

Abstract
This is the first installment of 2 articles that discuss the biology and pathophysiology of wound healing, review the role that growth factors play in this process, and describe current ways of growth factor delivery into the wound bed. Part 1 discusses the latest advances in clinicians’ understanding of the control points that regulate wound healing. Importantly, biological similarities and differences between acute and chronic wounds are considered, including the signaling pathways that initiate cellular and tissue responses after injury, which may be impeded during chronic wound healing.
Keywords: acute wound healing, drug delivery and wounds, wound care strategies
Acute and chronic wounds affect millions of people in the United States and around the world. In recent decades, clinicians have gained a better understanding of the mechanisms of normal wound repair process and causes of delays in healing. This progress has led to significant improvement in the quality of life of affected patients. This article reviews the latest insights and opportunities for wound repair science and innovations in wound care.
PHASES OF NORMAL WOUND HEALING
Acute wounds are a common health problem, with 11 million people affected1 and approximately 300,000 people hospitalized yearly in the United States.2 Typically, acute wound healing is a well-organized process leading to predictable tissue repair where platelets, keratinocytes, immune surveillance cells, microvascular cells, and fibroblasts play key roles in the restoration of tissue integrity.3,4 The wound repair process can be divided into 4 temporarily and spatially overlapping phases: coagulation, inflammation, formation of granulation tissue (proliferative phase), and remodeling or scar formation phase (Figure 1A-C).
Figure 1. MECHANISMS OF NORMAL WOUND HEALING.

Normal wound healing processes can be divided into 4 overlapping phases: coagulation (not shown), inflammatory phase (A), proliferative phase/granulation tissue formation (B), and remodeling phase (C). During coagulation and inflammatory phases (A) of the healing, blood-borne cells—neutrophils, macrophages, as well as platelets—play critical roles. These cells provide growth factors and provisional matrices that are necessary for recruitment of epidermal and dermal cells into the wound bed. The proliferative phase (B) starts at approximately 3 days after injury and is characterized by increased levels of keratinocyte and fibroblast proliferation, migration, and ECM synthesis in response to autocrine, paracrine, and juxtacrine growth factors. Angiogenesis/neovascularization also occurs during this phase. Because of the presence of blood vessels, the tissue has a granular appearance (granulation tissue). Finally, at approximately 1 to 2 weeks after injury, differentiated fibroblastic cells (myofibroblasts) that present within the granulation tissue begin to remodel extracellular matrix (C). Extracellular matrix remodeling followed by apoptosis of resident cells leads to the formation of an acellular scar.
Coagulation/Inflammatory Phase
Immediately after injury, platelets adhere to damaged blood vessels, initiate a release reaction, and begin a hemostatic reaction, giving rise to a blood-clotting cascade that prevents excessive bleeding and provides provisional protection for the wounded area (Figure 1A). As has been well studied, blood platelets release well over a dozen growth factors, cytokines, and other survival or apoptosis-inducing agents.5 Key components of the platelet release reaction include platelet-derived growth factor (PDGF) and transforming growth factors A1 and 2 (TGF-A1 and TGF-2), which attract inflammatory cells, such as leukocytes, neutrophils, and macrophages.3 As leukocytes are phagocytic cells, they release reactive oxygen species (ROS) that are antimicrobial and proteases that clear the wound of foreign bodies and bacteria. Resolution of the inflammatory phase is accompanied by apoptosis of inflammatory cells, which occurs gradually within a few days after wounding. The mechanism for resolution of inflammation is currently unknown. However, studies suggest that anti-inflammatory cytokines, such as TGF-A1 and interleukin 1, and bioactive lipids, such as cyclopentenone prostaglandin, lipoxins, and resolvins, take part in this process.6,7 The exact role of these entities during inflammatory phase resolution is under investigation.
Proliferative Phase: Granulation Tissue Formation
As the inflammatory phase subsides, the proliferative phase of repair begins (Figure 1A, B). At this stage, growth factors produced by remaining inflammatory cells and migrating epidermal and dermal cells act in autocrine, paracrine, and juxtacrine fashion to induce and maintain cellular proliferation while initiating cellular migration; all these events are required for the formation of granulation tissue while supporting epithelialization.3,4 As dermal and epidermal cells migrate and proliferate within the wound bed, there is a frank requirement for an adequate blood supply for nutrient delivery, gas, and metabolite exchange. Therefore, for wound healing to progress normally, a robust angiogenic response must be initiated and sustained.
Wound healing angiogenesis begins immediately after injury when local hypoxia, secondary to injury-induced blood vessel disruption, occurs. This event fosters the production of proangiogenic factors. Vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF-2), and PDGF,8 initially released by platelets and then by resident cells within the wound bed, are all central mediators of injury-induced angiogenic induction. In response, endothelial cells degrade basement membrane, migrate toward the wound site, proliferate, and form cell-cell contacts and eventually new blood vessels.3,9 More recently, it has been revealed that endothelial progenitor cells (EPCs) are also required for wound revascularization.10–12 Normally, EPCs reside in the bone marrow and are recruited into the circulation in response to injury. Subsequently, EPCs are engrafted into the remodeling microvasculature, taking residence adjacent to endothelial cells bordering the injury site. Endothelial progenitor cell mobilization is mediated by nitric oxide, VEGF, and matrix metalloproteinases (MMP), particularly MMP-912; EPC engraftment and possibly differentiation occur in response to stomal cell–derived factor 1> and, as has become apparent more recently, insulinlike growth factor (IGF).13 Although more research needs to be done to further elucidate the mechanisms of EPC recruitment and homing, it is clear that these progenitor cells are necessary for normal wound healing– associated neovasculogenesis and injury repair. In fact, key signaling intermediates responsible for coordinating/regulating wound healing angiogenesis and vasculogenesis may be dysfunctional during diabetes.14 Indeed, diabetic patients prone to the development of chronic wounds11 may exhibit deficiencies in either EPC bone marrow release or peripheral tissue homing and engraftment. Thus, therapies aimed at correcting EPC-linked deficiencies may prove beneficial for treating diabetes-induced chronic wounds.11
Matrix Remodeling and Scar Formation
Reestablishment of a normal blood supply provides a favorable microenvironment for epidermal and dermal cell migration and proliferation (Figure 1C). In turn, this leads to wound re-epithelialization and restoration of epidermal integrity. Fibroblasts proliferate within the wound and synthesize extra-cellular matrix (ECM) forming granulation tissue perfused with newly formed blood vessels. Simultaneously, provisional matrix mainly consisting of collagen III, fibrin, fibronectin, and hyaluronic acid is progressively substituted with ECM mainly containing collagen I.3 Next, wound contraction and matrix remodeling occur3 (Figure 1C). Contraction is mainly achieved by differentiated fibroblasts or myofibroblasts that, in response to TGF-A, tissue tension, and the presence of certain matrix proteins (such as ED-A fibronectin and tenascin C), acquire smooth muscle actin–containing stress fibers. Fibroblast-induced contractile forces are then transmitted to the ECM via cytoskeleton-associated and ECM receptor–dependent mechanocoupling focal adhesion complexes, that is, integrin receptors.15 Another mechanism leading to wound contraction is fibroblast motility with consequent matrix reorganization.16 This dynamic and reciprocal process involves slow cycles of ECM synthesis and degradation both occurring in a stromal- or fibroblastic cell–dependent manner. Here, matrix-remodeling enzymes, particularly MMPs, play important roles in remodeling the local matrix microenvironment in support of several healing responses, including cellular migration, proliferation, and angiogenic induction. Finally, apoptosis of fibroblastic cells occurs, leading to the formation of a relatively acellular scar tissue whose tensile strength is comparable with unwounded skin (Figure 1C).
Although the importance of apoptosis in granulation tissue remodeling and scar formation is widely accepted, the triggers of apoptosis are not well understood.17 It has been suggested that TGF-A, tumor necrosis factor, and surprisingly FGF-2 (that normally is considered a stimulator of cell proliferation) can lead to an increase in the number of apoptotic cells during the final phase of healing.18,19 Inability of dermal cells, particularly myofibroblasts to undergo timely apoptosis, has been linked to wound healing pathologies, including the hypertrophic scar and keloid formation.20 Clinicians’ improved understanding of the role of apoptosis in normal and pathological wound healing may initiate novel approaches for their treatment and/or prevention.
CHRONIC WOUNDS: CLASSIFICATION AND MOLECULAR MECHANISMS OF CHRONICITY
With the estimated number of older adults 65 years or older in the United States almost doubling (from 35 million to 53 million people) by 2030,21 and the estimated risk of developing diabetes for children born in 2000 as high as 35%,22 the anticipated risks of diabetes and age-associated nonhealing chronic wounds continue to increase dramatically. In fact, annual chronic wound care costs now exceed $1 billion in the United States alone,23 and represents ~2% of total EU financial resources24 (Table 1). Importantly, the majority of the chronic wounds begin as minor traumatic injuries. Penetrating injuries, insect bites, or even simple scratches of dry skin that would normally heal within a few days/weeks can lead to formation of a nonhealing wound in patients with underlying pathologies,25 such as diabetes-induced and nondiabetic neuropathies.
Table 1.
MAJOR TYPES OF CHRONIC WOUNDS
| Wound Type | Pathology | No. of Affected Patients | Cost of Treatment | Total Annual Cost |
|---|---|---|---|---|
| Venous ulcers | Venous insufficiency, thrombosis, varicosis | 400,000–600,000 | $5000–$10,000 | $1.9 billion to $2.5 billion94 |
| Arterial ulcers | Macroangiopathy, atherosclerosis, arterial insufficiency |
100,00033 | $9000–$16,000 | |
| Diabetic ulcers | Neuropathy, microangiopathy, hyperglycemia |
2 million (http://www.feetnet.com/statistics.php) | $6000/patient | $150 million95 |
| Pressure ulcers | Immobility, excessive pressure | 1.3 million to 3 million | Up to $70,00096 | $3.5 billion to $7.0 billion annually96 |
Chronic wounds can be classified into vascular ulcers (eg, venous and arterial ulcers), diabetic ulcers, and pressure ulcers(Table 1). Some common features shared by each of these include a prolonged or excessive inflammatory phase,7 persistent infections,26 formation of drug-resistant microbial biofilms,27 and the inability of dermal and/or epidermal cells to respond to reparative stimuli. In aggregate, these pathophysiologic phenomena result in the failure of these wounds to heal (Figure 2). The underlying pathologies, however, deviate in different types of chronic wounds.
Figure 2. NORMAL VERSUS CHRONIC WOUND HEALING.

Microenvironment within a normal wound bed (left) is characterized by the presence of numerous growth factors, a well-organized ECM, and responsive cell populations. Matrix synthesis, here, exceeds its degradation, and MMP activity is regulated by the presence of MMP inhibitors (TIMPs). Angiogenesis and neovascularization of normal wounds proceed in a timely manner via well-regulated sprouting of existing blood vessels and recruitment of endothelial progenitor cells (EPC), respectively. Finally, unlike their chronic counterparts, acute wounds are generally characterized by low bacterial burden. Chronic wounds (right) often have high incidence of bacterial biofilms, leading to persistent inflammation, excessive proteolysis, and degradation of critical growth factors, receptors, and/or ECM. Cells residing within these wounds are unable to proliferate and/or migrate effectively perhaps because of the absence of functional receptors or appropriate promigratory matrix substrates. Impaired angiogenesis and neovascularization, both hallmarks of chronic wounds, result in insufficient oxygen and nutrient supply for the cells residing within the wound bed, which leads to further wound bed mutilation and impaired healing.
Venous ulcers display profound pathological changes that arise secondary to venous valvular incompetence in the deep and superficial veins. This, in turn, leads to a constant blood backflow resulting in an increase in venous pressure. Pressure-induced changes in blood vessel wall permeability then lead to leakage of fibrin and other plasma components into the perivascular space. Accumulation of fibrin has direct and negative effects on wound healing. It down-regulates collagen synthesis,28 leads to formation of pericapillary fibrin cuffs that create a barrier for normal vessel function, and traps blood-derived growth factors.29,30 In the 1980s and 1990s, the cuffs were considered as continuous obstructions preventing free blood-dermis oxygen exchange.30 Recently, however, using confocal microscopy, it has been demonstrated that fibrin deposits surrounding dermal veins are patchlike and discontinuous.31 This finding questions the barrier role of fibrin cuffs and suggests the presence of other yet unknown factors contributing to low oxygen tension found in venous ulcers and surrounding tissues. Identification of these factors may reveal novel targets for therapeutic interventions and treatment of venous ulcers.
Arterial ulcers are less common than chronic venous wounds. They occur because of arterial insufficiency caused by atherosclerosis or embolism that can lead to narrowing of arterial lumen and ischemia, which prevents timely healing of minor traumatic injuries.32 Unlike venous ulcers, which generally arise between the knee and the ankle, arterial leg wounds may present at any spot distal to arterial perfusion such as a tip of a toe. It is estimated that arterial ulcers affect 100,000 Americans annually.33 Unlike venous ulcers that often can be improved with therapeutic compression, chronic wounds linked to arterial insufficiency can be treated successfully only after the restoration of arterial function via revascularization.32 Current options for limb revascularization are rather limited and include reconstructive surgery (angioplasty) or pharmaceutical interventions. Because failure of wound revascularization almost inevitably leads to limb amputation in arterial ulcer sufferers, novel techniques allowing for restoration of blood supply to the wound bed, including stem cell therapies, are now under investigation.34
Pressure ulcers develop as a result of prolonged unrelieved pressure and shearing force applied to skin and the underlying muscle tissue leading to a decrease in oxygen tension, ischemiareperfusion injury, and tissue necrosis. Pressure ulcers are common in patients with compromised mobility and decreased sensory perception (neuropathies)35 and are exacerbated in individuals with arterial and venous insufficiencies described above.
Complications of aging and diabetes can lead to and exacerbate vascular pathologies related to both arterial and venous insufficiencies and worsen pressure ulcers. Other abnormalities leading to development of chronic wounds in diabetic patients (also called diabetic ulcers) include neuropathy, often linked to vascular impairment, deficiencies in muscle metabolism, and a number of microvascular pathologies often caused by hyperglycemia.4 Macroscopic pathologies seen in chronic, particularly diabetic, wounds often are linked to cellular phenotypic abnormalities, including low mitogenic/motogenic potential and inability to respond to environmental cues. Thus, a better understanding of these cellular changes may aid in the development of better treatment options.
Although all of the wounds described previously may have different origins, each wound is characterized by a chronically inflamed wound bed and a failure to heal (Figures 2 and 3). Excessive recruitment of inflammatory cells to the wound bed often triggered by infection and cell extravasation is facilitated by disproportionate expression of vascular cell adhesion molecule 1 and interstitial cell adhesion molecule 1 by resident endothelial cells. Inflammatory cells accumulated inside the chronic wound produce various ROS that damage structural elements of the ECM and cell membranes and lead to premature cell senescence36 (Figure 2). In addition to these direct negative effects, ROS together with proinflammatory cytokines induce production of serine proteinases and MMPs that degrade and inactivate components of the ECM and growth factors necessary for normal cell function.7 Inactivation of proteinase inhibitors by proteolytic degradation augments this process. Therefore, although the production of growth factors is often increased in chronic compared with acute wounds, their quantity and bio-availability are significantly decreased.37,38
Figure 3. PHYSIOLOGIC IMBALANCE: A KEY FEATURE OF CHRONIC WOUNDS.
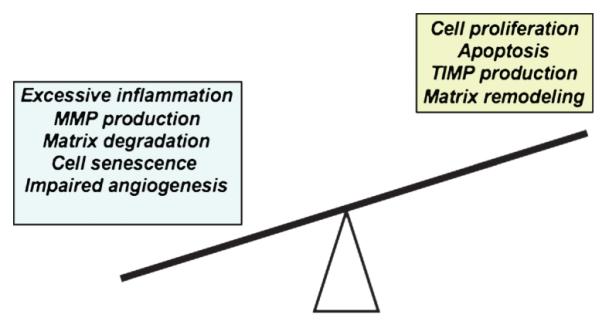
Inflammation, MMP production, matrix degradation, and cell senescence/apoptosis are all elevated in chronic wounds. These processes cannot be overcome because of insufficient levels of cell proliferation, ECM synthesis, production of TIMPs, and impaired angiogenesis/ neovascularization. This imbalance leads to inability of chronic wounds to heal.
Unlike acute wounds, which generally heal without significant interventions, all types of chronic wounds represent major challenges for patients and caregivers. It is now understood that the inability of the chronic wound to heal is caused by both cellular and molecular abnormalities occurring within the wound bed (Figure 4). However, proper diagnosis of wound etiology, selection of effective treatment, and prevention of wound reoccurrence remain a problem for chronic wound sufferers and healthcare providers.32 Next, the authors discuss the role of tissue microenvironment in normal and pathological wound healing.
Figure 4. CHRONIC WOUND CONUNDRUM.
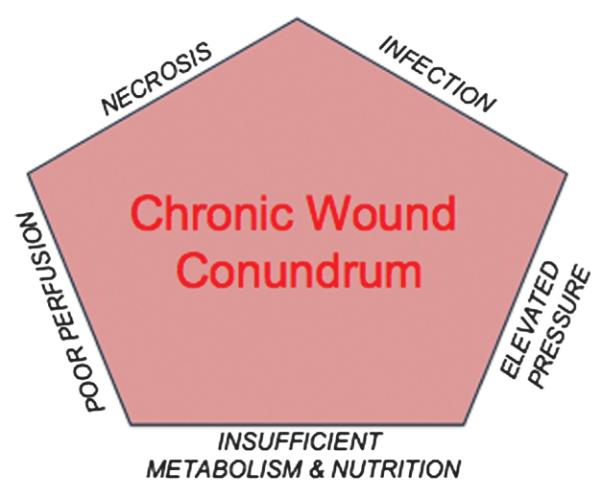
Diagrammatic representation of those physiologic functions that are perturbed or disequilibrated during chronic wound healing.
Phenotypic Abnormalities in Chronic Wound Cells
The phenotypic abnormalities of epidermis- and dermis-derived cells residing in chronic wounds include lower density of growth factor receptors and lower mitogenic potential preventing them from responding properly to environmental cues. For instance, fibroblasts, isolated from patients with chronic diabetic, chronic nondiabetic wounds, or patients with venous insufficiency, have lower mitogenic response to PDGF-AB, IGF, bFGF, and epidermal growth factor applied separately or in combination. These findings are likely due to a decrease in receptor density.39–41 Furthermore, fibroblasts isolated from leptin receptor–deficient diabetic mice, as well as derived from patients with chronic venous insufficiency, have reduced motility, compared with normal fibroblasts.42,43 These cellular abnormalities impede the formation of granulation tissue and ECM deposition, leading to formation of nonhealing wounds.
Keratinocytes derived from chronic ulcers have also been reported to possess a “chronic wound–associated” phenotype.44 Overexpressing the proliferation marker Ki67, these cells up-regulate expression of several cell cycle–associated genes, such as CDC2 and cyclin B1, suggesting a hyperproliferative status.45 However, these chronic wound–derived keratinocytes exhibit impaired migratory potential. The mechanisms of this impairment are not completely understood but have been linked to decreased production of laminin 332 (formerly known as laminin 5), which is an important epithelial ECM component and substrate for injury-induced keratinocyte migration.44 In addition, these cells possess an increased activation of A-catenin/c-myc pathway46 and do not express markers of differentiation, particularly keratin 10 and keratin 2. Finally, several genes encoding a variety of growth factors are down- or up-regulated; for example, VEGF, epiregulin, and TGF-A2 expression are decreased, whereas PDGF and platelet-derived endothelial growth factor encoding genes are up-regulated.45 Decreased growth factor production directly confirms the impaired state of the keratinocytes residing within a chronic wound and inability to fully participate in repair processes, whereas up-regulation of key growth factor genes enables a sustained proliferative capacity, suggesting that this could be an “entry point” for therapeutic intervention. Motogenic stimuli together with activators of keratinocyte differentiation, such as recently described hyperforin,47 may be able to induce phenotypic changes and transform the chronic wound keratinocytes into competent cells necessary for epithelialization. Similarly, modern transduction techniques48 could be used to improve growth factor responsiveness of cells residing in chronic wounds by increasing the density of growth factor receptors. These are just a few examples of how improved clinical understanding of cellular pathologies might lead to development of novel therapeutic modalities, which could be used for treatment of chronic wounds and are discussed in more detail in the subsequent sections.
Perturbations in the ECM Microenvironment That Contribute to Sustaining Wound Chronicity
The microenvironment of the chronic wound bed is heralded by a matrix (Figure 3). Information about the differences in chemical composition of the ECM found in chronic and acute wounds is scarce and controversial. It is known, however, that deposition of a number of matrix components is different in chronic as compared with acute wounds. For example, chronic wounds are characterized by prolonged49 or insufficient expression of fibronectin, chondroitin sulfate, and tenascin, which gives rise to impaired cellular proliferation and migration.50 Recently, reduced production of laminin 332—a basement membrane component that serves as a haptotactic substrate for postinjury keratinocyte motility44—was found to be one of the reasons for impaired reepithelialization and wound healing. Changes of the ECM, including posttranslational modification of key structural components, can also negatively influence cellular responses to injury. For instance, matrix glycation (Figure 2) is often seen in diabetic patients and is likely to be responsible for or linked to premature cell senescence, apoptosis, inhibition of cell proliferation, migration, and angiogenic sprout formation.51 Glycation adds to matrix instability and disrupts matrix assembly and interactions between collagen and its binding partners, including heparan sulfate proteoglycans.52,53 High glucose has also been shown to stimulate MMP production by fibroblasts, macrophages, and endothelial cells, thus contributing to a “vicious” cycle of matrix degradation detrimental for cell survival and therefore wound healing.54,55 Matrix instability that occurs because of glycation and insufficient intermolecular cross-linking seen under hypoxic conditions56 and excessive matrix degradation by MMPs are also detrimental to the healing process. Matrix instability prevents normal cell-matrix interactions necessary for cell survival and function and, ultimately, injury repair. Therefore, inhibition of matrix degradation, addition of exogenous matrices, and induction of matrix synthesis by resident cells all provide therapeutic opportunities. As an example, collagen-based dressings (Promogran; Systagenix, Quincy, Massachusetts) have been used in clinical trials, successfully decreasing the levels of matrix-degrading enzymes and improving healing.57 To date, protease inhibitors have not been used to treatchronic wounds clinically. However, synthetic bisphosphonates are under investigation for application in wound care.58 Further research is needed to delineate the therapeutic potential of both protease inhibitors and/or ECM-based preparations for chronic wound care.
Biofilms and the Chronic Wound Bed
Infection is an extrinsic factor that causes delay of wound healing (Figure 4), contributing to wound chronicity, morbidity, and mortality.59 High bacterial counts of greater than 105 viable bacteria or any number of A-hemolytic streptococci are considered detrimental. Bacterial toxins (as well as live bacteria) induce excessive inflammatory responses60 and tissue damage that can lead to abscess, cellulites, osteomyelitis, or limb loss (diabetic patients).26 Furthermore, recruited inflammatory cells, as well as bacteria, produce a number of proteases (including MMPs), which degrade the ECM and growth factors present within the wound bed (Figure 2). Bacteria that colonize chronic wounds often form polymicrobial communities called biofilms.61 These complex structures are composed of microbial cells embedded in secreted polymer matrix, which provides optimal environment for bacterial cell survival, enabling their escape from host immune surveillance/defense and resistance to antibiotic treatment.62 Although biofilms are prevalent in chronic wounds4,61 and significantly delay re-epithelialization in animal models,63 it remains unclear precisely how they delay healing. Increased bacterial survival and enhanced production of virulence factors are likely explanations. Nonetheless, it is possible that extracellular biofilm components possess or display a toxic phenotype for host cell functionality and therefore impede healing. Recently, it has been demonstrated that hindering biofilm formation by RNAIII-inhibiting peptide64 reverses wound-healing delays induced by bacterial biofilms.63 Better understanding of the precise mechanisms by which bacterial biofilms delay repair processes together with optimizing methods for biofilm detection and prevention may enhance opportunities for chronic wound beds to actively heal.
CHRONIC WOUND CARE
Successful treatment of a particular chronic wound requires a detailed understanding of the molecular and cellular components present within each wound bed. Currently, chronic (and acute) wounds of different etiologies are treated using a multistep approach based on contemporary knowledge of wound healing and known by the acronym TIME.65 First, nonviable tissues (T) from within and around a wound are removed using surgical debridement or debriding agents, such as bacterial collagenase. Second, infection and inflammation (I) are minimized with antibiotics and anti-inflammatory preparations. Next, moisture (M) imbalance is corrected, generally with carefully selected dressings. Finally, epithelialization (E) and granulation tissue formation are promoted by the application of specific therapies, such as growth factors.
The use of TIME strategy is not always sufficient, however, and some wounds remain nonresponsive to current therapies. Therefore, refined methods that will enable personalized therapeutics represent intriguing options that should be considered and/or developed. To this end, a novel wound diagnosis technology called “bar coding” of the wound has been proposed.66 It uses sampling of chronic wound fluids and/or collection of tissue biopsies that allow for identification of markers of wound chronicity, such as growth factors and their receptors, MMPs, members of the A-catenin/c-myc pathway, and keratinocyte differentiation markers. Wound bar coding can be used for both guiding wound debridement and treatment regimens. Necessary levels of dead tissue removal can be determined by the presence of A-catenin/c-myc–positive cells that, although they can proliferate, cannot migrate or differentiate and therefore have to be removed.
In addition, treatment strategies can be adjusted based on the needs of individual patients or bar-coding results. For example, if cells residing within a wound express low levels of growth factors, growth factor receptors, and high levels of MMPs as determined by enzyme-linked immunosorbent assay, growth factor delivery methods or patient-derived “engineered” cells could help restore the wound microenvironment to a healing phenotype. Thus, treatment for each wound would be carefully selected on an individual basis.
Wound Bed Preparation
Removal of nonviable tissue or wound debridement is beneficial for wound healing. Methods of debridement used in clinical practice include surgical, autolytic, biological, and enzymatic.67 Surgical, also known as sharp debridement, is performed by excising necrotic tissue with surgical tools. Autolytic debridement involves either careful removal of spontaneously separated necrotic tissues or the use of moisture-retaining dressings to induce eschar softening prior to its removal. Biological debridement uses larvae of the green blowfly species (maggot therapy); during enzymatic debridement, naturally occurring matrix-degrading enzymes are used.67–69 Both surgical and autolytic debridement strategies nondiscriminately remove dead and viable tissues, are very laborious, and can be painful. Although biological debridement is more selective, larvae are hard to store and may be unpleasant for the patients and medical personnel. Therefore, this method is not universally used.
Enzymatic debriding agents are also selective toward necrotic tissues, usually stable for several months upon refrigeration (or even at room temperature), and more aesthetically appealing.67 Until recently, 2 enzymatic debriding preparations were clinically used in the United States—papain-based and collagenase-based products (Table 2).
Table 2.
ENZYMATIC DEBRIDING AGENTS
| Enzyme/Source | Specificity | Trade Name(s) | FDA Approval Status |
|---|---|---|---|
| Papain/Carica papaya | Denatured proteins containing cysteine residues (including growth factors) |
Accuzyme, Allanfil, Allanzyme, Ethezyme, Gladase, Kovia, Panafil, Pap Urea, Ziox |
Not approved, manufacturing discontinued as of November 24, 2008 |
| Collagenase/C histolyticum | Denatured and native collagen I, II, III, IV, and V |
Santyl, Iruxol, Novuxol | Approved by FDA/European Medicines Agency |
| Bromelain/Ananas comosus | Necrotic cutaneous tissue | Debrase | Currently under investigation, Orphan Drug Status granted by FDA/European Committee for Proprietary Medicinal Products |
Papain—a cysteine protease derived from papaya fruit— degrades collagen and fibrin, the major components of eschar; as papain can degrade only denatured protein, urea is often added to the enzyme preparations, such as Accuzyme (Health-point Biotherapeutics, Fort Worth, Texas) to ensure protein denaturation.67 Although different preparations of papain were clinically used for more than 100 years and are relatively effective in removing necrotic tissues, the Food and Drug Administration (FDA) never approved the papain-containing products. Moreover, their benefit for wound healing and the effects on the cells inside the wound are questionable. In fact, papain-urea induces inflammatory response within the wound bed67 and decreases proliferation of cells,70 thus impeding the healing process. Recently, because of the safety concerns, the use of these debriding agents was discontinued.71
Another promising debriding agent is bacterial collagenase (clostridiopeptidase A). It is obtained from a Gram-positive bacterium Clostridium histolyticum and is an active ingredient in a number of debriding agents, including Santyl (Healthpoint Biotherapeutics), Iruxol, and Novuxol (Knoll Nordmark Arzneimittel, Uetersen, Germany). Clostridial collagenase, unlike its mammalian counterpart (MMP-1), can degrade both native and denatured collagens I, II, III, IV, and V.72 It efficiently clears the wound of necrotic tissues and decreases bacterial burden. More important, preparations of bacterial collagenase enhance migration and proliferation of keratinocytes, endothelial cells, and fibroblasts by releasing both ECM- and cell surface–bound growth factors and bioactive matrix fragments.70,73–80 Therefore, in addition to clinically relevant wound debridement, treatment of chronic wounds with preparations of bacterial collagenase will likely help to achieve efficient angiogenesis, epithelialization, and ultimately efficient healing81 (Figure 5). Further randomized controlled studies are necessary in order to unequivocally determine the wound-healing potential of clostridial collagenase.82
Figure 5. CONTROL OF WOUND HEALING: A ROLE FOR BACTERIAL COLLAGENASE?

Bacterial collagenase clinically used for wound debridement stimulates both endothelial and epithelial responses to injury. Degradation of the ECM in close proximity to the cells’ enzyme allows for efficient cell migration. The release of growth factors and liberation of biologically active matrix fragments that can interact with and activate cellular receptors increase the motogenic and mitogenic potential of the cells within the wound bed, promoting the healing responses. Similar to naturally occurring ECM fragments released by bacterial collagenase in vivo, synthetic matrix-derived peptides identified and tested in the laboratory can enhance cellular responses to injury. Therefore, the authors propose that the peptides could be used in combination or as an alternative to the bacterial products, which foster wound healing in vivo.
Another product that is under investigation for use as enzymatic debriding agent is bromelain. At the time of this writing, bromelain was being tested in a phase III clinical trial and was granted Orphan Drug Status by FDA/European Committee for Proprietary Medicinal Products. This enzymatic preparation extracted from pineapple stems and/or flowers is marketed as bromelain (Debridase, Debrase; MediWound Ltd, Industrial Zone Yavne, Israel). Typically, it is applied once for relatively short periods (4 hours), resulting in thorough debridement. In a small clinical study where the enzyme was used to debride deep partial- and full-thickness burns, no detrimental effects were detected on surrounding healthy tissues and no significant adverse effects.83 This study, however, does not provide any insight into the mechanism of action of bromelain or whether it was beneficial for the healing process or graft success.
Although significant progress has been made in clinicians’ un-derstanding of the mechanisms by which enzymatic debriding agents improve healing of chronic wounds, the delivery of debriding enzymes to the wound bed remains somewhat out-dated. For example, bromelain is supplied in the form of sterile lyophilized powder and has to be mixed immediately before application in a proprietary hydrating gel provided as a “kit” (Debrase). Because of short application time, secondary dressings are not necessary in this case. Bacterial collagenase, on the other hand, supplied premixed in petrolatum or emulsion ointments, has to be applied repeatedly and requires secondary dressings. Although this delivery system in general is considered relatively efficient and safe, it has been reported that contact dermatitis, in some cases, may develop upon exposure to both the active ingredient (clostridiopeptidase) or the excipients.84,85 To the authors’ knowledge, the only study published in peer-reviewed literature describing a different method of collagenase delivery to an in vivo wound was by a group from the Netherlands. In this report, a biodegradable hydrophilic film containing bacterial collagenase was used. Although collagenase activity in this delivery system was preserved, the formulation was unstable (at least at room temperature).86 Thus, more work remains to be done to developan efficient system for the delivery of enzymatic debriding agents. Recently, it has been shown that bacterial collagenase (as well as human MMPs) can be bound to collagen or oxidized regenerated cellulose/collagen sponge.87 This, together with the notion that components of the matrix degraded by bacterial collagenase have stimulatory effects on cells within the wound bed—epithelial, endothelial, inflammatory cells, and fibroblasts—suggest that matrix-based wound dressings may serve to deliver the enzyme into a wound bed.
Infection Control
Although proper wound debridement helps to control bacterial growth,88 it is not always sufficient, and additional antibiotics may be required. Antimicrobial preparations used in chronic wound care include topical antiseptics, topical antibacterials, and systemic antibiotics, all recently reviewed.62,89,90 Many preparations described in these reviews can effectively control bacterial growth; however, they can be toxic for host tissues. Currently, there is no conclusive evidence that one antibiotic or antiseptic is superior to any other achieving efficient elimination of infection and decreasing time to healing. Many modern antimicrobial therapies26 can effectively target planktonic bacteria, which may be beneficial for wound healing.91,92 However, biofilm-producing microorganisms remain a major challenge.26 Novel techniques, including photodynamic therapy93 and silver-containing dressings,62 could successfully eliminate planktonic, biofilm-associated, and multidrug-resistant bacteria.
SUMMARY
The normal wound healing process can be divided into 4 overlapping phases: coagulation, inflammation, formation of granulation tissue (proliferative phase), and remodeling or scar formation. During the coagulation phase, blood-clotting events prevent excessive bleeding and provide interim protection of the wounded area. Progression of the inflammatory phase leads to the recruitment of leukocytes, neutrophils, and macrophages; the production of growth factors; and the activation of dermal and epidermal cells. Completion of the proliferative phase of wound healing leads to formation of ECM-rich, vascularized granulation tissue. Finally, ECM remodeling and cell apoptosis lead to the formation of scar tissue with physical properties that are comparable with unwounded skin.
Chronic wounds are classified into vascular ulcers (venous and arterial ulcers), diabetic ulcers, and pressure ulcers. The majority of chronic wounds are characterized by a prolonged or excessive inflammatory phase, persistent infections, and the inability of dermal or epidermal cells to respond to reparative stimuli.
Phenotypic abnormalities common in chronic wound–derived cells include the lower density of growth factor receptors and lowered mitogenic/motogenic potential preventing them from responding properly to environmental cues. And, modifications of the ECM in chronic wounds include its glycation (diabetic patients) and excessive or insufficient production.
Infection, particularly in the form of biofilms, is an important factor that contributes to wound chronicity, morbidity, and mortality. Optimized methods for the detection and prevention of biofilm formation could lead to transforming chronic wound care.
A multistep approach based on the current understanding of wound healing mechanisms and known by the acronym TIME is used to treat the majority of chronic and acute wounds. Recent advances in understanding of the molecular and cellular components present within each wound bed may enable personalized diagnosis and therapy tailored to a particular patient’s needs and therefore lead to better therapeutic outcomes.
Removal of nonviable tissue (debridement) is critical for the successful healing of acute and chronic wounds. Enzymatic debridement using clostridial collagenase removes nonviable tissues and promotes epithelialization, angiogenesis, and wound healing.
Finally, wound infection control can be achieved using topical antiseptics, topical antibacterials, and systemic antibiotics. Novel methods such as photodynamic therapy and silver-containing dressings will enable eradication of multidrug-resistant and biofilm-associated bacteria.
REFERENCES
- 1.Singer AJ, Dagum AB. Current management of acute cutaneous wounds. N Engl J Med. 2008;359:1037–46. doi: 10.1056/NEJMra0707253. [DOI] [PubMed] [Google Scholar]
- 2.Hostetler SG, Xiang H, Gupta S, Sen C, Gordillo JM. Discharge patterns of injury-related hospitalizations with an acute wound in the United States. Wounds. 2006;18:340–51. [Google Scholar]
- 3.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 4.Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–43. doi: 10.1016/S0140-6736(05)67700-8. [DOI] [PubMed] [Google Scholar]
- 5.Weyrich AS, Zimmerman GA. Platelets: signaling cells in the immune continuum. Trends Immunol. 2004;25:489–95. doi: 10.1016/j.it.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 6.Gilroy DW, Lawrence T, Perretti M, Rossi AG. Inflammatory resolution: new opportunities for drug discovery. Nat Rev Drug Discov. 2004;3:401–16. doi: 10.1038/nrd1383. [DOI] [PubMed] [Google Scholar]
- 7.Eming SA, Krieg T, Davidson JM. Inflammation in wound repair: molecular and cellular mechanisms. J Invest Dermatol. 2007;127:514–25. doi: 10.1038/sj.jid.5700701. [DOI] [PubMed] [Google Scholar]
- 8.Humar R, Kiefer FN, Berns H, Resink TJ, Battegay EJ. Hypoxia enhances vascular cell proliferation and angiogenesis in vitro via rapamycin (mTOR)-dependent signaling. FASEB J. 2002;16:771–80. doi: 10.1096/fj.01-0658com. [DOI] [PubMed] [Google Scholar]
- 9.Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442–7. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- 10.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 11.Liu ZJ, Velazquez OC. Hyperoxia, endothelial progenitor cell mobilization, and diabetic wound healing. Antioxid Redox Signal. 2008;10:1869–82. doi: 10.1089/ars.2008.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Leone AM, Valgimigli M, Giannico MB, et al. From bone marrow to the arterial wall: the ongoing tale of endothelial progenitor cells. Eur Heart J. 2009;30(8):890–9. doi: 10.1093/eurheartj/ehp078. [DOI] [PubMed] [Google Scholar]
- 13.Maeng YS, Choi HJ, Kwon JY, et al. Endothelial progenitor cell homing: prominent role of the IGF2-IGF2R-PLCbeta2 axis. Blood. 2009;113:233–43. doi: 10.1182/blood-2008-06-162891. [DOI] [PubMed] [Google Scholar]
- 14.Gallagher KA, Liu ZJ, Xiao M, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. 2007;117:1249–59. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol. 2007;127:526–37. doi: 10.1038/sj.jid.5700613. [DOI] [PubMed] [Google Scholar]
- 16.Ehrlich HP, Keefer KA, Myers RL, Passaniti A. Vanadate and the absence of myofibroblasts in wound contraction. Arch Surg. 1999;134:494–501. doi: 10.1001/archsurg.134.5.494. [DOI] [PubMed] [Google Scholar]
- 17.Rai NK, Tripathi K, Sharma D, Shukla VK. Apoptosis: a basic physiologic process in wound healing. Int J Low Extrem Wounds. 2005;4:138–44. doi: 10.1177/1534734605280018. [DOI] [PubMed] [Google Scholar]
- 18.Desmouliere A, Redard M, Darby I, Gabbiani G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am J Pathol. 1995;146:56–66. [PMC free article] [PubMed] [Google Scholar]
- 19.Akasaka Y, Ono I, Yamashita T, Jimbow K, Ishii T. Basic fibroblast growth factor promotes apoptosis and suppresses granulation tissue formation in acute incisional wounds. J Pathol. 2004;203:710–20. doi: 10.1002/path.1574. [DOI] [PubMed] [Google Scholar]
- 20.Seifert O, Mrowietz U. Keloid scarring: bench and bedside. Arch Dermatol Res. 2009;301:259–72. doi: 10.1007/s00403-009-0952-8. [DOI] [PubMed] [Google Scholar]
- 21.Gosain A, DiPietro LA. Aging and wound healing. World J Surg. 2004;28:321–6. doi: 10.1007/s00268-003-7397-6. [DOI] [PubMed] [Google Scholar]
- 22.Narayan KMV, Boyle JP, Thompson TJ, Sorensen SW, Williamson DF. Lifetime risk for diabetes mellitus in the United States. JAMA. 2003;290:1884–90. doi: 10.1001/jama.290.14.1884. [DOI] [PubMed] [Google Scholar]
- 23.Ramsey SD, Newton K, Blough D, et al. Incidence, outcomes, and cost of foot ulcers in patients with diabetes. Diabetes Care. 1999;22:382–7. doi: 10.2337/diacare.22.3.382. [DOI] [PubMed] [Google Scholar]
- 24.Bitsch M, Saunte DM, Lohmann M, Holstein PE, Jorgensen B, Gottrup F. Standardised method of surgical treatment of chronic leg ulcers. Scand J Plast Reconstr Surg Hand Surg. 2005;39:162–9. doi: 10.1080/02844310510006196. [DOI] [PubMed] [Google Scholar]
- 25.Shai A, Halevy S. Direct triggers for ulceration in patients with venous insufficiency. Int J Dermatol. 2005;44:1006–9. doi: 10.1111/j.1365-4632.2005.02317.x. [DOI] [PubMed] [Google Scholar]
- 26.Edwards R, Harding KG. Bacteria and wound healing. Curr Opin Infect Dis. 2004;17(2):91–6. doi: 10.1097/00001432-200404000-00004. [DOI] [PubMed] [Google Scholar]
- 27.Wolcott RD, Rhoads DD, Dowd SE. Biofilms and chronic wound inflammation. J Wound Care. 2008;17:333–41. doi: 10.12968/jowc.2008.17.8.30796. [DOI] [PubMed] [Google Scholar]
- 28.Pardes JB, Takagi H, Martin TA, Ochoa MS, Falanga V. Decreased levels of alpha 1(I) procollagen mRNA in dermal fibroblasts grown on fibrin gels and in response to fibrinopeptide B. J Cell Physiol. 1995;162(1):9–14. doi: 10.1002/jcp.1041620103. [DOI] [PubMed] [Google Scholar]
- 29.Higley HR, Ksander GA, Gerhardt CO, Falanga V. Extravasation of macromolecules and possible trapping of transforming growth factor-beta in venous ulceration. Br J Dermatol. 1995;132:79–85. doi: 10.1111/j.1365-2133.1995.tb08629.x. [DOI] [PubMed] [Google Scholar]
- 30.Walker DJ. Venous stasis wounds. Orthop Nurs. 1999;18(5):65–74. 95. [PubMed] [Google Scholar]
- 31.Kobrin KL, Thompson PJ, van de Scheur M, Kwak TH, Kim S, Falanga V. Evaluation of dermal pericapillary fibrin cuffs in venous ulceration using confocal microscopy. Wound Repair Regen. 2008;16:503–6. doi: 10.1111/j.1524-475X.2008.00396.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bonham PA. Assessment and management of patients with venous, arterial, and diabetic/ neuropathic lower extremity wounds. AACN Clin Issues. 2003;14:442–56. doi: 10.1097/00044067-200311000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Sieggreen MY, Kline RA. Arterial insufficiency and ulceration: diagnosis and treatment options. Nurse Pract. 2004;29(9):46–52. doi: 10.1097/00006205-200409000-00007. [DOI] [PubMed] [Google Scholar]
- 34.Grey JE, Harding KG, Enoch S. Venous and arterial leg ulcers. BMJ. 2006;332:347–50. doi: 10.1136/bmj.332.7537.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Defloor T. The risk of pressure sores: a conceptual scheme. J Clin Nurs. 1999;8:206–16. doi: 10.1046/j.1365-2702.1999.00254.x. [DOI] [PubMed] [Google Scholar]
- 36.Ben-Porath I, Weinberg RA. The signals and pathways activating cellular senescence. Int J Biochem Cell Biol. 2005;37:961–76. doi: 10.1016/j.biocel.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 37.Mast BA, Schultz GS. Interactions of cytokines, growth factors, and proteases in acute and chronic wounds. Wound Repair Regen. 1996;4:411–20. doi: 10.1046/j.1524-475X.1996.40404.x. [DOI] [PubMed] [Google Scholar]
- 38.Lauer G, Sollberg S, Cole M, et al. Expression and proteolysis of vascular endothelial growth factor is increased in chronic wounds. J Invest Dermatol. 2000;115(1):12–8. doi: 10.1046/j.1523-1747.2000.00036.x. [DOI] [PubMed] [Google Scholar]
- 39.Loot MA, Kenter SB, Au FL, et al. Fibroblasts derived from chronic diabetic ulcers differ in their response to stimulation with EGF, IGF-I, bFGF and PDGF-AB compared to controls. Eur J Cell Biol. 2002;81:153–60. doi: 10.1078/0171-9335-00228. [DOI] [PubMed] [Google Scholar]
- 40.Seidman C, Raffetto JD, Marien B, Kroon C, Seah CC, Menzoian JO. bFGF-induced alterations in cellular markers of senescence in growth-rescued fibroblasts from chronic venous ulcer and venous reflux patients. Ann Vasc Surg. 2003;17:239–44. doi: 10.1007/s10016-003-0004-3. [DOI] [PubMed] [Google Scholar]
- 41.Vasquez R, Marien BJ, Gram C, Goodwin DG, Menzoian JO, Raffetto JD. Proliferative capacity of venous ulcer wound fibroblasts in the presence of platelet-derived growth factor. Vasc Endovascular Surg. 2004;38:355–60. doi: 10.1177/153857440403800408. [DOI] [PubMed] [Google Scholar]
- 42.Raffetto JD, Mendez MV, Marien BJ, et al. Changes in cellular motility and cytoskeletal actin in fibroblasts from patients with chronic venous insufficiency and in neonatal fibroblasts in the presence of chronic wound fluid. J Vasc Surg. 2001;33:1233–41. doi: 10.1067/mva.2001.113297. [DOI] [PubMed] [Google Scholar]
- 43.Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am J Pathol. 2003;162:303–12. doi: 10.1016/S0002-9440(10)63821-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Usui ML, Mansbridge JN, Carter WG, Fujita M, Olerud JE. Keratinocyte migration, proliferation, and differentiation in chronic ulcers from patients with diabetes and normal wounds. J Histochem Cytochem. 2008;56:687–96. doi: 10.1369/jhc.2008.951194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stojadinovic O, Pastar I, Vukelic S, et al. Deregulation of keratinocyte differentiation and activation: a hallmark of venous ulcers. J Cell Mol Med. 2008;12(6B):2675–90. doi: 10.1111/j.1582-4934.2008.00321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stojadinovic O, Brem H, Vouthounis C, et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. 2005;167:59–69. doi: 10.1016/s0002-9440(10)62953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Muller M, Essin K, Hill K, et al. Specific TRPC6 channel activation, a novel approach to stimulate keratinocyte differentiation. J Biol Chem. 2008;283:33942–54. doi: 10.1074/jbc.M801844200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Okwueze MI, Cardwell NL, Pollins AC, Nanney LB. Modulation of porcine wound repair with a transfected ErbB3 gene and relevant EGF-like ligands. J Invest Dermatol. 2007;127:1030–41. doi: 10.1038/sj.jid.5700637. [DOI] [PubMed] [Google Scholar]
- 49.Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol. 1998;111:850–7. doi: 10.1046/j.1523-1747.1998.00381.x. [DOI] [PubMed] [Google Scholar]
- 50.Agren MS, Steenfos HH, Dabelsteen S, Hansen JB, Dabelsteen E. Proliferation and mitogenic response to PDGF-BB of fibroblasts isolated from chronic venous leg ulcers is ulcer-age dependent. J Invest Dermatol. 1999;112:463–9. doi: 10.1046/j.1523-1747.1999.00549.x. [DOI] [PubMed] [Google Scholar]
- 51.Kuo PC, Kao CH, Chen JK. Glycated type 1 collagen induces endothelial dysfunction in culture. In Vitro Cell Dev Biol Anim. 2007;43:338–43. doi: 10.1007/s11626-007-9058-9. [DOI] [PubMed] [Google Scholar]
- 52.Reigle KL, Di Lullo G, Turner KR, et al. Non-enzymatic glycation of type I collagen diminishes collagen-proteoglycan binding and weakens cell adhesion. J Cell Biochem. 2008;104:1684–98. doi: 10.1002/jcb.21735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao H, Zakhaleva J, Chen W. Cells and tissue interactions with glycated collagen and their relevance to delayed diabetic wound healing. Biomaterials. 2009;30:1689–96. doi: 10.1016/j.biomaterials.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Death AK, Fisher EJ, McGrath KC, Yue DK. High glucose alters matrix metalloproteinase expression in two key vascular cells: potential impact on atherosclerosis in diabetes. Atherosclerosis. 2003;168:263–9. doi: 10.1016/s0021-9150(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 55.Lee SJ, Bae SS, Kim KH, et al. High glucose enhances MMP-2 production in adventitial fibroblasts via Akt1-dependent NF-kappaB pathway. FEBS Lett. 2007;581:4189–94. doi: 10.1016/j.febslet.2007.07.058. [DOI] [PubMed] [Google Scholar]
- 56.Dalton SJ, Whiting CV, Bailey JR, Mitchell DC, Tarlton JF. Mechanisms of chronic skin ulceration linking lactate, transforming growth factor-beta, vascular endothelial growth factor, collagen remodeling, collagen stability, and defective angiogenesis. J Invest Dermatol. 2007;127:958–68. doi: 10.1038/sj.jid.5700651. [DOI] [PubMed] [Google Scholar]
- 57.Lobmann R, Zemlin C, Motzkau M, Reschke K, Lehnert H. Expression of matrix metalloproteinases and growth factors in diabetic foot wounds treated with a protease absorbent dressing. J Diabetes Complications. 2006;20:329–35. doi: 10.1016/j.jdiacomp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 58.Rayment EA, Dargaville TR, Shooter GK, George GA, Upton Z. Attenuation of protease activity in chronic wound fluid with bisphosphonate-functionalised hydrogels. Biomaterials. 2008;29:1785–95. doi: 10.1016/j.biomaterials.2007.12.043. [DOI] [PubMed] [Google Scholar]
- 59.Bader MS. Diabetic foot infection. Am Fam Physician. 2008;78(1):71–9. [PubMed] [Google Scholar]
- 60.Ovington L. Bacterial toxins and wound healing. Ostomy Wound Manage. 2003;49(7A Suppl):8–12. [PubMed] [Google Scholar]
- 61.James GA, Swogger E, Wolcott R, et al. Biofilms in chronic wounds. Wound Repair Regen. 2008;16(1):37–44. doi: 10.1111/j.1524-475X.2007.00321.x. [DOI] [PubMed] [Google Scholar]
- 62.Martin JM, Zenilman JM, Lazarus GS. Molecular microbiology: new dimensions for cutaneous biology and wound healing. J Invest Dermatol. 2009;130:38–48. doi: 10.1038/jid.2009.221. [DOI] [PubMed] [Google Scholar]
- 63.Schierle Clark F., De la Garza Mauricio, Mustoe Thomas A., Galiano Robert D. Staphylococcal biofilms impair wound healing by delaying reepithelialization in a murine cutaneous wound model. Wound Repair Regen. 2009;17:354–9. doi: 10.1111/j.1524-475X.2009.00489.x. [DOI] [PubMed] [Google Scholar]
- 64.Cirioni O, Ghiselli R, Minardi D, et al. RNAIII-inhibiting peptide affects biofilm formation in a rat model of staphylococcal ureteral stent infection. Antimicrob Agents Chemother. 2007;51:4518–20. doi: 10.1128/AAC.00808-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schultz GS, Sibbald RG, Falanga V, et al. Wound bed preparation: a systematic approach to wound management. Wound Repair Regen. 2003;11(Suppl 1):S1–28. doi: 10.1046/j.1524-475x.11.s2.1.x. [DOI] [PubMed] [Google Scholar]
- 66.Tomic-Canic M, Ayello EA, Stojadinovic O, Golinko MS, Brem H. Using gene transcription patterns (bar coding scans) to guide wound debridement and healing. Adv Skin Wound Care. 2008;21:487–92. doi: 10.1097/01.ASW.0000323563.59885.1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ayello EA, Cuddigan JE. Debridement: controlling the necrotic/cellular burden. Adv Skin Wound Care. 2004;17:66–75. doi: 10.1097/00129334-200403000-00012. [DOI] [PubMed] [Google Scholar]
- 68.Attinger CE, Janis JE, Steinberg J, Schwartz J, Al-Attar A, Couch K. Clinical approach to wounds: debridement and wound bed preparation including the use of dressings and wound-healing adjuvants. Plast Reconstr Surg. 2006;117(7 Suppl):72S–109S. doi: 10.1097/01.prs.0000225470.42514.8f. [DOI] [PubMed] [Google Scholar]
- 69.Turkmen A, Graham K, McGrouther DA. Therapeutic applications of the larvae for wound debridement. J Plast Reconstr Aesthet Surg. 2009;63:184–8. doi: 10.1016/j.bjps.2008.08.070. [DOI] [PubMed] [Google Scholar]
- 70.Riley KN, Herman IM. Collagenase promotes the cellular responses to injury and wound healing in vivo. J Burns Wounds. 2005;4:e8. [PMC free article] [PubMed] [Google Scholar]
- 71.Food and Drug Administration [Last accessed April 13, 2012];FDA warns companies to stop marketing unapproved ophthalmic balanced salt solution drug products and topical drug products containing papain. 2008 Sep 23; http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/2008/ucm116956.htm?utm_source=fdaSearch&utm_medium=website&utm_term= papain&utm_content=5.
- 72.Mandl I. Bacterial collagenases and their clinical applications. Arzneimittelforschung. 1982;32(10a):1381–4. [PubMed] [Google Scholar]
- 73.Postlethwaite AE, Kang AH. Collagen-and collagen peptide-induced chemotaxis of human blood monocytes. J Exp Med. 1976;143:1299–1307. doi: 10.1084/jem.143.6.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Postlethwaite AE, Seyer JM, Kang AH. Chemotactic attraction of human fibroblasts to type I, II, and III collagens and collagen-derived peptides. Proc Natl Acad Sci U S A. 1978;75(2):871–5. doi: 10.1073/pnas.75.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Herman IM. Molecular mechanisms regulating the vascular endothelial cell motile response to injury. J Cardiovasc Pharmacol. 1993;22(Suppl 4):S25–36. doi: 10.1097/00005344-199322004-00005. [DOI] [PubMed] [Google Scholar]
- 76.Herman IM. Stimulation of human keratinocyte migration and proliferation in vitro: insights into the cellular responses to injury and wound healing. Wounds. 1996;8:33–42. [Google Scholar]
- 77.Radice M, Brun P, Bernardi D, Fontana C, Cortivo R, Abatangelo G. Clostridial collagenase releases bioactive fragments from extracellular matrix molecules. J Burn Care Rehabil. 1999;20:282–91. doi: 10.1097/00004630-199907000-00003. [DOI] [PubMed] [Google Scholar]
- 78.Demidova-Rice TN, Herman IM. Identification and characterization of novel wound-healing peptides derived from extracellular matrices. Wound Rep Regen; Abstracts of 19th Annual Meeting of the Wound Healing Society SAWC/WHS Joint Meeting.2009. p. A22. [Google Scholar]
- 79.Shi L, Ermis R, Garcia T, Telgenhoff D, Aust D. Abstracts of 19th Annual Meeting of the Wound Healing Society SAWC/WHS Joint Meeting. Wound Rep Regen. 2009;17(2):A34. [Google Scholar]
- 80.Demidova-Rice TN, Geevarghese A, Herman IM. Bioactive peptides derived from vascular endothelial cell extracellular matrices promote microvascular morphogenesis and wound healing in vitro. Wound Repair Regen. 2011;19:59–70. doi: 10.1111/j.1524-475X.2010.00642.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Falanga V. Wound bed preparation and the role of enzymes: a case for multiple actions of the therapeutic agents. Wounds. 2002;14:47–57. [Google Scholar]
- 82.Ramundo J, Gray M. Enzymatic wound debridement. J Wound Ostomy Continence Nurs. 2008;35:273–80. doi: 10.1097/01.WON.0000319125.21854.78. [DOI] [PubMed] [Google Scholar]
- 83.Rosenberg L, Lapid O, Bogdanov-Berezovsky A, et al. Safety and efficacy of a proteolytic enzyme for enzymatic burn debridement: a preliminary report. Burns. 2004;30:843–50. doi: 10.1016/j.burns.2004.04.010. [DOI] [PubMed] [Google Scholar]
- 84.Foti C, Conserva A, Casulli C, Scrimieri V, Pepe ML, Quaranta D. Contact dermatitis with clostridiopeptidase A contained in Noruxol ointment. Contact Dermatitis. 2007;56:361–2. doi: 10.1111/j.1600-0536.2006.01056.x. [DOI] [PubMed] [Google Scholar]
- 85.Tam CC, Elston DM. Allergic contact dermatitis caused by white petrolatum on damaged skin. Dermatitis. 2006;17:201–3. doi: 10.2310/6620.2006.06010. [DOI] [PubMed] [Google Scholar]
- 86.Mekkes JR, Zeegelaar JE, Westerhof W. Quantitative and objective evaluation of wound debriding properties of collagenase and fibrinolysin/desoxyribonuclease in a necrotic ulcer animal model. Arch Dermatol Res. 1998;290:152–7. doi: 10.1007/s004030050281. [DOI] [PubMed] [Google Scholar]
- 87.Metzmacher I, Ruth P, Abel M, Friess W. In vitro binding of matrix metalloproteinase-2 (MMP-2), MMP-9, and bacterial collagenase on collagenous wound dressings. Wound Repair Regen. 2007;15:549–55. doi: 10.1111/j.1524-475X.2007.00263.x. [DOI] [PubMed] [Google Scholar]
- 88.Payne WG, Salas RE, Ko F, et al. Enzymatic debriding agents are safe in wounds with high bacterial bioburdens and stimulate healing. Eplasty. 2008;8:e17. [PMC free article] [PubMed] [Google Scholar]
- 89.Howell-Jones RS, Wilson MJ, Hill KE, Howard AJ, Price PE, Thomas DW. A review of the microbiology, antibiotic usage and resistance in chronic skin wounds. J Antimicrob Chemother. 2005;55:143–9. doi: 10.1093/jac/dkh513. [DOI] [PubMed] [Google Scholar]
- 90.Lipsky BA. New developments in diagnosing and treating diabetic foot infections. Diabetes Metab Res Rev. 2008;24(Suppl 1):S66–71. doi: 10.1002/dmrr.828. [DOI] [PubMed] [Google Scholar]
- 91.Imegwu O, Chang TH, Steinberg JJ. Staphylococcus aureus peptidoglycan ameliorates cyclophosphamide-induced impairment of wound healing. Wound Repair Regen. 1997;5:364–72. doi: 10.1046/j.1460-9568.1997.50411.x. [DOI] [PubMed] [Google Scholar]
- 92.Levenson SM, Kan-Gruber D, Gruber C, Molnar J, Seifter E. Wound healing accelerated by Staphylococcus aureus. Arch Surg. 1983;118:310–20. doi: 10.1001/archsurg.1983.01390030042007. [DOI] [PubMed] [Google Scholar]
- 93.Di Poto A, Sbarra MS, Provenza G, Visai L, Speziale P. The effect of photodynamic treatment combined with antibiotic action or host defence mechanisms on Staphylococcus aureus biofilms. Biomaterials. 2009;30:3158–66. doi: 10.1016/j.biomaterials.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 94.Etufugh CN, Phillips TJ. Venous ulcers. Clin Dermatol. 2007;25:121–30. doi: 10.1016/j.clindermatol.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 95.Reiber GE. Diabetic foot care. Financial implications and practice guidelines. Diabetes Care. 1992;15(Suppl 1):29–31. doi: 10.2337/diacare.15.1.s29. [DOI] [PubMed] [Google Scholar]
- 96.Reddy M, Gill SS, Rochon PA. Preventing pressure ulcers: a systematic review. JAMA. 2006;296:974–84. doi: 10.1001/jama.296.8.974. [DOI] [PubMed] [Google Scholar]