

Abstract
Posttranslational modification (PTM) of antigen is a way to break T-cell tolerance to self-antigens and promote autoimmunity. However, the precise mechanisms by which modifications would facilitate autoimmune T-cell responses and how they relate to particular autoimmune-associated MHC molecules remain elusive. Celiac disease is a T-cell mediated enteropathy with a strong HLA association where the immune response is directed mainly against deamidated cereal gluten peptides that have been modified by the enzyme transglutaminase 2. The disease is further characterized by autoantibodies to transglutaminase 2 that have extraordinary high disease specificity and sensitivity. There have been important advances in the knowledge of celiac disease pathogenesis, and these insights may be applicable to other autoimmune disorders where posttranslational modification plays a role. This insight gives clues for understanding the involvement of PTMs in other autoimmune diseases.
Introduction
As a model, celiac disease is conducive to study as the tissue targeted by the immune process is easily accessible and the onset of pathogenesis can be controlled by the administration of gluten. The links between the causative antigen, the HLA molecules required for pathogenesis, and the enzyme involved in posttranslational modification have been extensively analyzed. The notion that observations made in the celiac disease model may help gain insights into the role of posttranslational modification and the basis for association with particular MHC molecules in other autoimmune disorders serves as the foundation for this paper. Even though the causative antigen in celiac disease is foreign, we will argue that observations of celiac disease are relevant to autoimmunity. The reasoning is based on genetic observations demonstrating sharing of a large number of susceptibility loci between various autoimmune disorders and celiac disease [1]. Moreover, key features of the pathogenesis of celiac disease, as summarized in Figure 1, are shared with other autoimmune disorders, notably with rheumatoid arthritis.
Figure 1.
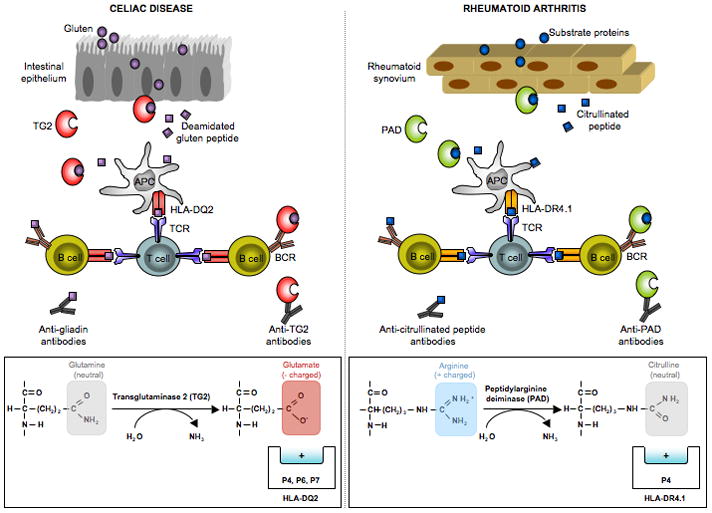
Key features of the pathogenesis of celiac disease that have parallels in rheumatoid arthritis. (i) In both diseases there are antibodies to posttranslationally modified antigens. In celiac disease there are antibodies to deamidated gliadin (gluten) [36] and in rheumatoid arthritis there are antibodies to citrullinated antigens [37,38]. (ii) In both diseases there are enzymes that mediate the posttranslational modifications of antigen. In celiac disease, transglutaminase 2 (TG2) converts certain glutamine residues to glutamate [39,40], and in rheumatoid arthritis various isoforms of peptidyl arginine deiminase (PAD) convert arginine residues to citrulline [41]. (iii) In both diseases, there are antibodies specific for these enzymes that mediate the polypeptide modifications [42–44]. (iv) In both diseases, the posttranslational modifications create antigens that are better suited to bind the HLA molecules associated with the disease. In celiac disease, which is associated with HLA-DQ2 and HLA-DQ8, glutamate serves as an anchor residue at positions P4, P6 and P7 for peptides binding to HLA-DQ2 [15,16] and as an anchor residue at positions P1 and P9 for peptides binding to HLA-DQ8 [18]. In rheumatoid arthritis, the P4 pocket of the disease associated HLA-DR4.1 molecule is positively charged [45] and thus will repel peptides with positively charged arginine residues at the P4 position. Peptides with neutrally charged citrulline at this position can be accommodated, however [46].
HLA and non-HLA genes predisposing to celiac disease
Celiac disease has a strong HLA association with HLA-DQ2 (DQA1*05,DQB1*02; a.k.a. HLA-DQ2.5) encoded in cis or trans and with HLA-DQ8 (DQ*03,DQB1*03:02). The very few celiac disease patients who do not carry HLA-DQ2.5 or HLA-DQ8, carry HLA-DQ molecules with one but not both of the DQA1*05 or DQB1*02 alleles found in HLA-DQ2.5, i.e. HLA-DQ7 (DQA1*05, DQB1*03:01) or HLA-DQ2.2 (DQA1*02:01,DQB1*02) [2]. HLA in celiac disease is a necessary, but not sufficient factor for disease development. Most individuals who express HLA-DQ2.5 or HLA-DQ8 never develop celiac disease. Contribution by non-HLA susceptibility loci is obviously also important for disease development. So far 39 non-HLA regions implicated in celiac disease development have been identified [3]**. Many of these are shared with other autoimmune diseases, in particular type 1 diabetes [4].
Enzymatic mechanism of transglutaminase 2 (TG2)
Transglutaminase 2 (TG2) belongs to a family of enzymes which are involved in crosslinking reactions [5]. The enzyme is targeting specific glutamine residues of polypeptides. As a first step in the enzymatic reaction, the glutamine residue makes a thiolester bond to the active site cysteine of TG2. This enzyme-substrate intermediate then reacts with a primary amine group, such as a lysine residue, and an isopeptide bonded covalent crosslink is formed in a process termed transamidation. Alternatively, the enzyme-substrate intermediate can react with water, and then the glutamine residue is converted to glutamate in a process termed deamidation. A deamidated product can also be formed by TG2-mediated hydrolysis of the isopeptide bond in the crosslink. TG2 is present both in the cytosol and the extracellular space. It is synthesized without a leader sequence, and by a poorly understood mechanism, it is transported to the extracellular environment. In the cytosol, it binds to GTP and exists in a closed, catalytically inactive conformation [6] (Figure 2). Extracellularly TG2 binds calcium, changes to an open conformation [7], and exerts its catalytic action.
Figure 2.
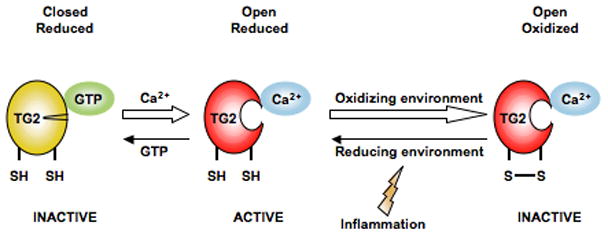
Transglutaminase 2 (TG2) is only active in an open conformation in a reduced state. In presence of GTP and in the absence of Ca2+ (i.e. intracellular environment), TG2 is in a reduced, closed state and the enzyme is inactive. Upon release to the extracellular environment with low GTP and high Ca2+, TG2 takes on an open conformation and is active. Usually there are oxidizing conditions in the extracellular environment and TG2 becomes inactivated in its open confirmation by the formation of a vicinal disulphide bond in the enzyme. Upon creation of reducing conditions, which has been shown to occur in lymph nodes after immunization, the disulphide bond is reduced and the enzyme can again take an active open conformation.
Regulation of TG2 activity by environmental factors
TG2 is ubiquitously present in tissues. There is evidence that most extracellular TG2 is inactive under normal physiological conditions, but abundant TG2 activity can be detected in vitro around the wound in a cultured fibroblast scratch assay and in vivo by stimulation with the toll-like receptor 3 ligand, polyinosinic-polycytidylic acid (poly(I:C)) [8]*. TG2 is rapidly inactivated in the extracellular environment by oxidation through formation of a vicinal disulphide bond outside of the active site, but reductive treatment can reactivate the catalytic activity of TG2 [9]**. Thus, TG2 may retain catalytic activity under reducing conditions in vivo, and more importantly, a change to a more reductive environment could potentially rescue TG2 from an inactive state (Figure 2). Little is known about the redox state in tissues in vivo, but a reductive environment after immunization, particularly in mesenteric lymph nodes after intraperitoneal immunization, has been reported [10]*. An ongoing immune response to any antigen may thus facilitate TG2 activity and an immune response to deamidated gluten.
Gluten T-cell epitopes
More than 15 different wheat gluten T-cell epitopes recognized in the context of HLA-DQ2.5 or HLA-DQ8 have been identified over the last 15 years. Recently epitopes from barley and rye were also characterized [11]*. The great majority of these T-cell epitopes contain glutamine residues that are targeted by TG2 converted to glutamate residues by a deamidation reaction. In general T cells of celiac disease patients recognize deamidated epitopes with greater efficiency than native ones. This effect of PTM appears to be stronger for HLA-DQ2.5 restricted epitopes than for HLA-DQ8 restricted epitopes, as many of the HLA-DQ2.5 restricted epitopes are not recognized in their native form whereas some HLA-DQ8 restricted epitopes are equally well recognized as native peptides. The reason for this difference may be related to the ability of HLA-DQ8 to recruit T cells with a negative charge that have the ability to stabilize native peptides bound to HLA-DQ8 [12]**.
Factors determining the selection of gluten T-cell epitopes
Despite the existence of many gluten epitopes by T cells of celiac disease patients, available data suggest that selection of these epitopes is anything but a random process. Gluten proteins are extremely heterogenous. In a single wheat cultivar several hundred distinct gluten proteins of more than 200 residues are expressed. In addition there is variation between various cultivars. Many different sequences are thus represented in gluten proteins. Three processes have been identified as being important for the selection of T-cell epitopes in gluten:
Proteolytic stability: The gluten epitopes localize in regions that are rich in proline residues [13]. These proline-rich gluten fragments are resistant to proteolysis by gastrointestinal digestion [14]. In terms of adaptive immunity to gluten, the increased integrity of these fragments in the gastrointestinal tract will likely lead to higher local concentrations of peptides that can bind to MHC class II molecules in the mucosa and hence lead to a better stimulation of CD4+ T cells in the lamina propria.
HLA binding: Determinant selection by MHC is well known to be an important factor for selection of antigenic epitopes, particularly for infectious agents. This also appears to be the case for selection of gluten epitopes which have to conform to certain binding motifs. Both HLA-DQ2.5 and HLA-DQ8 prefer negatively charged anchor residues, and notably these are residues that have been introduced by PTM. However, the negatively charged anchor residues are differentially located along the binding grooves of the two MHC molecules. HLA-DQ2.5 prefers negatively charged anchors at positions P1, P4 and P6 [15–17] whereas DQ8 prefers negatively charged anchors at positions P1 and P9 [18,19]. A comprehensive analysis of T-cell epitope usage in HLA-DQ2.5 or HLA-DQ8 bearing celiac disease patients revealed that the gluten T-cell epitopes presented by HLA-DQ2.5 or HLA-DQ8 were largely non-overlapping [20]. In addition to binding specificity, the ability to form kinetically stable peptide-MHC complexes appears to be a key factor for the in vivo generation of T-cell responses and development of celiac disease. The evidence for this notion comes from studies comparing the disease risk of two variants of HLA-DQ2 [21]**. HLA-DQ2.2, in contrast to HLA-DQ2.5, confers a low risk for celiac disease. The binding motif of HLA-DQ2.2 is very similar to that of HLA-DQ2.5 [22,23] except for an additional anchor position at P3 [22]. When gluten reactive T- cell clones obtained from HLA-DQ2.5 carrying patients were stimulated with HLA-DQ2.5 restricted epitopes in the context of HLA-DQ2.5 or HLA-DQ2.2, there was not much difference in the efficiency of presentation of the two MHC molecules, prompting the question as to why HLA-DQ2.2 carrying subjects are not at high risk for developing celiac disease [24,25]. An explanation for this conundrum was uncovered with the observation that, in contrast to HLA-DQ2.2, HLA-DQ2.5 can form kinetically stable complexes with the epitopes recognized by HLA-DQ2.5 patients [21]**. A polymorphism at DQ α22 controls this ability by which tyrosine of HLA-DQ2.5, but not phenylalanine of HLA-DQ2.2, can make a water mediated hydrogen bond to the peptide main chain.
Transglutaminase 2 specificity: TG2 demonstrates little specificity in targeting of lysine residues, but it is fairly particular in it’s targeting of glutamine residues. The enzyme preferentially targets residues within Gln-X-Pro sequences [26,27]. The specificity affects the generation of gluten T-cell epitopes. Notably, there is a correlation between how frequently T-cell epitopes are recognized by celiac disease patients and their propensity to be targeted as substrates for TG2 [28]. The notion that TG2 is a very important factor, perhaps in fact the most important factor involved in the selection of gluten T-cell epitopes in celiac disease patients, is supported by an experiment using TG2 to select peptides from a very complex mixture of gluten peptides [29]**. A proteolytic digest of gluten consisting of several thousand different peptides was incubated with a biotinylated primary amine and the TG2 enzyme. Peptides, which were targeted by TG2 and crosslinked with the biotin compound, were purified and sequenced. Very strikingly, of 31 selected peptides, more than 75% contained celiac disease related T-cell epitopes.
Effect of gluten peptide deamidation for the generation of stable peptide-MHC complexes
Studies on the differential risk of HLA DQ2.5 and HLA-DQ2.2 for celiac disease highlighted the importance of stable peptide-MHC complexes for generating in vivo T cell responses to gluten. It may well be that a major effect of deamidation in vivo also relates to peptide-MHC stability. The off-rate of a naturally occurring 33-mer fragment of digested gliadin was found to increase seven-fold upon deamidation [30]. Similar differences between native and deamidated peptides were observed for an HLA-DQ8 restricted epitope [12]** and may also be true for other gluten T-cell epitopes.
The impact of gluten deamidation on T-cell recognition
In addition to playing a critical role by allowing the generation of gluten peptides bound more avidly by MHC and hence the selection of T-cell epitopes, deamidation may also be involved in defining the T-cell receptor (TCR) repertoire that is selected in the response to gluten. In particular, this may be the case for HLA-DQ8, which can promote a response of similar amplitude to native and deamidated gluten peptide through the recruitment of T cells that have a negative charge in their TCR. This negative charge in the TCR stabilizes the binding of native peptide to HLA-DQ8 by providing the negative charge lacking in the peptide [12]**. In contrast, deamidated peptides do not impose this type of constraint on the T cells, and hence recruit a different TCR repertoire. Importantly, because T cells with a negative charge in their TCR recruited upon immunization with the native peptide are cross-reactive to the deamidated peptide, they can also be recruited once deamidation occurs. Consequently, anti-gluten T-cell responses can occur initially without TG2 activation. This T-cell response can create the inflammatory environment that leads to TG2 activation, which creates deamidated gluten peptides that in turn recruit a second set of T cells. The combination of HLA-DQ8 and TG2 thus creates the conditions for an immune response of maximal magnitude.
For many HLA-DQ2 restricted T-cell epitopes, the extent of the dependence on deamidation for T-cell recognition goes beyond what is explained by improved HLA binding of the deamidated peptides. This suggests that TCR interaction maybe involved as well. Evidence for this theory was obtained in a detailed study of the T-cell response in celiac disease patients to an immunodominant HLA-DQ2 restricted epitope from α-gliadin [31]*. A semipublic response to this epitope was found in the patients with conserved usage of V α and Vβ TCR chains. Furthermore, there was a conservation of a non-germline encoded arginine residue in the CDR3β loop of the TCRs which appeared essential for epitope recognition. This suggests that there can be an in vivo selection of TCRs specific for posttranslationally modified antigen in celiac disease, and that non-germline encoded TCR sequences may be implicated in the recognition of the modified residue. The latter mechanism has particular relevance to autoimmunity, as this type of recognition cannot be evolutionarily selected against, even if it leads to augmented recognition of modified autoantigens and autoimmunity.
Conclusion: Relevance to other autoimmune disorders
The ability of PTMs to cause break of tolerance has been widely suggested [32,33], the idea being that, given the correct MHC context, the creation of neoepitopes would allow for the recruitment of T cells that have not been negatively selected in the thymus (Figure 3). It is important to point out that creation of neoepitopes in the absence of inflammation would probably lead to peripheral tolerance to the neoantigen, because they would be presented by resting immature antigen-presenting cells that can not provide co-stimulation (Figure 3C). It is likely that tissue enzymes that induce the PTMs are, similarly to TG2 [8]**, not constitutively active and that the conditions that induce their activation are also conditions that would promote maturation of antigen-presenting cells and hence create the conditions for T-cell differentiation and expansion (Figures 3D and 3E). Under such a scenario, HLA molecules that can accommodate the posttranslationally modified self-peptides would present the peptides to T cells and promote the autoreactive immune response (Figure 3E). In addition to creating the conditions for the priming of autoreactive T cells, PTMs in the context of the right HLA may play a role in the amplification of the autoimmune response by expanding the potential TCR repertoire as described for HLA-DQ8 and gluten [12]*. Several observations suggest that PTMs similar to those occurring in celiac disease may also play a role in the pathogenesis of type 1 diabetes. HLA-DQ8 and HLA-DQ2, which are the main predisposing MHC molecules for both diseases, have a preference for negatively charged peptides. This suggests that with its ability to introduce negatively charged residues in proteins, TG2 could play a role in the pathogenesis of type 1 diabetes. In accordance, we have evidence that TG2 becomes activated as insulitis develops in non-obese diabetic mice (NOD) (BJ personal data), The primary autoantigens for type 1 diabetes in human and mice remain poorly defined. Interestingly, early pancreatic T cell infiltrates in NOD mice are primarily comprised of T cells with a negative charge in their TCR [34], akin to the T cells that recognize native gluten peptides. It is therefore tempting to speculate that the initial immune response against pancreatic self-antigens is directed against non-negatively charged self-peptides, and that upon inflammation and TG2 activation neoantigens of higher affinity for the HLA-DQ2 or HLA-DQ8 are generated that in turn will expand and amplify the immune response. Whether or not the autoantigen chromogranin A that does not bind optimally to I-Ag7 [35], can be modified by TG2 to acquire a higher affinity for I-Ag7, the mouse homolog of HLA-DQ8, remains to be determined.
Figure 3.
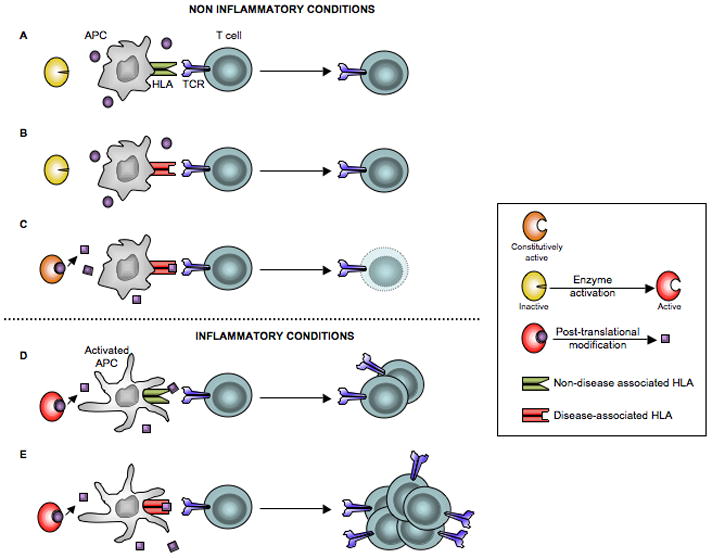
Depiction of various conditions and how these relate to T-cell activation that lead to disease development. Notably, inflammatory conditions may lead to induction of active posttranslationally modifying enzymes and antigen presenting cells that prime a disease-inducing T-cell response. A) Inactive enzyme, non-activated antigen presenting cells expressing HLA molecules that are not associated with disease are not permissive for T cell activation. B) Inactive enzyme, non-activated antigen presenting cells expressing HLA molecules that are associated with disease do not lead to T cell activation. C) Constitutively active enzyme in a non-inflamed tissue generates neoepitopes with increased affinity for the disease-associated HLA molecule. However, because peptides are presented by immature antigen presenting cells that cannot provide co-stimulation, T-cell deletion instead of T-cell activation occurs. D) Active enzyme, activated antigen presenting cells expressing HLA molecules that are not associated with disease do not lead to T cell activation. E) Active enzyme, activated antigen presenting cells that express HLA molecules associated with disease together constitute factors permissive for T-cell activation.
Identification of autoantigens in autoimmune disorders is central to understanding the pathogenesis of these disorders. Because of constraints related to human studies and the difficulty in obtaining human samples (especially longitudinally), the identification of primary and relevant autoantigens has been arduous. Lessons learned from celiac disease can inspire a strategy for the identification of autoantigenic peptides in diseases with a strong HLA association where there is also evidence for involvement of posttranslationally modifying enzymes (Figure 4). The main principle behind this strategy is to match the modifications imposed by the posttranslational modifying enzyme with the binding specificity of the disease associated HLA molecules, one should be able to identify the relevant PTM and candidate self-antigens whose relevance can be further tested by additional studies in human and mouse.
Figure 4.
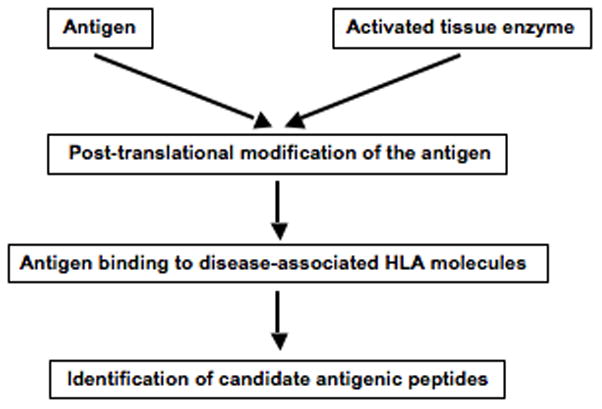
A proposed scheme for the identification of candidate peptide antigens in diseases with strong HLA associations and with evidence for the involvement of posttranslationally modifying tissue enzymes. Digests of relevant antigens or target tissue homogenates can be subjected to modification by activated tissue enzymes. Posttranslationally modified peptides are characterized by mass spectrometry, and peptides that harbor modifications that affect binding to the disease associated HLA molecules are identified. These peptides are tested for binding to the relevant HLA molecules for the closer identification of candidate antigenic peptides.
Highlights.
PTMs play a critical role in the pathogenesis of celiac disease.
PTMs select T-cell epitopes and TCR repertoire, and determine HLA association.
Conditions driving activation of enzymes mediating PTMs also mature APC allowing expansion of autoreactive T cells.
The redox status of the tissue plays a role in TG2 activation.
PTMs and HLA association can be used to identify self-antigens.
Acknowledgments
Funding by the Research Council of Norway, the European Research Council, the South-Eastern Norway Regional Health Authority (to L.M.S.) and National Institutes of Health (grant R01 DK-67180 to B.J.) is acknowledged. We thank V. Abadie for discussion and preparation of the figures, and B. Sally and H. Fehlner-Peach for critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Trynka G, Wijmenga C, van Heel DA. A genetic perspective on coeliac disease. Trends Mol Med. 2010;16:537–550. doi: 10.1016/j.molmed.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Karell K, Louka AS, Moodie SJ, Ascher H, Clot F, Greco L, Ciclitira PJ, Sollid LM, Partanen J. HLA types in celiac disease patients not carrying the DQA1*05-DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol. 2003;64:469–477. doi: 10.1016/s0198-8859(03)00027-2. [DOI] [PubMed] [Google Scholar]
- **3.Dubois PC, Trynka G, Franke L, Hunt KA, Romanos J, Curtotti A, Zhernakova A, Heap GA, Adany R, Aromaa A, et al. Multiple common variants for celiac disease influencing immune gene expression. Nat Genet. 2010;42:295–302. doi: 10.1038/ng.543. The most comprensive genome wide association study of celiac disease identifying 39 non-HLA susceptibility loci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Smyth DJ, Plagnol V, Walker NM, Cooper JD, Downes K, Yang JH, Howson JM, Stevens H, McManus R, Wijmenga C, et al. Shared and distinct genetic variants in type 1 diabetes and celiac disease. N Engl J Med. 2008;359:2767–2777. doi: 10.1056/NEJMoa0807917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol. 2003;4:140–156. doi: 10.1038/nrm1014. [DOI] [PubMed] [Google Scholar]
- 6.Liu S, Cerione RA, Clardy J. Structural basis for the guanine nucleotide-binding activity of tissue transglutaminase and its regulation of transamidation activity. Proc Natl Acad Sci U S A. 2002;99:2743–2747. doi: 10.1073/pnas.042454899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pinkas DM, Strop P, Brunger AT, Khosla C. Transglutaminase 2 undergoes a large conformational change upon activation. PLoS Biol. 2007;5:e327. doi: 10.1371/journal.pbio.0050327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **8.Siegel M, Strnad P, Watts RE, Choi K, Jabri B, Omary MB, Khosla C. Extracellular transglutaminase 2 is catalytically inactive, but is transiently activated upon tissue injury. PLoS ONE. 2008;3:e1861. doi: 10.1371/journal.pone.0001861. This paper shows that TG2 is not constitutively active in the intestinal muocsa and can be activated in vivo upon TLR3-induced tissue injury. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **9.Stamnaes J, Pinkas DM, Fleckenstein B, Khosla C, Sollid LM. Redox regulation of transglutaminase 2 activity. J Biol Chem. 2010;285:25402–25409. doi: 10.1074/jbc.M109.097162. This paper provides the molecular mechanism how oxidation inactivates TG2 and show that reductive conditions can reestablish catalytic activity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *10.Castellani P, Angelini G, Delfino L, Matucci A, Rubartelli A. The thiol redox state of lymphoid organs is modified by immunization: role of different immune cell populations. Eur J Immunol. 2008;38:2419–2425. doi: 10.1002/eji.200838439. Paper demonstrating that a local reduced microenvironment is generated during an immune response. [DOI] [PubMed] [Google Scholar]
- *11.Tye-Din JA, Stewart JA, Dromey JA, Beissbarth T, van Heel DA, Tatham A, Henderson K, Mannering SI, Gianfrani C, Jewell DP, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med. 2010;2:41ra51. doi: 10.1126/scitranslmed.3001012. This is the most recent papers of several that have identified epitopes of gluten recognized by T cells of celiac disease patients. This paper identifies new epitopes of barley and rye and establish which epitopes are most frequently recognized. [DOI] [PubMed] [Google Scholar]
- **12.Hovhannisyan Z, Weiss A, Martin A, Wiesner M, Tollefsen S, Yoshida K, Ciszewski C, Curran SA, Murray JA, David CS, et al. The role of HLA-DQ8 β57 polymorphism in the anti-gluten T-cell response in coeliac disease. Nature. 2008;456:534–538. doi: 10.1038/nature07524. This papers shows that β57 polymorphism in DQ8 impacts on the T cell repertoire and allows for the recruitment of T cells that are crossreactive for the native and posttranslationally modified antigens. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arentz-Hansen H, McAdam SN, Molberg, Fleckenstein B, Lundin KE, Jorgensen TJ, Jung G, Roepstorff P, Sollid LM. Celiac lesion T cells recognize epitopes that cluster in regions of gliadins rich in proline residues. Gastroenterology. 2002;123:803–809. doi: 10.1053/gast.2002.35381. [DOI] [PubMed] [Google Scholar]
- 14.Shan L, Molberg, Parrot I, Hausch F, Filiz F, Gray GM, Sollid LM, Khosla C. Structural basis for gluten intolerance in celiac sprue. Science. 2002;297:2275–2279. doi: 10.1126/science.1074129. [DOI] [PubMed] [Google Scholar]
- 15.Vartdal F, Johansen BH, Friede T, Thorpe CJ, Stevanovic S, Eriksen JA, Sletten K, Thorsby E, Rammensee HG, Sollid LM. The peptide binding motif of the disease associated HLA-DQ(α1*0501, β1*0201) molecule. Eur J Immunol. 1996;26:2764–2772. doi: 10.1002/eji.1830261132. [DOI] [PubMed] [Google Scholar]
- 16.van de Wal Y, Kooy YMC, Drijfhout JW, Amons R, Koning F. Peptide binding characteristics of the coeliac disease-associated DQ(α1*0501, β1*0201) molecule. Immunogenetics. 1996;44:246–253. doi: 10.1007/BF02602553. [DOI] [PubMed] [Google Scholar]
- 17.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwok WW, Domeier ML, Raymond FC, Byers P, Nepom GT. Allele-specific motifs characterize HLA-DQ interactions with a diabetes-associated peptide derived from glutamic acid decarboxylase. J Immunol. 1996;156:2171–2177. [PubMed] [Google Scholar]
- 19.Henderson KN, Tye-Din JA, Reid HH, Chen Z, Borg NA, Beissbarth T, Tatham A, Mannering SI, Purcell AW, Dudek NL, et al. A structural and immunological basis for the role of human leukocyte antigen DQ8 in celiac disease. Immunity. 2007;27:23–34. doi: 10.1016/j.immuni.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 20.Tollefsen S, Arentz-Hansen H, Fleckenstein B, Molberg, Raki M, Kwok WW, Jung G, Lundin KE, Sollid LM. HLA-DQ2 and -DQ8 signatures of gluten T cell epitopes in celiac disease. J Clin Invest. 2006;116:2226–2236. doi: 10.1172/JCI27620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **21.Fallang LE, Bergseng E, Hotta K, Berg-Larsen A, Kim CY, Sollid LM. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol. 2009;10:1096–1101. doi: 10.1038/ni.1780. This paper provides a molecular explanation for the differential risk of two HLA-DQ2 variants with celiac disease, and suggests that stable HLA binding of gluten peptides is essential for the making of a T-cell response in vivo. [DOI] [PubMed] [Google Scholar]
- 22.van de Wal Y, Kooy YC, Drijfhout JW, Amons R, Papadopoulos GK, Koning F. Unique peptide binding characteristics of the disease-associated DQ(α1*0501, β1*0201) vs the non-disease-associated DQ(α1*0201, β1*0202) molecule. Immunogenetics. 1997;46:484–492. doi: 10.1007/s002510050309. [DOI] [PubMed] [Google Scholar]
- 23.Johansen BH, Jensen T, Thorpe CJ, Vartdal F, Thorsby E, Sollid LM. Both alpha and beta chain polymorphisms determine the specificity of the disease-associated HLA-DQ2 molecules, with beta chain residues being most influential. Immunogenetics. 1996;45:142–150. doi: 10.1007/s002510050182. [DOI] [PubMed] [Google Scholar]
- 24.Vader W, Stepniak D, Kooy Y, Mearin L, Thompson A, van Rood JJ, Spaenij L, Koning F. The HLA-DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten-specific T cell responses. Proc Natl Acad Sci U S A. 2003;100:12390–12395. doi: 10.1073/pnas.2135229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiao SW, Bergseng E, Molberg O, Jung G, Fleckenstein B, Sollid LM. Refining the rules of gliadin T cell epitope binding to the disease-associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J Immunol. 2005;175:254–261. doi: 10.4049/jimmunol.175.1.254. [DOI] [PubMed] [Google Scholar]
- 26.Vader LW, de Ru A, van Der WY, Kooy YM, Benckhuijsen W, Mearin ML, Drijfhout JW, van Veelen P, Koning F. Specificity of tissue transglutaminase explains cereal toxicity in celiac disease. J Exp Med. 2002;195:643–649. doi: 10.1084/jem.20012028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleckenstein B, Molberg, Qiao SW, Schmid DG, Von Der MF, Elgstoen K, Jung G, Sollid LM. Gliadin T cell epitope selection by tissue transglutaminase in celiac disease. Role of enzyme specificity and pH influence on the transamidation versus deamidation process. J Biol Chem. 2002;277:34109–34116. doi: 10.1074/jbc.M204521200. [DOI] [PubMed] [Google Scholar]
- 28.Dorum S, Qiao SW, Sollid LM, Fleckenstein B. A quantitative analysis of transglutaminase 2-mediated deamidation of gluten peptides: implications for the T-cell response in celiac disease. J Proteome Res. 2009;8:1748–1755. doi: 10.1021/pr800960n. [DOI] [PubMed] [Google Scholar]
- **29.Dorum S, Arntzen MO, Qiao SW, Holm A, Koehler CJ, Thiede B, Sollid LM, Fleckenstein B. The preferred substrates for transglutaminase 2 in a complex wheat gluten digest are peptide fragments harboring celiac disease T-cell epitopes. PLoS One. 2010;5:e14056. doi: 10.1371/journal.pone.0014056. Paper providing evidence that TG2 is a very important factor for the selection of T-cell epitopes in celiac disease. It also presents an efficient method to identify T-cell epitopes in celiac disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia J, Sollid LM, Khosla C. Equilibrium and kinetic analysis of the unusual binding behavior of a highly immunogenic gluten peptide to HLA-DQ2. Biochemistry. 2005;44:4442–4449. doi: 10.1021/bi047747c. [DOI] [PubMed] [Google Scholar]
- *31.Qiao SW, Ráki M, Gunnarsen KS, Løset GÅ, Lundin KEA, Sandlie I, Sollid LM. Post-translational modification of gluten shapes T-cell receptor usage in celiac disease. J Immunol. 2011 doi: 10.4049/jimmunol.1101526. In press. This paper shows important aspects of the recognition of a deamidated gluten epitope at the TCR level by studying T cells from celiac disease patients. [DOI] [PubMed] [Google Scholar]
- 32.Doyle HA, Mamula MJ. Post-translational protein modifications in antigen recognition and autoimmunity. Trends Immunol. 2001;22:443–449. doi: 10.1016/s1471-4906(01)01976-7. [DOI] [PubMed] [Google Scholar]
- 33.Petersen J, Purcell AW, Rossjohn J. Post-translationally modified T cell epitopes: immune recognition and immunotherapy. J Mol Med (Berl) 2009;87:1045–1051. doi: 10.1007/s00109-009-0526-4. [DOI] [PubMed] [Google Scholar]
- 34.Baker FJ, Lee M, Chien YH, Davis MM. Restricted islet-cell reactive T cell repertoire of early pancreatic islet infiltrates in NOD mice. Proc Natl Acad Sci U S A. 2002;99:9374–9379. doi: 10.1073/pnas.142284899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stadinski BD, Delong T, Reisdorph N, Reisdorph R, Powell RL, Armstrong M, Piganelli JD, Barbour G, Bradley B, Crawford F, et al. Chromogranin A is an autoantigen in type 1 diabetes. Nat Immunol. 2010;11:225–231. doi: 10.1038/ni.1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Osman AA, Gunnel T, Dietl A, Uhlig HH, Amin M, Fleckenstein B, Richter T, Mothes T. B cell epitopes of gliadin. Clin Exp Immunol. 2000;121:248–254. doi: 10.1046/j.1365-2249.2000.01312.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girbal-Neuhauser E, Durieux JJ, Arnaud M, Dalbon P, Sebbag M, Vincent C, Simon M, Senshu T, Masson-Bessiere C, Jolivet-Reynaud C, et al. The epitopes targeted by the rheumatoid arthritis-associated antifilaggrin autoantibodies are posttranslationally generated on various sites of (pro)filaggrin by deimination of arginine residues. J Immunol. 1999;162:585–594. [PubMed] [Google Scholar]
- 38.Schellekens GA, de Jong BA, van den Hoogen FH, van de Putte LB, van Venrooij WJ. Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Clin Invest. 1998;101:273–281. doi: 10.1172/JCI1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Molberg, McAdam SN, Körner R, Quarsten H, Kristiansen C, Madsen L, Fugger L, Scott H, Roepstorff P, Lundin KEA, et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut-derived T cells. Nat Med. 1998;4:713–717. doi: 10.1038/nm0698-713. [DOI] [PubMed] [Google Scholar]
- 40.van de Wal Y, Kooy Y, van Veelen P, Pena S, Mearin L, Papadopoulos G, Koning F. Selective deamidation by tissue transglutaminase strongly enhances gliadin-specific T cell reactivity. J Immunol. 1998;161:1585–1588. [PubMed] [Google Scholar]
- 41.Vossenaar ER, Zendman AJ, van Venrooij WJ, Pruijn GJ. PAD, a growing family of citrullinating enzymes: genes, features and involvement in disease. Bioessays. 2003;25:1106–1118. doi: 10.1002/bies.10357. [DOI] [PubMed] [Google Scholar]
- 42.Dieterich W, Ehnis T, Bauer M, Donner P, Volta U, Riecken EO, Schuppan D. Identification of tissue transglutaminase as the autoantigen of celiac disease. Nat Med. 1997;3:797–801. doi: 10.1038/nm0797-797. [DOI] [PubMed] [Google Scholar]
- 43.Zhao J, Zhao Y, He J, Jia R, Li Z. Prevalence and significance of anti-peptidylarginine deiminase 4 antibodies in rheumatoid arthritis. J Rheumatol. 2008;35:969–974. [PubMed] [Google Scholar]
- 44.Halvorsen EH, Pollmann S, Gilboe IM, van der Heijde D, Landewe R, Odegard S, Kvien TK, Molberg O. Serum IgG antibodies to peptidylarginine deiminase 4 in rheumatoid arthritis and associations with disease severity. Ann Rheum Dis. 2008;67:414–417. doi: 10.1136/ard.2007.080267. [DOI] [PubMed] [Google Scholar]
- 45.Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 1997;7:473–481. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- 46.Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E. Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol. 2003;171:538–541. doi: 10.4049/jimmunol.171.2.538. [DOI] [PubMed] [Google Scholar]