all implicating increased stress leading to apoptosis of CECs.4,5
Oxidative stress has been shown to play a major role in the chronic degenerative process of corneal endothelium and CEC apoptosis seen in FECD.6–8 In a previous study,6 a higher than normal level of oxidative DNA damage marker 8-OHdG, as well as decreased expression of multiple antioxidants such as peroxiredoxins, thioredoxin reductase, metallothionein, and superoxide dismutases, were detected in FECD CECs, which led to oxidant–antioxidant imbalance. The downregulated antioxidants contained an antioxidant responsive element (ARE) in their proximal promoter regions and were ARE-dependent for their activation by the nuclear factor erythroid 2–related factor 2 (Nrf2) transcription factor. Previously, we identified a decrease in Nrf2 protein levels and transcriptional downregulation of heme oxygenase-1 (HMOX-1), one of the major Nrf2-regulated genes, in FECD endothelium, indicating that suboptimal Nrf2-regulated defense leads to the transcriptional downregulation of the antioxidants and the resulting oxidant–antioxidant imbalance in FECD.6
Numerous studies have shown that Nrf2 is a multiorgan protector and a master transcription regulator of cellular defense against oxidative stress.9 It is a member of the cap'n'collar family of basic leucine zipper transcription factors,10 which, upon oxidative stress, translocates from the cytoplasm to the nucleus and induces constitutive expression of antioxidant genes.11 Nrf2 level and activity in a cell can be regulated at several points: transcription, degradation, translocation, and protein stabilization. The transcription of the Nrf2 gene is regulated by a positive feedback loop through the ARE element in the promoter.12,13 Nrf2 degradation is controlled by cytoskeleton-associated Kelch-like ECH-associated protein 1 (Keap1), which, by binding Nrf2, sequesters and degrades it in the cytoplasm via a ubiquitin-dependent pathway, which mainly involves Cullin-based E3 ligases (Cul3).14 The cytoplasmic stability and eventual translocation of Nrf2 to the nucleus is controlled by one of its stabilizers, DJ-1.15 DJ-1 is a ubiquitous and multifunctional protein. In addition to being a transcriptional coactivator of Nrf2, it is suggested to function as a redox-regulated chaperone,16 an atypical peroxiredoxin-like peroxidase,17 a protector against UV-induced cell death,18 and a mitochondrial stabilizer.19 DJ-1 was first identified as an oncogene,20 and subsequent studies have shown its role in the pathogenesis of neurodegenerative disorders.21 Alteration of DJ-1 has been linked to Parkinson's disease (PD) as well as to Alzheimer's disease (AD); it is even considered to be a biomarker for PD.22
Under baseline conditions, Nrf2 is degraded in the cytoplasm and is transcriptionally inactive. Under oxidative stress, reactive oxygen species (ROS) induce modifications to the Nrf2–Keap1 complex that lead to Nrf2 dissociation from Keap1 and enable Nrf2 translocation to the nucleus to activate ARE. Nuclear translocation of Nrf2 is one of the most essential steps in gene-inducing and cytoprotective functions of Nrf2. Even though our preliminary studies point to the deficiency of the Nrf2-regulated pathway in FECD pathogenesis, we do not know the exact molecular mechanisms that induce Nrf2 decrease and the deficient Nrf2-regulated antioxidant defense.
In this study, we sought to investigate the mechanism of Nrf2 regulation in normal and FECD CECs in response to oxidative stress. We analyzed the levels of the main factors known to interact with Nrf2 protein and to affect its cytoplasmic and nuclear levels. This study presents evidence that in FECD, Nrf2 protein stability and ability to translocate to the nucleus are likely decreased due to a decrease in levels of its cytoplasmic stabilizer DJ-1. These findings point to the roles of DJ-1 and Nrf2 in the development of FECD and present potential therapeutic targets for decreasing endothelial susceptibility to oxidative stress.
Materials and Methods
Human Tissue
This study was conducted in accordance with the tenets of the Declaration of Helsinki and was approved by both Massachusetts Eye and Ear and Schepens Eye Research Institute Institutional Review Boards. Written, informed consent was obtained from patients undergoing corneal transplantation for FECD. After surgical removal, corneal specimens were immediately placed in corneal storage medium (Optisol-GS; Bausch & Lomb; Rochester, NY) at 4°C. Fresh, normal human corneal buttons were obtained from Tissue Banks International (Baltimore, MD) and National Disease Research Interchange (Philadelphia, PA) and used as controls. We used previously published criteria to determine donor tissue suitability.23 Descemet membrane (DM) with the corneal endothelial cells (DM-CECs) was dissected from the stroma of the corneal buttons under a dissecting microscope for use in the different experiments. The Table presents information regarding the tissue samples used. Normal donors were sex- and decade-matched with FECD CEC donors.
Table. .
Cornea Donor Information
|
|
Normal |
FECD |
| Age, y* | 66 ± 7 | 69 ± 12 |
| Sex (F/M) | 19/9 | 22/7 |
| Preservation time, days | 8.1 | 2.5 |
Average age in years with SD.
Real-Time Reverse Transcription–Polymerase Chain Reaction (Real-Time RT-PCR)
Total RNA was extracted from normal and FECD CEC-DM complexes using a commercial reagent (TRIzol; Invitrogen, Carlsbad, CA). Total RNA was extracted from HCECi 72 hours posttransfection (RNeasy Plus Mini Kit; Qiagen, Valencia, CA).
cDNA was prepared by reverse transcription with a commercially available kit (Promega, Madison, WI). Primers and probes for Nrf2, Keap1, HMOX-1, and DJ-1, as well as for the endogenous control β2-microglobulin (β2-MG), were commercially obtained (TaqMan; Applied Biosystems, Foster City, CA). Real-time PCR reactions were run on commercial detection systems (ABI Prism model 7900 HT sequence; Applied Biosystems; and Mastercycler ep realplex; Eppendorf AG, Hamburg, Germany). For data analysis, the comparative threshold cycle (CT) method was used, as previously established. Results were presented as the average, relative mRNA expression of the different genes normalized to β2-MG. SEM values were calculated.
Induction of Oxidative Stress
Oxidative stress was induced by incubating normal and FECD CEC-DM in tert-butyl hydroperoxide (tBHP; Sigma-Aldrich, St. Louis, MO), diluted in serum-free low-glucose Dulbecco's modified Eagle's medium (DMEM; Invitrogen) to a final concentration of 500 μM for 4 hours at room temperature. Controls were incubated in DMEM alone for 4 hours at 37°C without tBHP. This protocol was adopted based on previous experiments.7
Immunocytochemistry
Immunocytochemistry was performed on tissue samples as previously described.7 Briefly, after fixation, tissues were permeabilized and blocked in 2% bovine serum albumin. Tissues were stained with anti-Nrf2 (H-300) (1:100; Santa Cruz Biotechnology, Santa Cruz, CA) and incubated with secondary antibody FITC-conjugated donkey anti-rabbit IgG (1:100, Jackson ImmunoResearch Laboratories, West Grove, PA). TO-PRO-3 iodide (Molecular Probes, Eugene, OR) was used to stain the nuclei. Digital images were obtained using a spectral photometric confocal microscope (DM6000S with LCS 1.3.1 software; Leica, Solms, Germany).
Western Blot Analysis
Whole cell extracts were lyzed with the protein extraction buffer ER3 (Biorad, Hercules, CA) and 1 mM tributyl phosphine. Proteins were loaded onto 10% Bis-Tris NuPAGE gels (Invitrogen). Peptides were transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA) and nonspecific binding was blocked with 5% dry nonfat milk in PBS for 1 hour. Membranes were incubated overnight at 4°C with mouse monoclonal anti-Keap1 (R&D Systems, Minneapolis, MN) diluted 1:500, and goat polyclonal anti-DJ-1 (Novus Biologicals, Littleton, CO) diluted 1:1000. Mouse anti-β-actin (1:4000; Sigma-Aldrich) was used to normalize protein loading. Blots were rinsed, reblocked, and exposed for 1 hour to horseradish peroxidase (HRP)–conjugated goat anti-mouse IgG, 1:1500 for Keap1 blots, and HRP-conjugated donkey anti-goat IgG, 1:2500 for DJ-1 blots. After washing in 0.1% Triton X-100, antibody binding was detected with a chemiluminescent substrate (Thermo Scientific, Pittsburgh, PA). Densitometry was analyzed with ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD; available at http://rsb.info.nih.gov/ij/index.html), and protein content was normalized relative to β-actin content. Experiments were repeated a minimum of three times. Results were averaged and SEM values were calculated.
Immunoprecipitation and Analysis of DJ-1 Oxidative Modifications
Immortalized normal and FECD human corneal endothelial cell lines (HCECi and FECDi, respectively) were cultured in Chen's medium, as previously described.7,24,25 When cells reached confluence, culture medium was removed and cell lysis buffer (Cell Signaling Technology, Danvers, MA) containing a protease and phosphatase inhibitor cocktail (Thermo Scientific) was added. The supernatant was prepared by sonication of cells on ice and centrifugation at 14,000g at 4°C for 10 minutes and collected for protein assay (BCA; Thermo Scientific). The supernatant was used for Western blot analysis with anti-DJ-1 antibody (1:1000; Novus Biologicals) and anti-β-actin (1:5000; Sigma-Aldrich) as described above.
Anti-DJ-1 antibody (3 μg) (Novus Biologicals) was added to 0.6 mg of HECEi and FECDi cell lysates. The immunocomplexes were incubated by gentle rocking overnight at 4°C and then added to prewashed protein G magnetic beads (Cell Signaling Technology) for 30 minutes at room temperature. Pellets were collected after placing the samples in the magnetic separation rack for 30 seconds and dissolved in SDS sample buffer by heating for 5 minutes at 95°C. The supernatants were collected for Western blot analysis with anti-oxidized DJ-1 antibody (1:50; AbD Serotec, Oxford, UK) followed by incubation with HRP-conjugated secondary antibody (1:350, mouse anti C-MYC; AbD Serotec) as above. Anti-DNP antibody was used to detect carbonyl modification of DJ-1 according to the manufacturer's instructions (OxiSelect Protein Carbonyl Immunoblot Kit; Cell Biolabs, San Diego, CA). Briefly, transblotted membrane was prederivatized with dinitrophenolhydrazine followed by incubation with anti-DNP (1:1000) and HRP-conjugated secondary antibody (1:1000). Cul3 and DJ-1 interaction was detected using anti-Cul-3 antibody (1:100; Santa Cruz Biotechnology) and HRP-conjugated secondary antibody (1:2500) after the immunoprecipitation experiment, as described above.
DJ-1 siRNA Studies
HCECi cells were seeded in two-well chamber slides with a density of 1 × 105 cells per well and transfected with 50 nM of DJ-1 siRNA (Santa Cruz Biotechnology) using a commercial transfection reagent (Lipofectamine 2000; Invitrogen). Scrambled siRNA (Santa Cruz Biotechnology) was used as a control. At 72 hours posttransfection, cells were fixed and immunohistochemistry with anti-Nrf2 antibody was performed as described above. Images were acquired by a fluorescence microscope (Axioscope Mot 2; Carl Zeiss Meditec, Inc, Jena, Germany). DJ-1 knockdown efficiency was confirmed with Western blotting with anti-DJ-1 antibody as previously described.
Results
Downregulation of Nrf2 in FECD Does Not Occur at the Transcriptional Level
Our previous study identified a significantly decreased protein level of Nrf2 in FECD CECs as compared with normal.6 In this study, real-time PCR was performed first to determine whether transcription of Nrf2 and its regulators Keap1 and DJ-1 were altered in FECD to account for diminished protein levels. No difference was detected in levels of Nrf2 mRNA (n = 12, P = 0.17) and Keap1 mRNA (n = 6, P = 0.72) between normal and FECD CECs (Fig. 1). Interestingly, significant underexpression of DJ-1 mRNA in FECD CECs was detected as compared with normal. DJ-1 mRNA level was decreased 2.2-fold in FECD CECs as compared with normal (n = 12, P = 0.005) (Fig. 1). In correlation with mRNA levels, a significant reduction in DJ-1 protein synthesis was detected in FECD CECs as measured by Western blot. DJ-1 protein level was 4.2-fold lower in FECD CECs as compared with normal (n = 5, P = 0.0008) (Fig. 2). Since there was a decline in Nrf2 protein and its major transcriptional targets in FECD, the decrease in DJ-1 levels was indicative of potential deficiency in Nrf2 protein stability.
Figure 1. .

Relative mRNA expression of Nrf2 and its regulators Keap1 and DJ-1. Real-time PCR analysis of mRNA extracted from normal donor corneal endothelium and from age- and sex-matched FECD specimens obtained at time of keratoplasty. Results were expressed as fold-changes and were normalized to β2-microglobulin. Data showed transcriptional downregulation of DJ-1 in FECD, whereas Nrf2 and Keap1 mRNA were unaffected. Data are represented as mean ± SEM. *P < 0.05.
Figure 2. .

Decreased protein level of DJ-1. Left: representative bands of Western blot analysis of normal CECs and age- and sex-matched FECD specimens. β-Actin was used for normalization of protein loading. Jurkat cell lysate was used as a positive control. Right: averaged densitometric analysis showed a 4-fold decrease in DJ-1 level in FECD as compared with normal CECs. Data are represented as mean ± SEM. *P < 0.05.
Lack of Upregulation of DJ-1 in FECD CECs in Response to Oxidative Stress
Previously we showed that tBHP induces higher levels of oxidative DNA damage and apoptosis in FECD versus normal CEC specimens ex vivo.7 To determine whether increased susceptibility to oxidative stress in FECD stems from dysregulation of the antioxidant defense, ex vivo FECD and normal CECs were treated with tBHP, and DJ-1 and Keap1 levels were analyzed. In normal CECs, oxidative stress upregulated DJ-1 protein levels (n = 6, P = 0.03), unlike in FECD CECs, which showed unchanged levels of DJ-1 protein when treated and untreated conditions were compared (n = 6, P = 1.0) (Fig. 3A). FECD CECs exhibited significantly lower levels of DJ-1 in both untreated (P = 0.0014) and treated (P = 3.35E-06) conditions as compared with normal CECs.
Figure 3. .

Response of DJ-1 and Keap1 to oxidative stress. (A) Representative bands of Western blot analysis of DJ-1 levels in FECD CECs and normal control after incubation in 0 or 500 μM of tBHP. Averaged densitometric analysis showed an increase in DJ-1 level in response to oxidative stress in normal corneal endothelium but not in FECD. β-Actin was used for normalization of protein loading. (B) Representative bands of Western blot analysis of Keap1 levels in FECD CECs and normal control after incubation in 0 or 500 μM of tBHP. Averaged densitometric analysis detected increase in Keap1 levels in FECD as compared with normal CECs. β-Actin was used for normalization of protein loading. Data are represented as mean ± SEM. *P < 0.05.
There was an increase in Keap1 protein levels in FECD CECs as compared with normal CECs in both untreated (n = 6, P = 0.0279) and treated (n = 6, P = 0.0018) conditions (Fig. 3B). The treatment with prooxidants did not lead to a detectable change in Keap1 synthesis in either normal or FECD specimens.
Oxidative Modification and Degradation of DJ-1 Protein in FECD
It has been shown that FECDi display a higher release of ROS and have a heightened susceptibility to oxidative stress–induced apoptosis when compared with HCECi, replicating the differences seen in primary tissue samples.7 It has also been shown that, in pathologic conditions, oxidative stress–induced modifications of DJ-1 may destabilize and decrease functional DJ-1 protein.15,26–30 To assess the level of DJ-1 protein in HCECi and FCEDi, cell lysates were subjected to Western blot analysis. There was a decrease in DJ-1 protein in FECDi as compared with HCECi. To further investigate whether oxidative modification of DJ-1 causes enhanced DJ-1 degradation that in turn leads to decreased cellular levels, DJ-1 protein was immunoprecipitated (IP) with anti-DJ-1 antibody from cell lysates, followed by immunoblotting with antioxidized DJ-1 antibody, which specifically recognizes oxidized cysteine residue 106 of DJ-1. DJ-1 exists as a monomer at a molecular weight of 20 kDa, but oxidatively modified DJ-1 exists as a dimer with molecular mass weight of 40 kDa. Decreased levels of DJ-1 in FECDi, as compared with HCECi, correlated with a greater level of oxidized DJ-1 by IP analysis in FECDi (Fig. 4A). Similarly, FECDi exhibited an increase in carbonylation of DJ-1 protein as compared with HCECi (Fig. 4A).
Figure 4. .
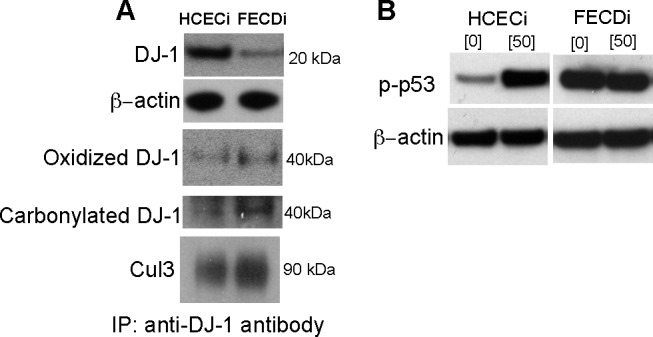
DJ-1 protein oxidative modification and degradation. Western blot and immunoprecipitation (IP)–Western blot analysis of cell lysates from HCECi and FECDi. Cell lysates were immunoprecipitated with anti-DJ-1 antibody. (A) The level of DJ-1 protein in HCECi and FECDi was compared by Western blotting. β-Actin was used for normalization of protein loading. Oxidized DJ-1 and carbonylated DJ-1 in HCECi and FECDi were IP-Western analyzed with anti-oxidized DJ-1 antibody and anti-2,4-dinitrophenol (DNP) antibody, respectively. Anti-Cul3 antibody was used for detection of DJ-1 degradation. (B) Phospho-p53 level was compared in HCECi and FECDi after 1 hour of 50 μm tBHP treatment. β-Actin was used for normalization of protein loading.
To determine the interaction of DJ-1 with Cul3, an immunoprecipitation experiment with anti-DJ-1 was performed and showed increased labeling with anti-Cul3 in FECDi as compared with HCECi cells (Fig. 4A). In addition, we determined that, similarly to native tissue samples, FECDi exhibited increased phospho-p53 levels as compared with HCECi, indicating that a decline in DJ-1 and increased degradation of DJ-1 might be correlated with this heightened sensitivity of CECs to p53-mediated apoptosis in FECD (Fig. 4B).
Lack of Nrf2 Movement and Localization in Nuclei in FECD CECs in Response to Oxidative Stress
Since DJ-1 has been shown to be involved in cellular stabilization of Nrf2 and “enhancement” of its nuclear localization, which is essential for transcriptional activation, FECD and normal specimens were investigated for nuclear localization of Nrf2 under baseline and stressed conditions. To investigate endothelial cell response to oxidative stress, localization of Nrf2 was compared between the CECs of normal and FECD donors. Both normal CECs and FECD CECs were subjected to 0.5 mM tBHP for 4 hours, and indirect immunofluorescence was performed to examine the localization of Nrf2 in CECs. Figure 5 presents confocal images of normal untreated (top row) and normal treated with tBHP (bottom row). In normal CECs, Nrf2 was primarily localized in the cytoplasm; but after treatment with tBHP, there was an accumulation of Nrf2 in the nuclei, suggesting Nrf2 movement from the cytoplasm to the nuclei in response to oxidative stress. Figure 6 shows confocal images of FECD CECs (top row) and FECD CECs treated with tBHP (bottom row). Compared with normal CECs, in FECD, diffuse cytoplasmic staining with Nrf2 was detected in the majority of cells without movement into the nuclei after treatment with tBHP. Negative controls consisted of normal and FECD CECs incubated with secondary antibody alone.
Figure 5. .

Nrf2 movement and localization in nuclei in normal endothelium in response to oxidative stress. Representative confocal images of normal corneal endothelial cell whole mounts are from normal (top row), normal treated with tBHP (bottom row) specimens. Cytoplasmic localization of Nrf2 (green) was present in normal endothelium before treatment with tBHP and nuclear localization after treatment with tBHP. TOPRO-3 was used for nuclei staining (blue). Images of negative controls incubated with only secondary antibody are shown in the right column. Original magnification was ×400 with 2× zoom.
Figure 6. .

Lack of Nrf2 translocation to the nuclei in FECD in response to oxidative stress. Representative confocal images of FECD corneal endothelial cell whole mounts at baseline (top row) and after tBHP treatment (bottom row). Diffuse but scant cytoplasmic staining of Nrf2 (green) in FECD was detected prior to and after treatment with tBHP. TOPRO-3 was used for nuclei staining (blue). Images of negative controls incubated with only secondary antibody are shown in the right column. Original magnification ×400 with 2× zoom.
To investigate the effect of DJ-1 downregulation on Nrf2 nuclear localization, HCECi cells were treated with DJ-1 siRNA and scrambled siRNA as control. After 72 hours of siRNA transfection, Western blot analysis revealed significant DJ-1 protein and mRNA downregulation in DJ-1 siRNA-treated cells as compared with cells treated with scrambled siRNA (Figs. 7A, 7C). Meanwhile, immunochemistry analysis detected decreased intensity of Nrf2 nuclear staining in DJ-1 siRNA-treated cells as compared with scrambled siRNA treated or untreated cells (Fig. 7B). In addition to decreased translocation of Nrf2 to the nucleus, there was a notable depletion in the expression of one of the major Nrf2 target genes, HMOX-1, in DJ-1 siRNA-treated cells as compared with scrambled siRNA treated or untreated cells (P = 0.002) (Fig. 7C).
Figure 7. .

Effect of DJ-1 downregulation on Nrf2 localization. (A) Western blot analysis of DJ-1 levels in HCECi cells transfected with DJ-1 siRNA and scrambled siRNA. β-Actin was used for normalization of protein loading. (B) Representative fluorescence microscopy images of Nrf2 localization in HCECi cells transfected with DJ-1 siRNA and scrambled siRNA. Diffuse cytoplasmic and nuclear staining of Nrf2 (green) is detected in cytoplasm and nuclei of no siRNA and scrambled siRNA samples. Lower intensity of Nrf2 nuclear staining is present in DJ-1 siRNA samples. DAPI was used for nuclei staining (blue). Original magnification was ×400. (C) Real-time PCR analysis of mRNA extracted from no siRNA treated, scrambled siRNA treated, and DJ-1 siRNA-treated HCECi. Results were expressed as fold-changes and were normalized to β2-microglobulin. Data showed transcriptional downregulation of DJ-1 and HMOX-1 in DJ-1 siRNA-treated HCECi as compared with no siRNA treated controls. Data are represented as mean ± SEM. *P < 0.05.
Discussion
Oxidative stress has been shown to be a major contributing factor in a wide variety of diseases, including FECD. Cellular responses to oxidative stress are major determinants of disease susceptibility, particularly in tissues that do not regenerate, such as corneal endothelial cells. Previously, we detected a deficiency in Nrf2 protein levels and Nrf2-dependent antioxidants in FECD, as well as a concomitant increase in oxidative DNA damage and apoptosis.6,31 During the search to determine which Nrf2 pathway component(s) was defective, accounting for the suboptimal Nrf2-regulated defense in FECD, we detected that FECD tissue exhibits a decline in one of its major stabilizers, DJ-1. We report herein that there is significantly decreased production of DJ-1 in FECD tissue and provide in vitro evidence that a decline in DJ-1 levels is accompanied by oxidative modification and enhanced degradation of DJ-1 protein in a FECD cell line. Although normal corneal endothelium upregulated DJ-1 under oxidative stress, FECD endothelium did not show a similar response, indicating a potential deficiency in the cytoprotective functioning of DJ-1 and its role in Nrf2 stabilization. As a result, there was diminished Nrf2 nuclear translocation in FECD as compared with normal endothelium. In addition, knockdown of DJ-1 in the corneal endothelial cell line resulted in decreased translocation of Nrf2 to the nucleus and significant downregulation of a major Nrf2 transcriptional target and ARE-dependent antioxidant, HMOX-1. The in vitro data corroborated findings detected in FECD tissue samples where DJ-1 downregulation was accompanied by diminished Nrf2-regulated antioxidant defense.
Studies have shown that, in pathologic states, oxidation of cysteine 106 (Cys106) leads to loss of biological properties of DJ-1, including antioxidative function.26,27 For example, it was detected that DJ-1 is irreversibly oxidized by carbonylation and cysteine oxidation in brains from PD and AD patients.21 Similarly, we detected oxidative modification at Cys106 and carbonyl modification of DJ-1 in FECD, posttranslational modifications that have been shown to occur during oxidative stress and target DJ-1 for proteosomal degradation, rendering it inactive.30 Since FECD has been shown to be an oxidative stress disorder,6 we speculate that the oxidant–antioxidant imbalance seen in FECD leads to irreversible oxidative modifications of DJ-1, its rapid degradation, which in turn affects the cytoplasmic stability of Nrf2 and impairs Nrf2 nuclear translocation (Fig. 8). Similar findings have been detected in a smoke-induced, chronic obstructive pulmonary disease (COPD) model in which Nrf2 protein levels, but not mRNA levels, were decreased.15 In COPD, oxidative modification and enhanced degradation of DJ-1 led to decreased cytoplasmic stabilization of Nrf2 and loss of Nrf2-regulated antioxidant defense in human lung epithelial cells exposed to smoke.15
Figure 8. .

Diagram of the role of DJ-1 in pathogenesis of FECD. Genetic and environmental factors may lead to activation of p53 pathways and CEC apoptosis by causing oxidant–antioxidant imbalance, oxidation, and degradation of DJ-1, deficient nuclear translocation of Nrf2, and further transcriptional downregulation of the antioxidant defense.
Since DJ-1 decline was present on the transcriptional level in FECD as well as on the protein level, we used the transcription element search system (http://www.cbil.upenn.edu/cgi-bin/tess) and detected the presence of a conserved sequence for Nrf2 binding in the DJ-1 promoter. It is possible that decline of DJ-1 on the mRNA level represents overall transcriptional decline of antioxidants due to deficient Nrf2-regulated defense and the resulting cellular oxidant antioxidant imbalance (Fig. 8). Further studies need to be performed to investigate specific factors involved in the transcriptional regulation of DJ-1, about which little is currently known.
Among other functions, DJ-1 is also a negative regulator of p53 transcriptional activity. When properly SUMOylated, DJ-1 translocates to the nucleus and represses p53-dependent gene transcription, protecting the cells from apoptosis.32,33 One of our previous studies detected increased levels of p53 in FECD,7 and the current study shows further correlation between oxidative modifications and degradation of DJ-1, as well as increased levels of phospho-p53 in FECD cell lines. These results suggest that the decline seen in DJ-1 in FECD may lead to a higher endothelial susceptibility to p53-regulated apoptosis (Fig. 8).
Interestingly, FECD samples showed upregulation of Keap1 under basal and prooxidant conditions. Keap1 binds Nrf2 in cytosol and targets it for cullin-dependent degradation. In the cell, under oxidative stress there is ongoing nuclear-cytoplasmic shuttling of Nrf2 in which Keap1 is disengaged from Nrf2, allowing nuclear translocation and gene expression to occur.34 While in cytosol, degradation of Nrf2 requires association with Keap1, which in turn associates with Cul3.14 Upregulation of Keap1 might indicate an enhanced Keap1 contribution toward Keap1-Cul3–dependent Nrf2 turnover, as evidenced by decreased Nrf2 protein but not mRNA levels.6
It is possible that factors other than DJ-1 and Keap1 affect Nrf2 protein cytoplasmic stabilization, translocation to the nucleus, and degradation. Phosphorylation of Nrf2 by several protein kinases has been postulated to influence Nrf2 activation and should be investigated in other studies.35,36 In addition, epigenetic factors whose role is largely unknown in pathogenesis of FECD should be considered. Recent data in cancer cells suggest that epigenetic mechanisms may play a role in the regulation of Keap1 and Nrf2 expression.37,38 It is important to note that the FECD cell line, even though removed from the diseased environment, maintained the characteristics of Nrf2 pathway dysregulation similar to that seen in diseased native tissue, suggesting that epigenetic factors persisted during culture and should be explored further in relation to Nrf2 regulation and pathogenesis of FECD.
In summary, this study suggests that decline in DJ-1 and its failure to upregulate in response to oxidative stress could increase Nrf2 cytoplasmic degradation and localization to the nucleus. Decline in transcriptional antioxidant activation increases FECD CEC susceptibility to oxidative stress and apoptosis. Although further studies are needed to understand more precisely the role of DJ-1 downregulation in FECD, this is the first study suggesting that DJ-1 could play a key role in FECD pathogenesis. These studies are significant, since targeting either the DJ-1 and/or Nrf2 pathway may provide a specific pharmacologic target for cytoprotective therapies for corneal endothelium.
Acknowledgments
The authors thank Roberto Pineda II, MD, Kathryn Colby, MD, PhD, Pedram Hamrah, MD, Peter Rapoza, MD, Michael Raizman, MD, Kathryn Hatch, MD, Jerome Kieval, MD, and Nicoletta Fynn-Tompson, MD, for donating specimens and Tissue Banks International for donating normal corneas.
Footnotes
Supported by an Eye Bank Association Award (TS), National Institutes of Health/National Eye Institute Grant R01 EY20581 (UVJ), and a Research to Prevent Blindness Award (UVJ).
These authors contributed equally to the work presented here and should therefore be regarded as equivalent authors.
Disclosure: M.S. Bitar, None; C. Liu, None; A. Ziaei, None; Y. Chen, None; T. Schmedt, None; U.V. Jurkunas, None
References
- 1.Wilson SE, Bourne WM. Fuchs' dystrophy. Cornea. 1988;7:2–18 [PubMed] [Google Scholar]
- 2.Riazuddin SA, Parker DS, McGlumphy EJ, et al. Mutations in LOXHD1, a recessive-deafness locus, cause dominant late-onset Fuchs Corneal Dystrophy. Am J Hum Genet. 2012;90:533–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Riazuddin SA, Zaghloul NA, Al-Saif A, et al. Missense mutations in TCF8 cause late-onset Fuchs corneal dystrophy and interact with FCD4 on chromosome 9p. Am J Hum Genet. 2010;86:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baratz KH, Tosakulwong N, Ryu E, et al. E2-2 protein and Fuchs's corneal dystrophy. N Engl J Med. 2010;363:1016–1024 [DOI] [PubMed] [Google Scholar]
- 5.Engler C, Kelliher C, Spitze AR, Speck CL, Eberhart CG, Jun AS. Unfolded protein response in Fuchs endothelial corneal dystrophy: a unifying pathogenic pathway? Am J Ophthalmol. 2010;149:194–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jurkunas UV, Bitar MS, Funaki T, Azizi B. Evidence of oxidative stress in the pathogenesis of Fuchs endothelial corneal dystrophy. Am J Pathol. 2010;177:2278–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Azizi B, Ziaei A, Fuchsluger T, Schmedt T, Chen Y, Jurkunas UV. p53-regulated increase in oxidative-stress--induced apoptosis in Fuchs endothelial corneal dystrophy: a native tissue model. Invest Ophthalmol Vis Sci. 2011;52:9291–9297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Z, Handa JT, Green WR, Stark WJ, Weinberg RS, Jun AS. Advanced glycation end products and receptors in Fuchs' dystrophy corneas undergoing Descemet's stripping with endothelial keratoplasty. Ophthalmology. 2007;114:1453–1460 [DOI] [PubMed] [Google Scholar]
- 9.Li N, Alam J, Venkatesan MI, et al. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol. 2004;173:3467–3481 [DOI] [PubMed] [Google Scholar]
- 10.Moi P, Chan K, Asunis I, Cao A, Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A. 1994;91:9926–9930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sun Z, Zhang S, Chan JY, Zhang DD. Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol Cell Biol. 2007;27:6334–6349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Purdom-Dickinson SE, Sheveleva EV, Sun H, Chen QM. Translational control of nrf2 protein in activation of antioxidant response by oxidants. Mol Pharmacol. 2007;72:1074–1081 [DOI] [PubMed] [Google Scholar]
- 13.Kwak MK, Itoh K, Yamamoto M, Kensler TW. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter. Mol Cell Biol. 2002;22:2883–2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi A, Kang MI, Okawa H, et al. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol. 2004;24:7130–7139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malhotra D, Thimmulappa R, Navas-Acien A, et al. Decline in NRF2-regulated antioxidants in chronic obstructive pulmonary disease lungs due to loss of its positive regulator, DJ-1. Am J Respir Crit Care Med. 2008;178:592–604 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Knobbe CB, Revett TJ, Bai Y, et al. Choice of biological source material supersedes oxidative stress in its influence on DJ-1 in vivo interactions with Hsp90. J Proteome Res. 2011;10:4388–4404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Andres-Mateos E, Perier C, Zhang L, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–14812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ren H, Fu K, Wang D, Mu C, Wang G. Oxidized DJ-1 interacts with the mitochondrial protein BCL-XL. J Biol Chem. 2011;286:35308–35317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCoy MK, Cookson MR. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy. 2011;7:531–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagakubo D, Taira T, Kitaura H, et al. DJ-1, a novel oncogene which transforms mouse NIH3T3 cells in cooperation with ras. Biochem Biophys Res Commun. 1997;231:509–513 [DOI] [PubMed] [Google Scholar]
- 21.Choi J, Sullards MC, Olzmann JA, et al. Oxidative damage of DJ-1 is linked to sporadic Parkinson and Alzheimer diseases. J Biol Chem. 2006;281:10816–10824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol. 2011;69:570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyce NC, Zhu CC. Human corneal endothelial cell proliferation: potential for use in regenerative medicine. Cornea. 2004;23:S8–S19 [DOI] [PubMed] [Google Scholar]
- 24.Griffith M, Osborne R, Munger R, et al. Functional human corneal equivalents constructed from cell lines. Science. 1999;286:2169–2172 [DOI] [PubMed] [Google Scholar]
- 25.He Y, Weng J, Li Q, Knauf HP, Wilson SE. Fuchs' corneal endothelial cells transduced with the human papilloma virus E6/E7 oncogenes. Exp Eye Res. 1997;65:135–142 [DOI] [PubMed] [Google Scholar]
- 26.Saito Y, Hamakubo T, Yoshida Y, et al. Preparation and application of monoclonal antibodies against oxidized DJ-1. Significant elevation of oxidized DJ-1 in erythrocytes of early-stage Parkinson disease patients. Neurosci Lett. 2009;465:1–5 [DOI] [PubMed] [Google Scholar]
- 27.Akazawa YO, Saito Y, Hamakubo T, et al. Elevation of oxidized DJ-1 in the brain and erythrocytes of Parkinson disease model animals. Neurosci Lett. 2010;483:201–205 [DOI] [PubMed] [Google Scholar]
- 28.Meulener MC, Xu K, Thomson L, Ischiropoulos H, Bonini NM. Mutational analysis of DJ-1 in Drosophila implicates functional inactivation by oxidative damage and aging. Proc Natl Acad Sci U S A. 2006;103:12517–12522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ooe H, Maita C, Maita H, Iguchi-Ariga SM, Ariga H. Specific cleavage of DJ-1 under an oxidative condition. Neurosci Lett. 2006;406:165–168 [DOI] [PubMed] [Google Scholar]
- 30.Wilson MA. The role of cysteine oxidation in DJ-1 function and dysfunction. Antioxid Redox Signal. 2011;15:111–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jurkunas UV, Rawe I, Bitar MS, et al. Decreased expression of peroxiredoxins in Fuchs' endothelial dystrophy. Invest Ophthalmol Vis Sci. 2008;49:2956–2963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan J, Ren H, Fei E, et al. Sumoylation is critical for DJ-1 to repress p53 transcriptional activity. FEBS Lett. 2008;582:1151–1156 [DOI] [PubMed] [Google Scholar]
- 33.Vasseur S, Afzal S, Tomasini R, et al. Consequences of DJ-1 upregulation following p53 loss and cell transformation. Oncogene. 2012;31:664–670 [DOI] [PubMed] [Google Scholar]
- 34.Theodore M, Kawai Y, Yang J, et al. Multiple nuclear localization signals function in the nuclear import of the transcription factor Nrf2. J Biol Chem. 2008;283:8984–8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Niture SK, Jain AK, Shelton PM, Jaiswal AK. Src subfamily kinases regulate nuclear export and degradation of transcription factor Nrf2 to switch off Nrf2-mediated antioxidant activation of cytoprotective gene expression. J Biol Chem. 2011;286:28821–28832 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 36.Keum YS, Yu S, Chang PP, et al. Mechanism of action of sulforaphane: inhibition of p38 mitogen-activated protein kinase isoforms contributing to the induction of antioxidant response element-mediated heme oxygenase-1 in human hepatoma HepG2 cells. Cancer Res. 2006;66:8804–8813 [DOI] [PubMed] [Google Scholar]
- 37.Muscarella LA, Parrella P, D'Alessandro V, et al. Frequent epigenetics inactivation of KEAP1 gene in non-small cell lung cancer. Epigenetics. 2011;6:710–719 [DOI] [PubMed] [Google Scholar]
- 38.Yu S, Khor TO, Cheung KL, et al. Nrf2 expression is regulated by epigenetic mechanisms in prostate cancer of TRAMP mice. PLoS One. 2010;5:e8579. [DOI] [PMC free article] [PubMed] [Google Scholar]