

Abstract
Purpose
Increased expression and/or activation of epidermal growth factor receptor (EGFR) is associated with tumor progression and poor prognosis in many cancers including head and neck squamous cell carcinoma (HNSCC). Src family kinases, including c-Src, mediate a variety of intra- or extracellular signals that contribute to tumor formation and progression. This study was undertaken to elucidate the role of c-Src in the growth and invasion of HNSCC and to determine the effects of combined targeting of EGFR and Src kinases in HNSCC cell lines.
Experimental design
HNSCC cells were engineered to stably express a dominant-active (DA) form of c-Src and investigated in cell growth and invasion assays. The biochemical effects of combined treatment with the Src inhibitor, AZD0530, a potent, orally active Src inhibitor with Bcr/Abl activity and the EGFR kinase inhibitor, gefitinib, were examined as well as the consequences of dual Src/EGFR targeting on the growth and invasion of a panel of HNSCC cell lines.
Results
HNSCC cells expressing DA c-Src demonstrated increased growth and invasion compared with vector-transfected controls. Combined treatment with AZD0530 and gefitinib resulted in greater inhibition of HNSCC cell growth and invasion compared with either agent alone.
Conclusions
These results suggest that increased expression and activation of c-Src promotes HNSCC progression where combined targeting of EGFR and c-Src may be an efficacious treatment approach.
Keywords: head and neck cancer, epidermal growth factor receptor, Src family kinases, protein kinase inhibitors
Introduction
Head and neck squamous cell carcinoma (HNSCC) is characterized by a marked propensity for local invasion and cervical lymph node metastasis that is associated with adverse outcomes. Despite advances in the treatment of HNSCC, up to 50% of patients with this disease develop locoregional recurrence, distant metastasis and/or a second primary cancer. For patients who experience treatment failure following first-line therapy for recurrent or metastatic disease, retrospective analysis has demonstrated a median overall survival of only 3 to 4 months even with treatment, underscoring the aggressiveness of this malignancy and the need for more effective therapies (1).
Signaling through the epidermal growth factor receptor (EGFR/HER1) has been implicated in various processes that contribute to cancer progression, including cell cycle regulation, inhibition of apoptosis, angiogenesis, tumor cell motility, invasion and metastasis (2, 3). Overexpression of EGFR occurs in a variety of solid tumors, including the majority of HNSCC (4–6). We reported previously that high levels of EGFR in HNSCC tumors are correlated with increased regional lymph node metastasis and decreased survival (7). Advanced stage (8), increased tumor size (8), increased recurrence (9), and decreased radiation sensitivity (10) have also been correlated with high tumor EGFR levels. Overexpression of EGFR in HNSCC tumors and the correlation with poor clinical outcome implicate EGFR in the development and progression of HNSCC, and suggest a role for EGFR as a target for cancer therapy. EGFR targeting using a monoclonal antibody approach in combination with radiation has demonstrated efficacy and served as the basis for the FDA approval of cetuximab for HNSCC (11). However, despite promising results in preclinical models, clinical trials using EGFR tyrosine kinase inhibitors as monotherapy in patients with advanced or metastatic HNSCC have shown relatively low therapeutic responses (4–10%) (12, 13).
The c-Src protein is a 60 kDa non-receptor tyrosine kinase that is emerging as a potential target for cancer therapy. c-Src regulates signals from multiple cell surface molecules, including integrins (14), growth factors (15), and G-protein-coupled receptors (16, 17). Elevated c-Src protein levels and/or kinase activity has been reported in a variety of cancers including HNSCC (18–20). The activation of c-Src has been reported to mediate several aspects of tumor growth and progression, including proliferation, migration, invasion, survival and angiogenesis (21–26). We demonstrated previously that c-Src is activated downstream of EGFR in HNSCC (20). The present study was designed to test the qhypothesis that increased c-Src activation contributes to HNSCC progression and that blockade of both c-Src and EGFR would enhance antitumor effects compared with targeting either pathway alone.
AZD0530 is a small-molecule Src inhibitor that blocks the ATP-binding site of Src kinases and is also an inhibitor of kinases with closely related structures, such as Abl (27). Phase I testing of AZD0530 is complete and phase II studies are underway in several solid tumors including HNSCC (28). We elected to use AZD0530, which has not been studied previously in HNSCC, to determine if dual inhibition of EGFR and Src was a rationale treatment approach that could ultimately be tested in patients with HNSCC.
Materials and Methods
Cells and reagents
Cell lines derived from HNSCC patients were used for these studies. The 1483 HNSCC cell line was derived from a tumor in the oropharynx (29). Cal-33 cell line was derived from a patient with squamous cell carcinoma of the tongue(30). The UM-22B cell line was derived from a metastatic cervical lymph node of a patient with a hypopharyngeal cancer (31), while the PCI-37B cell line was derived from a metastatic lymph node of a patient with laryngeal cancer (32). Cell lines were maintained in Dulbecco’s modified eagle medium (DMEM) supplemented with heat inactivated fetal bovine serum (FBS) at 37°C with 5% CO2. The Src inhibitor, AZD0530 and the EGFR specific tyrosine kinase inhibitor, gefitinib (ZD1839), were obtained from AstraZeneca. Both reagents were dissolved in dimethyl sulfoxide (DMSO) at 10 mM, stored at −20°C, and diluted with DMEM prior to use.
Transfection of HNSCC cells with dominant-active c-Src
HNSCC cells (1483) were transfected with a pUSEamp vector (Upstate Biotechnology Inc, Lake Placid, NY) containing mutant Src (Y529F) cDNA using LipofectAMINE 2000 Transfection Reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s recommendations. Stably transfected clones were selected for resistance to the neomycin analogue (400 µg/ml G418) (Invitrogen, Carlsbad, CA). Dominant-active (DA) c-Src was generated by mutation of tyrosine (Tyr) 529 to phenylalanine, which eliminated the inhibitory phosphotyrosine at position 529. HA-tagged DA c-Src HNSCC cells were also generated separately by transfection with a dominant-active Src construct engineered to contain an HA tag. Vector-transfected control clones were established by stable transfection of the cells with pUSEamp control vector.
Biochemical dose response to AZD0530
HNSCC cells were cultured in 10% FBS-containing media. When cells were 70–80% confluent, they were treated with AZD0530 (1–104 nM) or DMSO for 2 hours prior to lysis. Immunoblotting was performed with pY418 Src antibody, anti-c-Src antibody, pFAK Tyr 861 and total FAK.
Matrigel invasion assay
The invasive ability of HNSCC cells was measured using Matrigel-coated modified Boyden inserts with a pore size of 8 µm (Becton Dickinson). Cells were plated in triplicate at a density of 1 × 104 cells/well in serum-free DMEM with the control vehicle (DMSO), AZD0530 (at IC50 value), gefitinib (at IC50 value), or the combination of AZD0530 and gefitinib (both at IC50 values) in the insert. The lower well contained DMEM with 10% FBS. After 48 hours of treatment at 37°C in a 5% CO2 incubator, the cells in the insert were removed by wiping gently with cotton swabs. Cells on the reverse side of the insert were fixed and stained with Hema 3 (Fisher Scientifics, Pittsburgh, PA) according to the manufacturer’s instructions. Invading cells in 4–8 representative fields per insert were counted using light microscopy at 200X magnification.
Proliferation assay
HNSCC cells (PCI-37B, Cal-33 and 1483) were plated in triplicate at a density of 3 × 104 cells/well and allowed to seed overnight in a 12-well plate. Cells were then treated with the control vehicle (DMSO), AZD0530 (at IC50 value), gefitinib (at IC50 value), or the combination of AZD0530 and gefitinib (both at IC50 values) in DMEM with 10% FBS. At selected time points, cells were trypsinized, stained with trypan blue and vital cells were counted using a hemocytometer.
Western blotting
Cell lines were lysed in detergent containing 1% NP40, 150 mM NaCl, 1 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, 1 µg/ml leupeptin, and 1 µg/ml aprotinin, and protein levels were determined using the Bio-Rad protein assay method (Bio-Rad Laboratories, Hercules, CA). Forty µg of total protein was separated on 8% SDS-PAGE gels and transferred to nitrocellulose membranes using a semi-dry transfer machine (BioRad Laboratories, Hercules, CA). Membranes were blocked with 5% skim milk/Tris-buffered saline with Tween 20 solution for 1 hour at room temperature, and incubated with primary antibodies in 5% skim milk in TBS-T overnight at 4°C. After washing with TBS-T three times, membranes were incubated for 1 hour with horseradish peroxidase-conjugated secondary antibody (Bio-Rad Laboratories, Hercules, CA) 1:3000 diluted in 5% skim milk in TBS-T. The filters were rinsed with TBS-T three times, and the blot was developed using Luminol Regent (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) by autoradiography.
Antibodies used for blotting included an anti-phospho-Src polyclonal antibody (Tyr418; BioSource International, Camarillo, CA); c-Src (Santa Cruz Biotechnology, Inc., Santa Cruz, CA); HA (Covance, PA); β-actin (Ab-1) monoclonal mouse IgM (Oncogene Research Products, Boston, MA); phospho-p44/42 MAPK (Thr202/Tyr204), p44/42 MAPK, phospho-AKT (Ser473), AKT, phospho-STAT3 (Tyr705) and STAT3 (all from Cell Signaling Technology, Beverly, MA); phospho-FAK (Tyr861; BioSource International, Camarillo, CA); and FAK (Santa Cruz Biotechnology, Inc., Santa Cruz, CA).
Statistical analysis
For invasion and proliferation studies, the statistical significance of differences in the number of invading cells or viable cells was assessed by use of ANOVA or Wilcoxon-Mann-Whitney one-tailed exact test.
Results
Characterization of dominant-active c-Src HNSCC clones
To determine the role of enhanced c-Src activation in HNSCC, DA c-Src HNSCC clones were established by stable transfection of a representative cell line (1483) with a DA c-Src construct. We also generated HA-tagged DA c-Src clones in 1483 using a similar strategy. The C-terminus of c-Src contains the Tyr 529 residue, which can bind to the SH2 domain when phosphorylated. Interactions between the C-terminus and the SH2 domain, and between the kinase domain and the SH3 domain, cause the c-Src molecule to assume a closed configuration that obscures the kinase domain and reduces its potential for substrate interaction (33, 34). The kinase activating mutation in the DA c-Src construct that we used contains a substitution of phenylalanine for tyrosine at position 529 in the C-terminus. This Y529F mutation renders the SH1 kinase domain open by hindering the binding between the C-terminus and the SH2 domain of the c-Src molecule, which, in turn, enhances the potential for substrate interaction. After transfection, 1483 cells were cultured with 400 µg/ml G418 and G418-resistant clones were obtained. After cell lysis, protein extracts were fractionated on an 8% SDS-PAGE gel. Immunoblots were probed with pY418 c-Src antibody and c-Src to evaluate levels of phosphorylated and total c-Src, respectively. Compared to the vector-transfected controls, DA c-Src-transfected clones demonstrated a 2 to 3-fold increase in c-Src tyrosine phosphorylation at position 418 as well as increased total c-Src (Figures 1A and 1B). Expression and activation of c-Src in vector control clones were not different from levels detected in non-transfected parental 1483 cells (data not shown).
Figure 1. Characterization of dominant-active (DA) c-Src clones.

(A) HNSCC cells (1483) were transfected with a DA c-Src construct (Upstate Biotechnology) followed by isolation of stable clones in G418-containing media (400 µg/ml). Expression of Tyr 418 phosphorylation and total c-Src were examined by immunoblotting with pY418 Src antibody (BioSource International, Camarillo, CA) and anti-c-Src antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA). Actin is shown as a control for loading (Oncogene Research Products, Boston, MA). (B) Additional HA-tagged DA c-Src clones were generated in 1483 cells. A representative western blot showing increased expression of pY418 Src as well as total c-Src is shown, in conjunction with expression of the HA-tag. The experiment was repeated 3 times with similar results.
Increased c-Src enhances HNSCC cell invasion and proliferation
Decreased activity of c-Src by dominant-negative (DN) c-Src transfection or treatment with a Src inhibitor has been reported to abrogate invasion of cancer cells including HNSCC (17, 20, 35, 36). Breast cancer cells with higher EGFR and c-Src levels have been shown to have a more invasive phenotype (37). To determine whether increased c-Src could enhance invasion and/or proliferation in HNSCC cells, Matrigel invasion and growth assays were performed. In addition to HNSCC cells stably expressing DA c-Src, we also evaluated clones from the same cell line stably transfected with a DN c-Src construct as described previously (20). As shown in Figure 2, DA c-Src-transfected cells were 2.6-fold more invasive than vector-transfected control cells under these conditions (p=0.0143), while DN c-Src-transfected cells were 6-fold less invasive than vector-transfected control cells (p=0.0143). A similar trend was seen in another set of experiments using 1483 vector-transfected control clone #1 (1483 VC #1), with 1483 HA-DA c-Src clone #62 (data not shown).
Figure 2. Invasion of dominant-active and dominant-negative c-Src HNSCC cells.

Representative vector-transfected control, DN c-Src-transfected, and DA c-Src-transfected HNSCC (1483) clones (DA c-Src #6) were serum-starved for 4 days, and then plated at a density of 5× 103 cells/well in serum-free DMEM in Matrigel-coated inserts. Lower wells contained DMEM with 10% FBS. After 48 hours at 37°C in a 5% CO2 incubator, the cells in the insert were removed by wiping gently with a cotton swab. Cells on the reverse side of the insert were fixed and stained with Hema 3 (Fisher Scientific) according to the manufacturer’s instructions. Invading cells in 4–8 representative fields were counted using light microscopy at 200× magnification. The experiment was repeated 4 times with similar results.
To assess proliferation, cell counting experiments using trypan blue dye exclusion were performed. In 1% FBS, the growth of DA c-Src-transfected cells (DA c-Src #6) and DN c-Src-transfected cells was 77.7% higher (p=0.0043) and 44.6% lower (p=0.0422, respectively, than vector-transfected control cells (VC #3) (Figure 3A). In another experiment, the rate of cell proliferation was increased in DA c-Src #62 compared to 1483 vector-transfected control cells (VC #1) and DN transfected c-Src when grown in 1% FBS (Figure 3B, lower panel). Similar results were observed in another HNSCC cell line stably transfected with DA c-Src (Figures 3 C–D). These results suggest that HNSCC cell invasion and growth may be enhanced by increased expression and activation of c-Src.
Figure 3. Proliferation of dominant-active and dominant-negative c-Src HNSCC cells.
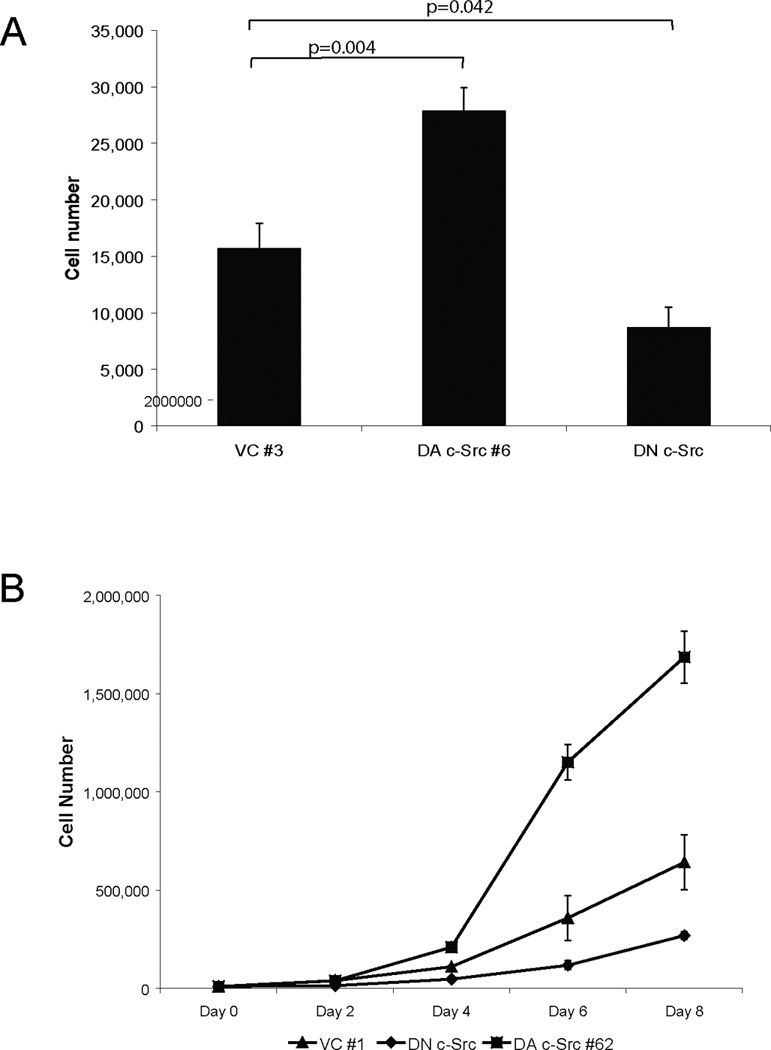
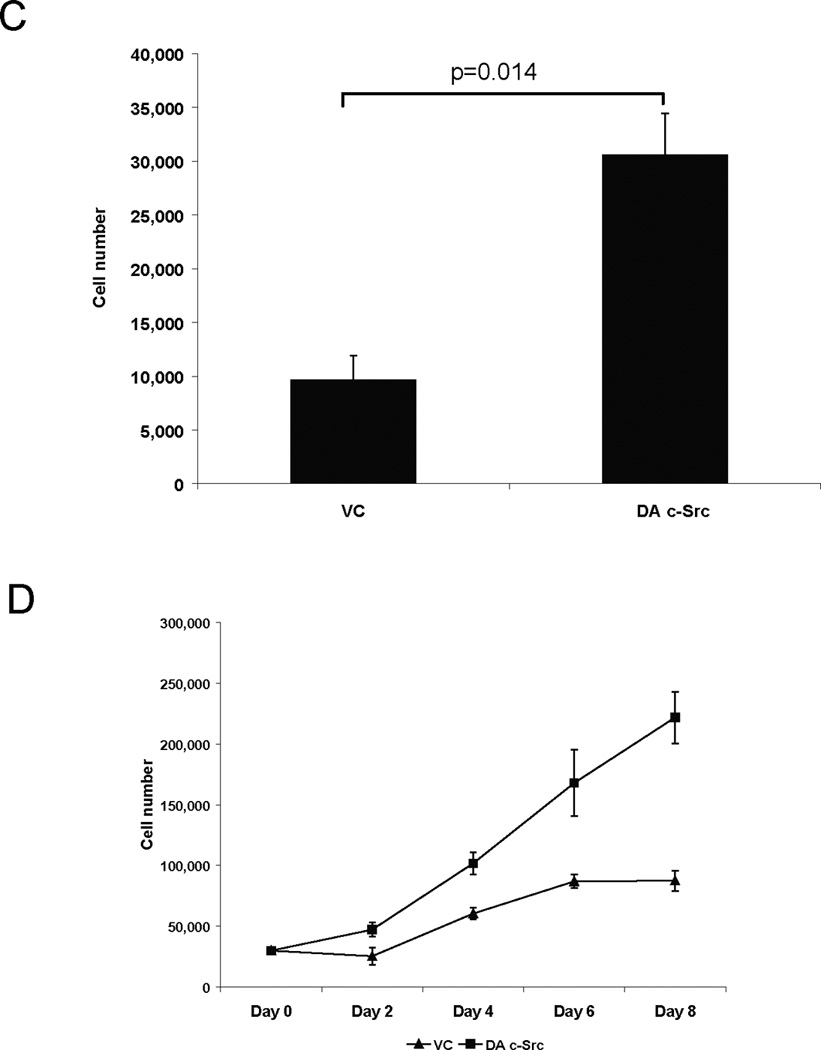
For growth experiments, (A) representative vector-transfected (VC #3), DN c-Src-transfected, and DA c-Src-transfected HNSCC (1483 DA c-Src #6) clones were plated in 48-well plates at a density of 5 × 103 cells/well in DMEM with 1% FBS. Cells were incubated at 37°C, 5% CO2 incubator for 3 days, followed by cell counts using trypan blue exclusion. Cumulative results are shown from 6 independent experiments, each performed in triplicate (upper panel). (B) In the lower panel, the growth kinetics of a DA c-Src transfected 1483 clone, DA c-Src clone #62 was compared over the course of 8 days with the 1483 vector-transfected control clone (VC #1) and to the DN-Src clone in low serum containing media (1% FBS). Cells (3 × 104) of each clone were seeded in triplicate in 12-well plates and allowed to adhere overnight. Cell counts were carried out using trypan blue dye over the next 8 days. All data values are represented as mean ± SEM. The experiment was repeated 3 times with similar results. (C) PCI-15B cells were stably transfected with DA c-Src or vector control and plated as above in DMEM with 1% of FBS. Cells were harvested on day 4 and counted with vital dye exclusion. (D) Cell counts on days 2, 4, 6 and 8 from 3 independent experiments are represented.
AZD0530 inhibits Src family kinase activity in vitro
AZD0530 is a dual-specific Src/Abl kinase inhibitor that has been reported to decrease activated Src in tamoxifen-resistant breast cancer cells (37). To determine the effect of AZD0530 on c-Src expression and activation in HNSCC cells, the expression of total and phosphorylated c-Src, and its downstream target, FAK was evaluated. UM-22B, 1483 and PCI-37B were treated with 1 to 104 nM AZD0530, or DMSO for 2 hours. After protein extraction, immunoblots were probed with the Src family kinase-specific pY418 antibody and an anti-c-Src antibody. AZD0530 inhibited the activity of Src family kinases in a dose-dependent manner and caused a substantial decrease of pY418 Src at 1 µM in the cell lines examined (Figure 4A). We also saw a concomitant decrease in expression levels of pFAK Tyr 861 (a Src dependent phosphorylation site) at this concentration of AZD0530 in these cells. Inhibition of c-Src activation by AZD0530 at a concentration of 1 µM is consistent with previous findings in breast cancer cells (37). The Cal-33 HNSCC cell line was more sensitive to AZD0530 than the other 3 HNSCC cell lines, with an IC50 value of 0.6 µM. AZD0530 at this concentration also decreased pY418 Src and pFAK Tyr 861 levels. In addition, expression of E-cadherin was increased by AZD0530 in this cell line (Figure 4B).
Figure 4. The effect of increasing doses of AZD0530 on expression and activation of c-Src and FAK in HNSCC cell lines.


(A) UM-22B, 1483 and PCI-37B cells were treated with AZD0530 (1–104 nM) or DMSO for 2 hours prior to lysis. Immunoblotting was performed with pY418 c-Src antibody, anti-c-Src antibody, pFAK Tyr 861 and total FAK. (B) Cal-33 cells were treated with increasing concentrations of AZD0530 (0.06–6µM) or DMSO for 2 hrs followed by immunoblotting for total and activated c-Src, E-cadherin and pFAK Y861. The experiment was performed 3 times with similar results.
Combined inhibition of c-Src and EGFR abrogates invasion of HNSCC cells
Src family kinases interact with many receptor tyrosine kinases including EGFR (38). EGFR has been reported to activate Src through a variety of mechanisms. We reported previously that Src family kinases also regulate gastrin-releasing peptide (GRP)-induced EGFR proligand cleavage, leading to downstream EGFR and MAPK activation in HNSCC, which contributes to cell proliferation and invasion (17) Gefitinib is an orally active synthetic quinazoline derivative that binds EGFR and inhibits the activation of the receptor in a reversible manner. To test whether the combination of AZD0530 and gefitinib enhanced the inhibition of HNSCC invasion compared with either treatment alone, Matrigel invasion assays were performed. HNSCC cell lines (1483, PCI-37B) were cultured in DMEM with 10% FBS. The IC50 values of gefitinib for various cell lines used in this study were initially determined by MTT and cell counts (Table 1). Cells were plated at a density of 1 × 104 cells/well in serum-free DMEM with the vehicle (DMSO), AZD0530 (1 µM), gefitinib (at IC50 value), or the combination of AZD0530 and gefitinib (both at IC50 values) in the insert. The lower well contained DMEM with 10% FBS as a chemoattractant. As shown in Figures 5A (1483) and 5B (PCI-37B), invasion of 1483 and PCI-37B cells treated with the combination of AZD0530 and gefitinib was decreased to 34.84% (±4.59%) and 28.39% (±4.71%) of the vehicle control respectively, which was lower than AZD0530 alone [48.73 (±3.30%) (p=0.034) and 51.94 % (±7.02%), (p=0.024) respectively] or gefitinib alone [71.51% (±4.95%) (p=0.003) and 69.85% (±3.82%) (p<0.001), respectively using ANOVA]. These results suggest that combined inhibition of Src and EGFR abrogates HNSCC invasion more than inhibition of either Src or EGFR alone.
Table 1.
| SCCHN Cell Line | IC50 gefitinib | IC50 AZD0530 |
|---|---|---|
| 1483 | 15 µM | 1 µM |
| UM-22B | 20 µM | 1 µM |
| PCI-15B | 10 µM | 1.3 µM |
| PCI-37B | 6.5 µM | 1 µM |
| Cal-33 | 6 µM | 0.6 µM |
Figure 5. Matrigel invasion assay of HNSCC cell lines treated with AZD0530 and/or gefitinib.
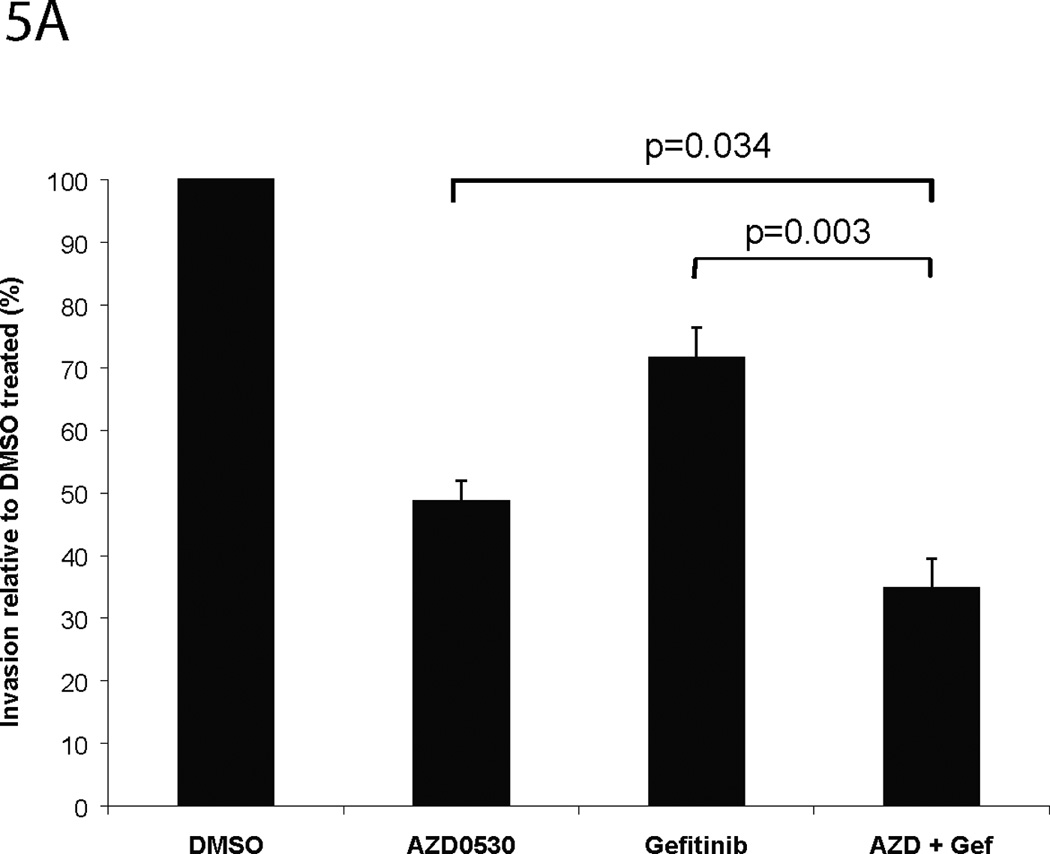

(A) 1483 and (B) PCI-37B cells were plated in Matrigel-coated invasion chamber at a density of 1 × 104 cells/well in serum-free DMEM containing DMSO, AZD0530 (1 µM), gefitinib (at IC50 value), or combination of AZD0530 and gefitinib (both at IC50 values). Lower wells contained DMEM with 10% FBS. After 48 hours of treatment at 37°C in a 5% CO2 incubator, the cells in the insert were removed by wiping gently with a cotton swab. Cells on the reverse side of the insert were fixed and stained with Hema 3 (Fisher Scientific) according to the manufacturer’s instructions. Invading cells in 4 representative fields each from 3 replicates were counted using light microscopy at 200× magnification. Cumulative data from at least 5 independent experiments for both cell lines are shown.
Combined inhibition of c-Src and EGFR abrogates growth of HNSCC cells
Src kinases are involved in multiple signaling pathways that contribute to cell cycle progression and survival. Inhibition of Src kinases has been reported to inhibit tumor cell adhesion, migration and invasion, however their anti-proliferative activity has been less consistently observed. AZD0530 was reported to have little effect on reducing the growth of breast cancer cells (37). Other Src kinase inhibitors including dasatinib and AP23846 were reported to inhibit proliferation of HNSCC and lung cancer cells (35), as well as pancreatic adenocarcinoma cells (39). To determine whether AZD0530 inhibited proliferation of HNSCC cells, we performed cell-counting experiments in several HNSCC cell lines (1483, PCI-37B and Cal-33) following treatment with AZD0530 and/or gefitinib (see Table 1 for IC50 values of each compound in each cell line). These IC50 values for gefitinib are comparable to values reported by others in HNSCC cells (40, 41). Cells were plated in 12-well plates at a density of 3 × 104 cells/well in DMEM with 10% FBS. After 24 hours, growth media was replaced with DMEM with 10% FBS containing DMSO, AZD0530 (at IC50 value), gefitinib (at IC50 value), or a combination of AZD0530 and gefitinib (both at IC50 values). Viable cells, after trypan blue staining, were counted on days 2, 4, 6, and 8 after a single treatment. Growth of 1483 cells was not abrogated by AZD0530 (Figure 6A). However, AZD0530 treatment resulted in growth inhibition of PCI-37B cells by 35% at 8 days after treatment (Figure 6B). Proliferation of Cal-33 cells was also inhibited by AZD0530 (Figure 6C). Even though AZD0530 did not affect growth of 1483 cells, the combination of AZD0530 and gefitinib significantly inhibited proliferation more than gefitinib alone or AZD0530 alone in all the three cell lines examined (Figure 6). These results suggest that, although HNSCC cells have variable sensitivity to the growth inhibitory effects of AZD0530 and gefitinib, the combination of gefitinib and AZD0530 inhibits the proliferation of HNSCC cells to a greater degree than EGFR or Src inhibition alone.
Figure 6. Proliferation assay of HNSCC cell lines treated with AZD0530 and/or gefitinib.



(A) 1483, (B) PCI-37B, and (C) Cal-33 cells were plated in 12-well plates at a density of 3 × 104 cells/well in DMEM with 10% FBS. After 24 hours, growth media was replaced with media containing DMSO, AZD0530 (at IC50 value), gefitinib (at IC50 value), or a combination of AZD0530 and gefitinib. On days 0, 2, 4, 6 and 8, viable cells were counted using vital dye exclusion. Four independent experiments (in triplicate) were performed for each cell line. Cell numbers on day 8 after treatment are shown.
Effect of AZD0530 and/or gefitinib on downstream signaling molecules
To begin to determine the antitumor mechanisms of AZD0530 and/or gefitinib we examined the expression of downstream molecules by immunoblotting in HNSCC cells (1483, PCI-37B and Cal-33) treated for 2 hours with AZD0530 (at IC50 value), gefitinib (at IC50 value), or a combination of AZD0530 and gefitinib (both at IC50 values) (Figure 7). Inhibition of Src activity by AZD0530 was confirmed, whereas gefitinib did not abrogate Src phosphorylation. Phosphorylation of MAPK was reduced by gefitinib or AZD0530 in 2 out of the 3 cell lines examined with no apparent change in the setting of combined therapy. AKT phosphorylation was modestly reduced by treatment with gefitinib or AZD0530 with no apparent enhancement following combined treatment. FAK phosphorylation was inhibited by AZD0530 but not by gefitinib. STAT5 phosphorylation was reduced by gefitinib and further reduced by combined treatment with AZD0530 in 2 of 3 cell lines examined. In the 3rd cell line (PCI-37B) pSTAT5 was completely abrogated by AZD0530 (Figure 7). These results suggest that reduction of pSTAT5 may be a marker of the combined effect of targeting EGFR and Src in HNSCC cells.
Figure 7. The effect of AZD0530 and/or gefitinib on expression of total and phosphorylated c-Src, MAPK, AKT, and FAK and STAT5.

HNSCC cells (1483, PCI-37B and Cal-33) were treated with DMSO, AZD0530 (at IC50 value), gefitinib (at IC50 value) or combination of AZD0530 and gefitinib (both at IC50 values) in DMEM with 10% FBS. After 2 hours, total protein was extracted prior to lysis and immunoblotting. Beta-tubulin is shown for loading control. The experiment was performed at least 2 times, with similar results.
Discussion
The results of the present study suggest that increased c-Src expression and/or phosphorylation promotes the invasion and growth of HNSCC cells, and that combined inhibition of c-Src and EGFR augments the inhibition of invasion and growth compared with blockade of either tyrosine kinase alone. These data provide a rationale for further investigation of the mechanism of combined EGFR and Src targeting in cancers that express increased levels of these proteins.
c-Src is a cellular homologue of v-Src, the oncogene product of the avian tumor virus Rous sarcoma virus (42). c-Src is expressed at low levels in most cell types and, in the absence of appropriate extracellular stimuli, maintained in an inactive conformation through phosphorylation of a negative-regulatory domain at Tyr 530. The transforming activity of v-Src is much higher than that of c-Src due to the absence of the negative-regulatory C-terminal domain and the presence of point mutations throughout the gene (43). A C-terminally truncated c-Src that exhibits constitutive catalytic activity similar to v-Src has been detected in a small subset of colon and endometrial cancers (44, 45). Other studies, however, have failed to detect such mutations in malignant colon tumors (46–48), suggesting that mutations that lead to activation of c-Src are rare. c-Src activity in metastatic liver lesions has been reported to be higher than levels detected in primary colon cancers (49). In transitional cell carcinoma of the bladder, c-Src activity appeared to increase as superficial tumors invaded into muscle (50), implicating c-Src in tumor progression.
There is little information on the expression and activation of c-Src in head and neck carcinogenesis. Increased expression of c-Src has been detected in areas of hyperproliferation in HNSCC as well as premalignant mucosal lesions (19). To begin to determine the consequences of Src activation in HNSCC, we engineered cells to stably express a DA form of c-Src. Dominant active c-Src HNSCC cells showed increased growth and invasion in vitro compared with vector-transfected control cells. Increased invasion by c-Src is consistent with previous reports using colon cancer cells (51, 52). Transfection of DA c-Src has been reported to result in increased mitosis, tumor growth and colony formation in an in vivo model using breast cancer cells (53), and decreased TGF-α-induced apoptosis in hepatocellular carcinoma cells (54), consistent with our proliferation assay results (Figure 3). Cancer cell migration is regulated by a variety of factors including integrins, matrix-degrading enzymes, cell–cell adhesion molecules and cell–cell communication. c-Src has been reported to mediate cancer cell invasion by several mechanisms, including FAK phosphorylation and loss of focal adhesions (55), R-Ras phosphorylation and subsequent inhibition of integrin activity and adhesion (56), decreased E-cadherin by tyrosine phosphorylation, ubiquitylation and endocytosis of E-cadherin (57), and/or decreased hyaluronic acid-dependent migration by interaction with CD44 (58). To this effect, we observed increased expression of E-cadherin in Cal-33 cells, with increasing doses of AZD0530, and a concomitant decrease in pFAK Tyr 861 levels (Figure 4B). pFAK Tyr 861 levels also decreased with increasing doses of AZD0530 in PCI-37B, UM-22B and 1483 treated cells (Figure 4A).
The central role of c-Src in many signaling transduction pathways combined with the frequent overexpression of c-Src in human epithelial cancers makes Src an attractive target for cancer therapy. Preclinical strategies for targeting c-Src include DN mutants (17, 20), small inhibitory RNA (siRNA) (59) and Src kinase inhibitors. The pyrazolopyrimidines PP1 and PP2 have been used extensively in in vitro models of Src inhibition, although concentrations up to 10 µM are often necessary to achieve complete Src kinase inhibition in cell culture (60). High concentrations of these inhibitors often inhibit “off-target” kinases, including CSK and p38 MAPK, as well (61). Because of the nonspecific inhibition of other kinases, it has been hard to delineate Src-specific effects of these reagents in the absence of supporting data from DN c-Src, siRNA, or gene knockout studies. A new generation of Src-specific inhibitors has been FDA-approved for selected hematologic malignancies (dasatinib) and/or is being tested in solid tumors (AZD0530 and dasatinib) (35, 39, 62–64). Dasatinib was reported to inhibit migration and invasion of HNSCC cells, and induce cell cycle arrest and apoptosis in some of the cell lines (35). This study also demonstrated downregulation of pSTAT5 in HNSCC cells following treatment with the Src inhibitor. Our finding of further reduction of pSTAT5 levels with dual EGFR/Src inhibition suggests that pSTAT5 may serve as a biomarker to identify patients or indicate therapeutic response. We reported previously that blockade of Src kinases using a second generation pyrrolopyrimidine compound, A419259, or a pyrido[2,3-d] pyrimidine derivative, PD180970, inhibited growth in HNSCC cells (20). In the present study, we used AZD0530, which blocks the ATP-binding site of Src kinases and is also an inhibitor of kinases with closely related structures, such as Abl (27). AZD0530 has been reported to inhibit the invasion of tamoxifen-resistant breast cancer cells at low concentrations (0.1 µM) but only modestly reduced cell growth at higher concentrations (1 µM) (37). We found that Src and FAK activation in HNSCC cells was inhibited by 1 µM AZD0530 in 3 of 4 cell lines examined. AZD0530 blocked the invasion of HNSCC cells at similar concentrations to that observed in breast cancer cells. However, HNSCC cells were more sensitive to the growth inhibitory effects of AZD0530 (Figure 6). These results suggest that while c-Src plays a central role in cancer invasion, the role of c-Src in mediating cell growth may depend upon the specific model under investigation.
Overexpression of EGFR and ligands in cancer tissue, the correlation between EGFR expression and poor clinical outcome, and the aberrant function of the EGFR network in cancer cells have implicated EGFR as a target for cancer therapy. EGFR has been shown to activate c-Src kinase through a variety of mechanisms, including the small GTPases Ras and Ral (65), and the recruitment of Shp2, which dephosphorylates the Csk-binding protein PAG, thereby preventing the access of Csk, a negative regulator of Src, to Src (66). Inhibition of EGFR has been reported to reduce Src activity to basal levels in colon cancer cell lines (67). We demonstrated previously that EGFR deficient murine embryonic fibroblasts showed decreased activation of c-Src following stimulation with GRP where c-Src was shown to mediate the cleavage of EGFR pro-ligand cleavage (17). In addition, overexpression of c-Src has reported to result in the accumulation of EGFR at the cell surface by promoting destruction of c-Cbl, which mediates EGFR ubiquitylation and sorting to endocytosis and degradation (68). In the present study, combined inhibition of EGFR and c-Src inhibited cell growth to a larger degree than treatment with a single inhibitor alone. These results suggest that the combined targeting of c-Src and EGFR may be a rational therapeutic approach in cancers that demonstrate increased EGFR and Src activation, including HNSCC.
Acknowledgements
This work was supported by grants CA77308, CA098372 and CA097190 (to JRG).
References
- 1.Leon X, Hitt R, Constenla M, et al. A retrospective analysis of the outcome of patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck refractory to a platinum-based chemotherapy. Clin Oncol (R Coll Radiol) 2005;17:418–424. doi: 10.1016/j.clon.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 2.Carpenter G, Cohen S. Epidermal growth factor. J Biol Chem. 1990;265:7709–7712. [PubMed] [Google Scholar]
- 3.Mendelsohn J, Baselga J. The EGF receptor family as targets for cancer therapy. Oncogene. 2000;19:6550–6565. doi: 10.1038/sj.onc.1204082. [DOI] [PubMed] [Google Scholar]
- 4.Rubin Grandis J, Melhem MF, Barnes EL, Tweardy DJ. Quantitative immunohistochemical analysis of transforming growth factor- alpha and epidermal growth factor receptor in patients with squamous cell carcinoma of the head and neck. Cancer. 1996;78:1284–1292. doi: 10.1002/(SICI)1097-0142(19960915)78:6<1284::AID-CNCR17>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 5.Santini J, Formento JL, Francoual M, et al. Characterization, quantification, and potential clinical value of the epidermal growth factor receptor in head and neck squamous cell carcinomas. Head Neck. 1991;13:132–139. doi: 10.1002/hed.2880130209. [DOI] [PubMed] [Google Scholar]
- 6.Dassonville O, Formento JL, Francoual M, et al. Expression of epidermal growth factor receptor and survival in upper aerodigestive tract cancer. J Clin Oncol. 1993;11:1873–1878. doi: 10.1200/JCO.1993.11.10.1873. [DOI] [PubMed] [Google Scholar]
- 7.Rubin Grandis J, Melhem MF, Gooding WE, et al. Levels of TGF-alpha and EGFR protein in head and neck squamous cell carcinoma and patient survival. J Natl Cancer Inst. 1998;90:824–832. doi: 10.1093/jnci/90.11.824. [DOI] [PubMed] [Google Scholar]
- 8.Chen BK, Ohtsuki Y, Furihata M, et al. Co-overexpression of p53 protein and epidermal growth factor receptor in human papillary thyroid carcinomas correlated with lymph node metastasis, tumor size and clinicopathologic stage. Int J Oncol. 1999;15:893–898. doi: 10.3892/ijo.15.5.893. [DOI] [PubMed] [Google Scholar]
- 9.Ang KK, Berkey BA, Tu X, et al. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002;62:7350–7356. [PubMed] [Google Scholar]
- 10.Gupta AK, McKenna WG, Weber CN, et al. Local recurrence in head and neck cancer: relationship to radiation resistance and signal transduction. Clin Cancer Res. 2002;8:885–892. [PubMed] [Google Scholar]
- 11.Bonner JA, Harari PM, Giralt J, et al. Radiotherapy plus cetuximab for squamous-cell carcinoma of the head and neck. N Engl J Med. 2006;354:567–578. doi: 10.1056/NEJMoa053422. [DOI] [PubMed] [Google Scholar]
- 12.Cohen EE, Rosen F, Stadler WM, et al. Phase II trial of ZD1839 in recurrent or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol. 2003;21:1980–1987. doi: 10.1200/JCO.2003.10.051. [DOI] [PubMed] [Google Scholar]
- 13.Soulieres D, Senzer NN, Vokes EE, et al. Multicenter phase II study of erlotinib, an oral epidermal growth factor receptor tyrosine kinase inhibitor, in patients with recurrent or metastatic squamous cell cancer of the head and neck. J Clin Oncol. 2004;22:77–85. doi: 10.1200/JCO.2004.06.075. [DOI] [PubMed] [Google Scholar]
- 14.Moro L, Dolce L, Cabodi S, et al. Integrin-induced epidermal growth factor (EGF) receptor activation requires c-Src and p130Cas and leads to phosphorylation of specific EGF receptor tyrosines. J Biol Chem. 2002;277:9405–9414. doi: 10.1074/jbc.M109101200. [DOI] [PubMed] [Google Scholar]
- 15.Tice DA, Biscardi JS, Nickles AL, Parsons SJ. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc Natl Acad Sci U S A. 1999;96:1415–1420. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luttrell LM, Hawes BE, van Biesen T, et al. Role of c-Src tyrosine kinase in G protein-coupled receptor- and Gbetagamma subunit-mediated activation of mitogen-activated protein kinases. J Biol Chem. 1996;271:19443–19450. doi: 10.1074/jbc.271.32.19443. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Q, Thomas SM, Xi S, et al. SRC family kinases mediate epidermal growth factor receptor ligand cleavage, proliferation, and invasion of head and neck cancer cells. Cancer Res. 2004;64:6166–6173. doi: 10.1158/0008-5472.CAN-04-0504. [DOI] [PubMed] [Google Scholar]
- 18.Rosen N, Bolen JB, Schwartz AM, et al. Analysis of pp60c-src protein kinase activity in human tumor cell lines and tissues. J Biol Chem. 1986;261:13754–13759. [PubMed] [Google Scholar]
- 19.van Oijen MG, Rijksen G, ten Broek FW, Slootweg PJ. Overexpression of c-Src in areas of hyperproliferation in head and neck cancer, premalignant lesions and benign mucosal disorders. J Oral Pathol Med. 1998;27:147–152. doi: 10.1111/j.1600-0714.1998.tb01931.x. [DOI] [PubMed] [Google Scholar]
- 20.Xi S, Zhang Q, Dyer KF, et al. Src kinases mediate STAT growth pathways in squamous cell carcinoma of the head and neck. J Biol Chem. 2003;278:31574–31583. doi: 10.1074/jbc.M303499200. [DOI] [PubMed] [Google Scholar]
- 21.Maa MC, Leu TH, McCarley DJ, Schatzman RC, Parsons SJ. Potentiation of epidermal growth factor receptor-mediated oncogenesis by c-Src: implications for the etiology of multiple human cancers. Proc Natl Acad Sci U S A. 1995;92:6981–6985. doi: 10.1073/pnas.92.15.6981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mukhopadhyay D, Tsiokas L, Zhou XM, et al. Hypoxic induction of human vascular endothelial growth factor expression through c-Src activation. Nature. 1995;375:577–581. doi: 10.1038/375577a0. [DOI] [PubMed] [Google Scholar]
- 23.Sheffield LG. C-Src activation by ErbB2 leads to attachment-independent growth of human breast epithelial cells. Biochem Biophys Res Commun. 1998;250:27–31. doi: 10.1006/bbrc.1998.9214. [DOI] [PubMed] [Google Scholar]
- 24.Ellis LM, Staley CA, Liu W, et al. Down-regulation of vascular endothelial growth factor in a human colon carcinoma cell line transfected with an antisense expression vector specific for c-src. J Biol Chem. 1998;273:1052–1057. doi: 10.1074/jbc.273.2.1052. [DOI] [PubMed] [Google Scholar]
- 25.Windham TC, Parikh NU, Siwak DR, et al. Src activation regulates anoikis in human colon tumor cell lines. Oncogene. 2002;21:7797–7807. doi: 10.1038/sj.onc.1205989. [DOI] [PubMed] [Google Scholar]
- 26.Ito H, Gardner-Thorpe J, Zinner MJ, Ashley SW, Whang EE. Inhibition of tyrosine kinase Src suppresses pancreatic cancer invasiveness. Surgery. 2003;134:221–226. doi: 10.1067/msy.2003.224. [DOI] [PubMed] [Google Scholar]
- 27.Green TP, Fennell M, Whittaker R, et al. Preclinical activity of AZD0530, a novel, oral, potent and selective inhibitor of the Src family kinases. European Journal of Cancer, Supplement. 2004;2(8) Abstract #361. [Google Scholar]
- 28.Tabernero J, Cervantes A, Hoekman K, et al. Phase I study of AZD0530, an oral potent inhibitor of Src kinase: First demonstration of inhibition of Src activity in human cancers; Journal of Clinical Oncology, 2007 ASCO Annual Meeting Proceedings Part I Vol 25, No 18S (June 20 Supplement); 2007. p. 2007. [Google Scholar]
- 29.Sacks PG, Parnes SM, Gallick GE, et al. Establishment and characterization of two new squamous cell carcinoma cell lines derived from tumors of the head and neck. Cancer Res. 1988;48:2858–2866. [PubMed] [Google Scholar]
- 30.Gioanni J, Fischel JL, Lambert JC, et al. Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: establishment, characterization and response to cytotoxic treatment. Eur J Cancer Clin Oncol. 1988;24:1445–1455. doi: 10.1016/0277-5379(88)90335-5. [DOI] [PubMed] [Google Scholar]
- 31.Jetten AM, Kim JS, Sacks PG, et al. Inhibition of growth and squamous-cell differentiation markers in cultured human head and neck squamous carcinoma cells by beta-all-trans retinoic acid. Int J Cancer. 1990;45:195–202. doi: 10.1002/ijc.2910450135. [DOI] [PubMed] [Google Scholar]
- 32.Heo DS, Snyderman C, Gollin SM, et al. Biology, cytogenetics, and sensitivity to immunological effector cells of new head and neck squamous cell carcinoma lines. Cancer Res. 1989;49:5167–5175. [PubMed] [Google Scholar]
- 33.Yamaguchi H, Hendrickson WA. Structural basis for activation of human lymphocyte kinase Lck upon tyrosine phosphorylation. Nature. 1996;384:484–489. doi: 10.1038/384484a0. [DOI] [PubMed] [Google Scholar]
- 34.Sicheri F, Kuriyan J. Structures of Src-family tyrosine kinases. Curr Opin Struct Biol. 1997;7:777–785. doi: 10.1016/s0959-440x(97)80146-7. [DOI] [PubMed] [Google Scholar]
- 35.Johnson FM, Saigal B, Talpaz M, Donato NJ. Dasatinib (BMS-354825) tyrosine kinase inhibitor suppresses invasion and induces cell cycle arrest and apoptosis of head and neck squamous cell carcinoma and non-small cell lung cancer cells. Clin Cancer Res. 2005;11:6924–6932. doi: 10.1158/1078-0432.CCR-05-0757. [DOI] [PubMed] [Google Scholar]
- 36.Nam S, Kim D, Cheng JQ, et al. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005;65:9185–9189. doi: 10.1158/0008-5472.CAN-05-1731. [DOI] [PubMed] [Google Scholar]
- 37.Hiscox S, Morgan L, Green TP, et al. Elevated Src activity promotes cellular invasion and motility in tamoxifen resistant breast cancer cells. Breast Cancer Res Treat. 2005:1–12. doi: 10.1007/s10549-005-9120-9. [DOI] [PubMed] [Google Scholar]
- 38.Bromann PA, Korkaya H, Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004;23:7957–7968. doi: 10.1038/sj.onc.1208079. [DOI] [PubMed] [Google Scholar]
- 39.Summy JM, Trevino JG, Lesslie DP, et al. AP23846, a novel and highly potent Src family kinase inhibitor, reduces vascular endothelial growth factor and interleukin-8 expression in human solid tumor cell lines and abrogates downstream angiogenic processes. Mol Cancer Ther. 2005;4:1900–1911. doi: 10.1158/1535-7163.MCT-05-0171. [DOI] [PubMed] [Google Scholar]
- 40.Frederick BA, Helfrich BA, Coldren CD, et al. Epithelial to mesenchymal transition predicts gefitinib resistance in cell lines of head and neck squamous cell carcinoma and non-small cell lung carcinoma. Mol Cancer Ther. 2007;6:1683–1691. doi: 10.1158/1535-7163.MCT-07-0138. [DOI] [PubMed] [Google Scholar]
- 41.Magne N, Fischel JL, Dubreuil A, et al. ZD1839 (Iressa) modifies the activity of key enzymes linked to fluoropyrimidine activity: rational basis for a new combination therapy with capecitabine. Clin Cancer Res. 2003;9:4735–4742. [PubMed] [Google Scholar]
- 42.Stehelin D, Varmus HE, Bishop JM, Vogt PK. DNA related to the transforming gene(s) of avian sarcoma viruses is present in normal avian DNA. Nature. 1976;260:170–173. doi: 10.1038/260170a0. [DOI] [PubMed] [Google Scholar]
- 43.Jove R, Hanafusa H. Cell transformation by the viral src oncogene. Annu Rev Cell Biol. 1987;3:31–56. doi: 10.1146/annurev.cb.03.110187.000335. [DOI] [PubMed] [Google Scholar]
- 44.Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–190. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- 45.Sugimura M, Kobayashi K, Sagae S, et al. Mutation of the SRC gene in endometrial carcinoma. Jpn J Cancer Res. 2000;91:395–398. doi: 10.1111/j.1349-7006.2000.tb00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang NM, Yeh KT, Tsai CH, Chen SJ, Chang JG. No evidence of correlation between mutation at codon 531 of src and the risk of colon cancer in Chinese. Cancer Lett. 2000;150:201–204. doi: 10.1016/s0304-3835(99)00398-5. [DOI] [PubMed] [Google Scholar]
- 47.Nilbert M, Fernebro E. Lack of activating c-SRC mutations at codon 531 in rectal cancer. Cancer Genet Cytogenet. 2000;121:94–95. doi: 10.1016/s0165-4608(00)00226-0. [DOI] [PubMed] [Google Scholar]
- 48.Laghi L, Bianchi P, Orbetegli O, et al. Lack of mutation at codon 531 of SRC in advanced colorectal cancers from Italian patients. Br J Cancer. 2001;84:196–198. doi: 10.1054/bjoc.2000.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91:53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benistant C, Chapuis H, Mottet N, et al. Deregulation of the cytoplasmic tyrosine kinase cSrc in the absence of a truncating mutation at codon 531 in human bladder carcinoma. Biochem Biophys Res Commun. 2000;273:425–430. doi: 10.1006/bbrc.2000.2948. [DOI] [PubMed] [Google Scholar]
- 51.Jones RJ, Avizienyte E, Wyke AW, et al. Elevated c-Src is linked to altered cell-matrix adhesion rather than proliferation in KM12C human colorectal cancer cells. Br J Cancer. 2002;87:1128–1135. doi: 10.1038/sj.bjc.6600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Brunton VG, Ozanne BW, Paraskeva C, Frame MC. A role for epidermal growth factor receptor, c-Src and focal adhesion kinase in an in vitro model for the progression of colon cancer. Oncogene. 1997;14:283–293. doi: 10.1038/sj.onc.1200827. [DOI] [PubMed] [Google Scholar]
- 53.Myoui A, Nishimura R, Williams PJ, et al. C-SRC tyrosine kinase activity is associated with tumor colonization in bone and lung in an animal model of human breast cancer metastasis. Cancer Res. 2003;63:5028–5033. [PubMed] [Google Scholar]
- 54.Park SS, Eom YW, Kim EH, et al. Involvement of c-Src kinase in the regulation of TGF-beta1-induced apoptosis. Oncogene. 2004;23:6272–6281. doi: 10.1038/sj.onc.1207856. [DOI] [PubMed] [Google Scholar]
- 55.Fincham VJ, Frame MC. The catalytic activity of Src is dispensable for translocation to focal adhesions but controls the turnover of these structures during cell motility. Embo J. 1998;17:81–92. doi: 10.1093/emboj/17.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zou JX, Liu Y, Pasquale EB, Ruoslahti E. Activated SRC oncogene phosphorylates R-ras and suppresses integrin activity. J Biol Chem. 2002;277:1824–1827. doi: 10.1074/jbc.M103133200. [DOI] [PubMed] [Google Scholar]
- 57.Behrens J, Vakaet L, Friis R, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bourguignon LY, Zhu H, Shao L, Chen YW. CD44 interaction with c-Src kinase promotes cortactin-mediated cytoskeleton function and hyaluronic acid-dependent ovarian tumor cell migration. J Biol Chem. 2001;276:7327–7336. doi: 10.1074/jbc.M006498200. [DOI] [PubMed] [Google Scholar]
- 59.Trevino JG, Summy JM, Lesslie DP, et al. Inhibition of SRC expression and activity inhibits tumor progression and metastasis of human pancreatic adenocarcinoma cells in an orthotopic nude mouse model. American Journal of Pathology. 2006;168:962–972. doi: 10.2353/ajpath.2006.050570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hanke JH, Gardner JP, Dow RL, et al. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 61.Bain J, McLauchlan H, Elliott M, Cohen P. The specificities of protein kinase inhibitors: an update. Biochem J. 2003;371:199–204. doi: 10.1042/BJ20021535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sekharam M, Nasir A, Kaiser HE, Coppola D. Insulin-like growth factor 1 receptor activates c-SRC and modifies transformation and motility of colon cancer in vitro. Anticancer Res. 2003;23:1517–1524. [PubMed] [Google Scholar]
- 63.Yezhelyev MV, Koehl G, Guba M, et al. Inhibition of SRC tyrosine kinase as treatment for human pancreatic cancer growing orthotopically in nude mice. Clin Cancer Res. 2004;10:8028–8036. doi: 10.1158/1078-0432.CCR-04-0621. [DOI] [PubMed] [Google Scholar]
- 64.Golas JM, Lucas J, Etienne C, et al. SKI-606, a Src/Abl inhibitor with in vivo activity in colon tumor xenograft models. Cancer Res. 2005;65:5358–5364. doi: 10.1158/0008-5472.CAN-04-2484. [DOI] [PubMed] [Google Scholar]
- 65.Goi T, Shipitsin M, Lu Z, et al. An EGF receptor/Ral-GTPase signaling cascade regulates c-Src activity and substrate specificity. Embo J. 2000;19:623–630. doi: 10.1093/emboj/19.4.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhang SQ, Yang W, Kontaridis MI, et al. Shp2 regulates SRC family kinase activity and Ras/Erk activation by controlling Csk recruitment. Mol Cell. 2004;13:341–355. doi: 10.1016/s1097-2765(04)00050-4. [DOI] [PubMed] [Google Scholar]
- 67.Mao W, Irby R, Coppola D, et al. Activation of c-Src by receptor tyrosine kinases in human colon cancer cells with high metastatic potential. Oncogene. 1997;15:3083–3090. doi: 10.1038/sj.onc.1201496. [DOI] [PubMed] [Google Scholar]
- 68.Bao J, Gur G, Yarden Y. Src promotes destruction of c-Cbl: implications for oncogenic synergy between Src and growth factor receptors. Proc Natl Acad Sci U S A. 2003;100:2438–2443. doi: 10.1073/pnas.0437945100. [DOI] [PMC free article] [PubMed] [Google Scholar]