

Abstract
The zinc metalloprotease ZMPSTE24 plays a critical role in nuclear lamin biology by cleaving the prenylated and carboxylmethylated 15-amino acid tail from the C-terminus of prelamin A to yield mature lamin A. A defect in this proteolytic event, caused by a mutation in the lamin A gene (LMNA) that eliminates the ZMPSTE24 cleavage site, underlies the premature aging disease Hutchinson-Gilford Progeria Syndrome (HGPS). Likewise, mutations in the ZMPSTE24 gene that result in decreased enzyme function cause a spectrum of diseases that share certain features of premature aging. Twenty human ZMPSTE24 alleles have been identified that are associated with three disease categories of increasing severity: mandibuloacral dysplasia type B (MAD-B), severe progeria (atypical ‘HGPS’) and restrictive dermopathy (RD). To determine whether a correlation exists between decreasing ZMPSTE24 protease activity and increasing disease severity, we expressed mutant alleles of ZMPSTE24 in yeast and optimized in vivo yeast mating assays to directly compare the activity of alleles associated with each disease category. We also measured the activity of yeast crude membranes containing the ZMPSTE24 mutant proteins in vitro. We determined that, in general, the residual activity of ZMPSTE24 patient alleles correlates with disease severity. Complete loss-of-function alleles are associated with RD, whereas retention of partial, measureable activity results in MAD-B or severe progeria. Importantly, our assays can discriminate small differences in activity among the mutants, confirming that the methods presented here will be useful for characterizing any new ZMPSTE24 mutations that are discovered.
INTRODUCTION
ZMPSTE24 is a zinc metalloprotease required for the maturation of the nuclear scaffold protein, lamin A (1–4). Lamin A, together with lamin C and the B-type lamin proteins, form the meshwork that underlies the nuclear envelope. Lamins provide structure and shape to the nucleus, and play many critical roles in nuclear function. These roles include, but are not limited to, binding of heterochromatin, regulation of transcription factors, positioning of nuclear pore complexes and facilitation of the DNA damage response (5–8).
Lamin A and the B-type lamins are synthesized as precursors that terminate in a CAAX motif (where C is cysteine, A is often an aliphatic amino acid and X is any residue). CAAX proteins are post-translationally modified in a series of ordered reactions beginning with farnesylation of the CAAX cysteine, followed by endoproteolytic removal of the terminal AAX residues, and ending with carboxylmethylation of the terminal farnesyl-cysteine (Supplementary Material, Fig. S1 (right), steps 1–3) (4,9). For reasons that are not fully understood, the lamin A precursor protein, called prelamin A, undergoes a surprising additional step in which the 15-amino acid modified tail is removed, resulting in mature lamin A that does not retain the hydrophobic modifications (Supplementary Material, Fig. S1 (right), step 4) (10–13). In contrast, B-type lamins do not undergo this final cleavage event, and retain their lipid-modified and carboxylmethylated tail (5). Lamin C, a short A-type lamin splice isoform generated from the LMNA gene, lacks the CAAX motif altogether and thus does not undergo post-translational modification. The zinc metalloprotease ZMPSTE24 mediates the last cleavage step in the lamin A maturation pathway (1–4). ZMPSTE24 is a membrane-associated enzyme that is predicted to contain seven transmembrane segments and is localized to the ER membrane and the inner nuclear membrane (14,15). ZMPSTE24 contains a consensus zinc metalloprotease motif, consisting of a HEXXH catalytic site, located in a loop predicted to reside in the cytosol/nucleoplasm (1,16,17). Farnesylated substrates such as prelamin A are accessible to the HEXXH domain for proteolytic cleavage.
The failure to cleave the farnesylated and carboxylmethylated tail from the prelamin A precursor due to LMNA mutations that remove the ZMPSTE24 cleavage site can severely impact human health and longevity. For example, the classical premature aging disease, Hutchinson-Gilford Progeria Syndrome (HGPS), is most commonly caused by a dominant mutation (c. 1824C>T; p. G608G) in LMNA that activates a cryptic mRNA splice site and leads to an internal in-frame deletion of 50 amino acids that removes the ZMPSTE24 cleavage site within prelamin A (18,19). The resulting mutant lamin A protein (called progerin) cannot be cleaved. HGPS is a childhood disease characterized by an onset of premature aging symptoms by age 2 years that include loss of hair, thin skin and progressive cardiovascular disease from which the children succumb at an average age of 13 years (20,21). Two significantly more severe cases of HGPS have been documented in which alternative mutations (c. 1968+1G>A and c. 1821G>A) serve as more potent activators of the cryptic splice site, and result in the generation of more progerin and death at an earlier age (22). Retention of the hydrophobic farnesyl and carboxlymethyl modifications disrupts the normal function of lamin A (and/or allows it to acquire a new function), conferring a dominant-negative phenotype. The amount of toxic progerin in cells appears to correlate with the severity of disease outcomes (22). In a mouse progeria model, preventing progerin farnesylation by genetic alteration of the progerin CAAX motif or treatment with a farnesyltransferase inhibitor lessens disease phenotypes (23–25).
Mutations in ZMPSTE24 are associated with diseases that share features of progeria. The aberrant form of prelamin A that results from decreased ZMPSTE24 activity permanently retains its farnesylated and carboxylmethylated tail, similar to progerin. The zmpste24−/− mouse exhibits a severe progeroid disorder that is completely alleviated in a mouse heterozygous for lamin A (zmpste24–/– lmna+/–), which reduces the load of toxic prelamin A by 50%. Thus, the amount of uncleaved, persistently prenylated prelamin A is thought to be the critical determinant of disease phenotypes (1,26–33).
Three distinct but related human diseases, with a gradation of severity, result from ZMPSTE24 mutations. These include, in order of increasing severity, the relatively mild disorder mandibuloacral dysplasia (MAD-B) (27–29,32,34–37), a severe form of progeria [sometimes called atypical HGPS and here designated AT ‘HGPS’ (33,38)], and the lethal neonatal disease restrictive dermopathy (RD) (29,30,39–44). These diseases are termed progeroid disorders because they share features in common with HGPS, such as thin skin (RD), lipodystrophy (MAD-B) and premature aging (AT ‘HGPS’) (27,29,33,38,43). The inheritance pattern of these diseases is autosomal recessive; thus, both chromosomal ZMPSTE24 copies are defective (29,43). It should be noted that an early report concluding that RD is digenic (45) was later corrected (29,43). Heterozygous carriers of disease alleles are unaffected. Thus, a single functional copy of ZMPSTE24 is sufficient to prevent disease (28–30,37,40).
Several of the ZMPSTE24 mutations that cause MAD-B have previously been assayed for activity using a qualitative yeast-based in vivo halo assay (27,28,46). However, a direct side-by-side comparison between the various human ZMPSTE24 disease mutations identified to date has not been performed, nor have any quantitative methods been used to assay these alleles. Herein we have utilized in vivo and in vitro yeast halo, mating and activity assays to quantitate and directly compare the activities of a panel of ZMPSTE24 disease mutants associated with each disease category (MAD-B, AT ‘HGPS’ and RD). We also examined a newly reported ZMPSTE24 allele potentially associated with metabolic syndrome (47). Using these assays, we demonstrate that mutant alleles associated with RD have no activity, while ZMPSTE24 alleles associated with MAD-B or AT ‘HGPS’ retain residual activity. Thus, ZMPSTE24 activity can be correlated to the severity of the disease state. Importantly, we report conditions using established activity assays that are able to reveal small differences in enzymatic activity. These assays will be useful for analyzing newly discovered mutations in ZMPSTE24.
RESULTS
Nineteen patients with homozygous or compound heterozygous mutations in ZMPSTE24 have been reported in the literature. These individuals have been diagnosed with MAD-B, AT HGPS or RD, as indicated (Fig. 1). In RD patients (Fig. 1, lines 10–19), both ZMPSTE24 mutations might reasonably be expected to represent ‘null’ alleles that cause complete loss of protein function. These include frameshift or nonsense mutations that cause early termination, or large internal in-frame deletions that could interfere with the membrane topology of ZMPSTE24. In contrast, MAD-B or AT ‘HGPS’ patients (Fig. 1, lines 1–8) generally have a predicted null allele in combination with a point mutation (or two point mutant alleles) that could potentially partially affect, but not eliminate, ZMPSTE24 enzymatic activity.
Figure 1.

Summary of the ZMPSTE24 disease alleles reported in cases of MAD, atypical HGPS and RD. Diseases are grouped in order of increasing severity, with MAD-B as the least severe and RD as most severe. The pairwise combination of specific mutations that have been attributed to each patient case is provided. Grey shading indicates mutants that do not retain the domain containing the HEXXH motif, which lies in positions 335–339 in ZMPSTE24. Mutants tested in this study are highlighted in yellow.
Our goal in this study was to quantitatively assess enzymatic activity across the spectrum of ZMPSTE24 alleles to determine whether the level of residual proteolytic activity, or lack thereof, correlates with disease severity. The individual disease mutants we examined in this study are highlighted in yellow in Figure 1. These include the substitution mutations P248L, W340R and N265S that result in MAD-B or AT ‘HGPS’ when present in combination with a possible null allele (such as W450X or L362F(fsX18), also tested herein) (Fig. 1, lines 1–4, 6 and 8), and L94P that causes MAD-B when homozygous (Fig. 1, line 5). We also tested the homozygous in-frame deletion mutation T159_L209del identified in a case of RD (Fig. 1, line 16). Taken together, the alleles we investigated in this study (including substitutions, frameshifts, internal in-frame deletions and null alleles) represent mutant ZMPSTE24 proteins that retain the catalytic zinc metalloprotease HEXXH motif at position 335–339 (Fig. 2 and Supplementary Material, Fig. S2). Additional mutants that are predicted to retain the catalytic domain (L462R, V402S(fsX1), L91_L209del, Q417X; Fig. 1, lines 7, 9, 13, 15) were not examined here; however, Q417X and V402S(fsX1) have been previously reported to lack activity by using either a yeast-based activity assay (V402S(fsX1)) or by examination of prelamin A processing (Q417X) (38,43).
Figure 2.
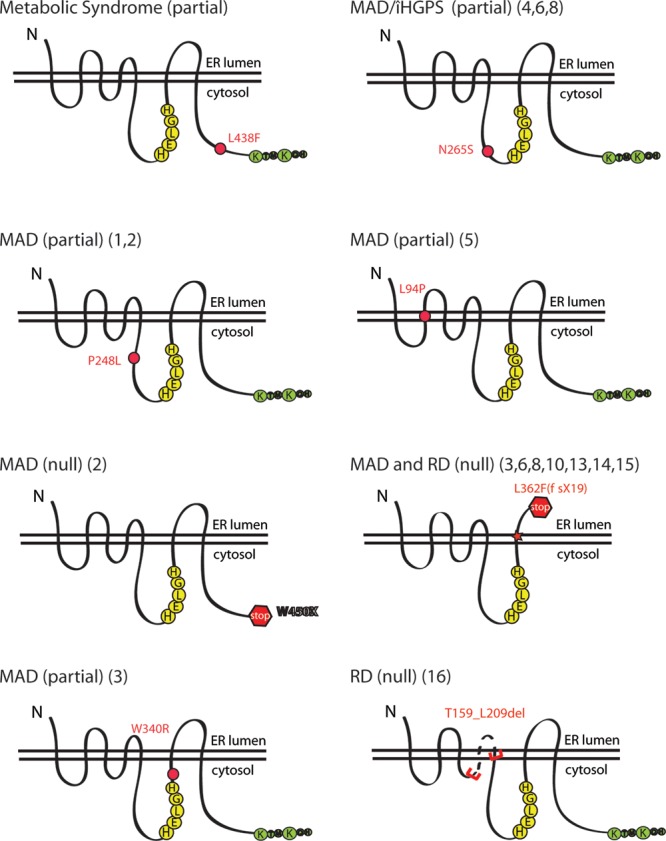
Schematic of the patient mutations analyzed in this study by in vivo and in vitro activity assays. ZMPSTE24 is predicted to contain seven transmembrane spans, with three cytosolic loops and a C-terminal cytosolic tail containing a dilysine ER retrieval motif (green). The loops are drawn roughly to scale. Representative schematics show the approximate location of the molecular lesions in the ZMPSTE24 mutants examined in this study. The HEXXH catalytic motif (HELGH) in human ZMPSTE24 is at positions 335–339 (yellow). The correlation to the disease/patient case as shown in Figure 1 is noted in parentheses.
The positions of the amino acid residues that are altered or the segments that are deleted by the ZMPSTE24 mutations tested here are schematized in Figure 2. In addition to the mutants highlighted in yellow in Figure 1, we also included a recently identified ZMPSTE24 mutation (L438F) that was detected in a patient with metabolic syndrome (47). This mutation represents an unusual case, as a second allelic mutation was not detected in the patient, a finding that seemingly contradicts the autosomal recessive nature of diseases associated with ZMPSTE24 mutations. Nevertheless, because L438F may be implicated in disease and may also provide useful structure–function information, we included this mutation in our studies.
In vivo a-factor halo assays can broadly distinguish active versus inactive ZMPSTE24 disease alleles
The yeast homolog of ZMPSTE24 is STE24, which is critical for the production of the Saccharomyces cerevisiae mating pheromone, a-factor (Supplementary Material, Fig. S1, left) (48). The Ste24 protein plays a redundant role with Rce1 in the proteolysis of the C-terminal three amino acids of the CAAX motif; Ste24 also performs a unique role in an N-terminal cleavage event required to produce mature, bioactive a-factor, similar to the role of ZMPSTE24 in lamin A processing (Supplementary Material, Fig. S1) (4,16,17,48–53). Importantly, the mammalian homologue of STE24, ZMPSTE24, can complement both of these Ste24 activities in a-factor biogenesis, as the heterologous expression of human ZMPSTE24 in a ste24Δ rce1Δ double mutant can restore export of a-factor (16). The a-factor exported from cells of the MATa mating type binds to receptors on cells of the opposite mating type (MATα) and stimulates mating. Thus, mating efficiency can be used as an indirect measure of Ste24/ZMPSTE24 activity (49,54). An alignment of the yeast and human enzymes is shown in Supplementary Material, Figure S2.
Initially, we used the well-established qualitative yeast-based halo assay to systematically compare the export of biologically active a-factor from the yeast strain SM3614 (MATa ste24Δ rce1Δ) expressing each ZMPSTE24 allele predicted to retain the HEXXH catalytic motif on a CEN URA3 plasmid (Fig. 3A). The halo assay has previously been used to qualitatively assess a few ZMPSTE24 disease alleles for activity (27,28,34); however, multiple side-by-side comparisons of a series of ZMPSTE24 alleles have not been performed with this assay. As a control for catalytically inactive ZMPSTE24, we included a mutation (H335A) in the histidine residue of the HEXXH catalytic motif. This mutation is predicted to eliminate activity, based on the evidence from yeast Ste24 HEXXH mutations (H297A, E298A and E298D), in which mating activity is completely lacking (48). For the halo assay, the MATa cells expressing ZMPSTE24 are spotted onto a lawn of tester cells of the opposite mating type (MATα) containing a mutation (sst2). The ‘supersensitizing’ sst2 mutation causes irreversible cell cycle arrest and thus death of cells in the lawn that come in contact with secreted a-factor (51). As a result, the diffusion of the secreted bioactive a-factor away from a spot of MATa cells results in a circular ‘kill zone’ in the underlying MATα sst2 lawn that is indicative of a-factor activity.
Figure 3.

The halo and mating assays reveal measurable differences in activity of ZMPSTE24 mutants. Results of the qualitative halo assay (A) and the plate mating assay (B) are shown. Results of a quantitative filter mating assay are graphically represented below (C). The details of how these tests were performed are discussed in the text. The mating percentages graphed in (C) are normalized to WT (100%). The values for each mutant tested were: H335A (1%), W340R (47%), P248L (25%), N265S (11%), L438F (6%), L94P (2%), W450X (1%), L362F(fsX18) (1%) and T159_L209del (1%).
As evident in Figure 3A, cells expressing wild-type ZMPSTE24 produce an a-factor halo, as expected, whereas cells expressing the H335A mutation, presumed to be important for catalytic activity, do not. Thus, the HEXXH motif is indeed critical for activity of human ZMPSTE24 (Fig. 3A, left, and results below), just as it is for yeast STE24 (48). Interestingly, five ZMPSTE24 alleles to the right of the bar (W340R, P248L, N265S, L438F and L94P) all produce an a-factor halo, which is indicative of at least some ZMPSTE24 activity, whereas a halo is lacking for the other alleles (W450X, L362F(fsX18), and T159_L209del). It is apparent that the a-factor halo assay is useful for discerning the differences between null (W450X, L362F(fsX18) and T159_L209del) and at least partially active alleles (W340R, P248L, N265S, L438F and L94P).
However, it is notable that small differences in activity between the point mutants or the wild-type protein are harder to discriminate and are not obvious using this halo assay. For instance, the W340R mutant cannot be distinguished from wild-type ZMPSTE24, but is indeed defective in activity, as shown below (Figs 3C and 4). Furthermore, P248L is indistinguishable from N265S, L438F and L94P in the halo assay, but shows significantly greater activity than the others in the more quantitative assays below. It should be noted that a very tiny halo is seen with the L362F(fsX18) mutant, and has previously been observed under conditions where a concentrated cell suspension was used (27). The mutation in this case results in a frameshift predicted to eliminate the entire C-terminus of ZMPSTE24. It is possible that the very low but detectable residual activity observed for this allele in the extremely sensitive a-factor halo assay may be due to a small amount of ‘read-through’ of the premature translational stop codon. Notably, almost no activity is detectable for this L362F(fsX18) allele by the quantitative assays (Figs 3C and 4) below.
Figure 4.

A coupled in vitro proteolysis activity assay reveals differences in specific activity for the ZMPSTE24 disease mutants. The specific activity of membrane preparations containing wild-type ZMPSTE24, the H335A catalytic site mutant and disease mutants were determined by the coupled proteolysis and methylation assay described in the text. Experiments were performed three times in triplicate for each ZMPSTE24 protein (for a total of nine trials).
In vivo mating assays reveal a range of activity differences associated with ZMPSTE24 disease alleles
The halo assay described above is hypersensitive, and gives a positive signal for any small residual activity. Due to this extreme sensitivity, the halo assay is not useful for the quantitation of low but measurable differences in enzyme activity that could result in significant differences in disease severity in humans. To refine and advance the methods that can be used to more quantitatively compare the biological activity of disease alleles of ZMPSTE24, we employed a yeast mating assay. Importantly, this assay can accurately reflect differences in a-factor export and can be modified, by varying the MATα tester strains or assay conditions that are used, to obtain optimal sensitivity in the range of interest (49,54).
We expressed wild-type ZMPSTE24 and each mutant allele on a plasmid in the MATa ste24Δ rce1Δ strain SM3614, and assayed the ability of the strain to mate by spotting the strains onto a lawn of tester MATα cells (Fig. 3B). To maximize our ability to discriminate the small but significant differences in activity of the ZMPSTE24 mutants analyzed here, we utilized a ‘stringent’ mating condition that relies on a MATα mating tester strain containing a far1 mutation (55). The far1 mutation lowers the efficiency of the mating response, and confers a different range of sensitivity to the mating assay that is better suited to the mutants studied here than the standard assay using a wild-type MATα mating tester strain that registers positive for any small amount of activity. In contrast, when far1 cells were used as the tester strain, many of the ZMPSTE24 mutant proteins tested here conferred a graded range of reduced mating efficiency compared with the wild-type (Fig. 3B). Importantly, small but significant differences in mating activity are revealed between the W340R, P248L, N265S, L438F and L94P substitution mutations.
To more accurately quantify the residual activities of the partially active alleles, we utilized a quantitative filter mating assay (Fig. 3C). For quantitative mating, strains are allowed to mate for a limited time (4 h) on a filter disc prior to plating on media selective for diploids. The number of diploids that form in proportion to the total number of cells provides a quantitative measure of mating efficiency (54,56). The results from the quantitative mating assay correlate well with our qualitative spot assay (compare Fig. 3C with B), and differences in mating efficiency can be reproducibly discerned for each of the point mutations in ZMPSTE24. W340R retains ∼47% of wild-type activity, while P248L (25%), N265S (11%), L438F (6%) and L94P (2%) all show an even further reduction in activity. The premature termination, frameshift and deletion mutants, W450X, L362F(fsX18) and T159_L209del, respectively, register essentially no activity under these conditions. Notably, the spot-mating tests (Fig. 3B) and the quantitative mating test (Fig. 3C) provided similar information. Interestingly, the W340R mutant shows significantly higher residual activity in the mating assay (∼50%) than the others.
Enzymatic activity of ZMPSTE24 disease alleles
The activity of mammalian ZMPSTE24 expressed in yeast can be determined in vitro using a well-established coupled assay (53,57–59). This coupled endoproteolysis and methylation assay indirectly measures the proteolysis of the C-terminal three amino acids of the CAAX motif (Supplementary Material, Fig. S1, step 2), by quantifying the resulting farnesyl-cysteine methyl esters that become available for carboxylmethylation by isoprenyl cysteine carboxyl methyltransferase (ICMT), also called Ste14 in yeast (Supplementary Material, Fig. S1, step 3). In this assay, crude membranes prepared from the ste24Δ rce1Δ strain (SM3614), expressing wild-type or mutant ZMPSTE24 alleles, are the source of enzyme. The membranes are incubated with the substrate, a synthetic farnesylated peptide containing an intact C-terminal CAAX motif. ZMPSTE24 present in the membrane preparations endoproteolytically removes the terminal AAX residues, exposing the farnesylated cysteine residue, which subsequently undergoes carboxylmethylation by the yeast ICMT Ste14 (added in excess to the reaction), using the radiolabeled methyl donor, [14C-methyl]- S-adenosyl methionine (SAM), provided in the reaction mix. The extent of carboxylmethylation of the synthetic peptide is quantified using the vapor diffusion assay to measure the amount of base-labile [14C] methanol. The amount of methylation is quantified and, in turn, serves as an indirect proxy for the endoproteolytic activity of ZMPSTE24. This assay provides a robust and reliable way to quantify ZMPSTE24 activity in a membrane preparation (53,57,59).
Using this coupled proteolysis assay, we measured the C-terminal proteolytic activity of yeast membranes containing wild-type ZMPSTE24, the null mutant H335A and the disease mutants. In nearly all cases, the membranes were prepared from the same strains as were used for the halo and mating assays above, which express ZMPSTE24 alleles on a CEN (single copy) plasmid. The L362F(fsX18) allele, which encodes a truncation product with 17 additional residues and is also referred to as Y379X, was expressed from a 2 μm (multi-copy) plasmid, in an effort to determine whether any activity could be detected from the mutant protein. The specific activity calculated for each of the ZMPSTE24 alleles is indicated in Table 1 (middle column) and is displayed as a bar graph in Figure 4. The percentage of activity of each allele, normalized to wild-type ZMPSTE24, is also shown in Table 1 (right column).
Table 1.
C-terminal proteolytic activity of wild-type and mutant ZMPSTE24 proteins
| ZMPSTE24 | Specific activitya | % wild-type |
|---|---|---|
| WT | 173.0 ± 6.2 | 100 |
| H335A | 0.7 ± 1.2 | 0.4 |
| W340R | 23.7 ± 1.4 | 13.7 |
| P248L | 7.9 ± 1.7 | 4.6 |
| N265S | 7.5 ± 1.5 | 4.3 |
| L438F | 1.8 ± 2.5 | 1.0 |
| L94P | 4.8 ± 1.6 | 2.8 |
| W450X | 0.6 ± 0.6 | 0.3 |
| L362F(fsX18) | 2.2 ± 0.5 | 1.3 |
| T159_L209del | −2.7 ± 3.2 | 0 |
aValues were derived from three separate experiments each performed in triplicate.
The W340R point mutant retains the most activity of all the disease alleles, while P248L, N265S and L94P retain moderately less activity. The least activity is associated with the HEXXH domain mutant control (H335A) and the L438F, W450X, L362F(fsX18) and T159_L209del mutations. As evident by comparison of the graphs in Figures 3C and 4, the results from the activity assays are strikingly similar to those obtained with the quantitative mating assays. Notably, both assays indicate that the disease alleles fall into three groups: significant residual activity (W340R), some residual activity (P248L, N265S, L94P) and little or no residual activity (W450X, L362F(fsX18) and T159_L209del). The L438F allele demonstrates partial residual activity by mating, but virtually no enzyme activity. Immunoblotting of the membranes used for the activity assays (Fig. 5) revealed a varying amount of ZMPSTE24 protein in the membrane preparations (Fig. 5, see legend), which may indicate that certain disease mutations render the protein unstable. Changes in protein stability would not be unexpected, as several of the mutations are drastic, deleting large internal sections of the protein or eliminating the C-terminus. Interestingly, however, the expression and/or stability of the substitution mutations are also somewhat variable, possibly reflecting varying degrees of stability.
Figure 5.

Expression of ZMPSTE24 mutant proteins when compared with the wild-type. Crude membranes prepared from yeast strains expressing WT and mutant His10HA3 ZMPSTE24, or empty vector (EV) were subjected to 10% SDS–PAGE, transferred to nitrocellulose membrane, probed with α-HA primary antibody and a goat α-mouse horseradish peroxidase-conjugated secondary antibody and detected by ECL. Varying amounts of crude membranes were added to each lane to optimize visualization of the signal. When compared with the WT, H335A catalytic site mutant, and most point mutants (left), the frameshift, deletion, nonsense and L94P mutations (right) required significantly more membranes for visualization. Amounts added to each lane are as follows: left panel: WT (0.5 µg), N265S (0.5 µg), H335A (0.5 µg), L438F (0.5 µg), P248L (1.0 µg), W340R (3.0 µg), EV (3.0 µg); right panel: WT (0.1 µg), L362F(fsX18) (2.0 µg), W450X (3.0 µg), L94P (12 µg), T159_L209del (12 µg) and EV (12 µg).
DISCUSSION
The range of disease severity caused by mutations in ZMPSTE24 is quite broad, with the neonatal lethal disorder RD on one end of the spectrum, severe progeria (AT ‘HGPS’) characterized by a short lifespan midway through the spectrum and the relatively milder disease MAD-B, in which the lifespan of patients extends into the third decade or later, on the other end of the spectrum (Fig. 6) (27,29,38,43). It has been proposed that the greater the severity of disease, the lower the level of residual ZMPSTE24 activity that is retained (1,27–33). In this study, we have directly compared the activities of a number of disease alleles of ZMPSTE24 expressed in yeast, using quantitative mating and enzyme assays, as well as qualitative a-factor halo and mating assays. We wished to determine whether the disease severity spectrum shown in Figure 6 reflects the residual activity of ZMPSTE24 disease alleles.
Figure 6.

Schematic of the ZMPSTE24-associated progeroid disease severity spectrum. The results presented here and in other studies support the model that a spectrum of progeroid diseases of increasing severity, ranging from MAD-B to atypical progeria to RD, appears to correlate with an increasing amount of uncleaved prelamin A resulting from a failure of ZMPSTE24 cleavage. It has yet to be established whether metabolic syndrome is also directly connected to defects in prelamin A maturation, hence the question mark. The L438F studied here has been reported to be associated with metabolic syndrome (47), but the second patient allele is not known, and whether this mutation is truly causative of metabolic syndrome remains to be established.
Our results support the hypothesis that disease severity correlates with the cumulative, residual activity of ZMPSTE24 encoded by a patient's two alleles. For instance, the combination of two alleles with molecular lesions predicted to be drastic, such as frameshifts, large internal deletions or early terminations, is associated with the lethal disease RD. Our studies confirm that the alleles L362F(fsX18) and T159_L209del show no residual activity. As can be seen in Figure 1, RD cases 10 and 16 are homozygous for these mutations, respectively. Therefore, RD appears to result when ZMPSTE24 activity is completely lacking. RD is lethal in utero or shortly after birth, representing an extreme example of the severity of defective ZMPSTE24 activity in which all of the lamin A present in patient cells is predicted to be in the persistently farnesylated, toxic form.
For the milder MAD-B disease (Fig. 1, lines 1–6), patient genotypes contain a null allele in combination with a substitution mutant (i.e. P248L, W340R, N265S) that has been predicted to retain partial function. Here, we provide evidence that these MAD-B alleles indeed show measurable residual activity, with W340R having significantly higher activity than others (Figs 3 and 4, and Table 1). One exception to the rule that MAD-B patients all have one complete null allele is shown in Figure 1, line 5, where the patient was homozygous for L94P (35). In this case, the L94P mutant would be predicted to be so minimally functional that even the two copies present in the patient would not suffice for normal function. Indeed, the L94P mutant is very severely reduced in activity in our assays (Figs 3 and 4, Table 1). Thus, the total enzymatic activity that the patient retained apparently fell below a critical threshold required for maintaining health and the absence of disease phenotypes. This mutant has been shown previously to cause prelamin A accumulation, and due to its location inside one of the transmembrane spans, was predicted to potentially interfere with membrane topology, such that the active site of ZMPSTE24 could be oriented on the inaccessible luminal side of the membrane (35). Taken together, our results suggest that most, if not all, MAD-B patients (Fig. 1, lines 1–7) must have at least one allele with residual activity. Thus, some, but not all, of the lamin A present in these MAD-B patient cells is predicted to be present in the aberrantly and persistently farnesylated form.
The range of diseases that are caused by mutations in ZMPSTE24 overlap in phenotype with HGPS, the classical premature aging disorder caused by mutations in LMNA that remove the ZMPSTE24 cleavage site in prelamin A through activation of a cryptic splice site (18–20). Somewhat confusingly, two cases of RD could also be attributed to mutations in the lamin A gene that are particularly robust activators of the cryptic splice site (22). The larger amount of progerin produced in these cases resulted in a disease that was phenotypically very similar to RD, but mapped to LMNA rather than ZMPSTE24. Conversely, a case of severe progeria was reported in which the patient had two null alleles of ZMPSTE24 (Fig. 1, patient 9). This individual was shown to have a ‘mitigating’ mutation in one copy of the LMNA gene that resulted in 50% of the prelamin A being prematurely truncated prior to the CAAX motif (38). This LMNA mutation reduces the amount of uncleaved and persistently farnesylated prelamin A that the cells produce, and is thought to explain the severe progeria phenotype observed in the patient, rather than the RD phenotype that might be expected from the null ZMPSTE24 genotype. Thus, the diagnosis of disease based on the phenotype alone can be potentially misleading, and both genes, ZMPSTE24 and LMNA, need to be examined for mutations. It is notable that there are two patients shown in Figure 1 (lines 6 and 8) that have the identical ZMPSTE24 genotypes (N265S and L362F(fsX18)), but different diseases, namely MAD-B and atypical progeria, respectively. It is possible that additional modifier genes, still unaccounted for, may also contribute to the severity of these progeroid diseases.
Metabolic syndrome, which encompasses several risk factors for cardiovascular disease, is often present in MAD patients (60). Interestingly, the heterozygous L438F mutation was found in a patient presenting only with metabolic syndrome (47). As the genetics of the ZMPSTE24 diseases would indicate that haploinsufficiency is not normally causative of disease, this is an unusual case. It should be noted that another case of heterozygosity in a patient with MAD has been reported (Fig. 1, case 7); however, the authors suggested that although a second mutation was not identified, it may nevertheless indeed be present, especially since the heterozygous parents of the patient were reported as phenotypically normal (37).
The methods of analysis presented in this study, involving expression of ZMPSTE24 disease alleles in yeast, combined with the performance of several simple quantitative assays, provides the basis for assessing new disease alleles that may be discovered in the future. Such information will be useful for future studies directed toward dissecting the enzymatic mechanism of proteolysis, which to date remains unknown. Interestingly, several lines of study have suggested that an understanding of the mechanism could be critical for predicting (and preventing) potential off-target drug effects. For instance, drug cocktails used to treat patients infected with HIV have often included aspartyl protease inhibitors, some of which have been experimentally demonstrated to inhibit ZMPSTE24 activity in vitro and in vivo (57,58). It remains unclear whether the side effects exhibited by patients who have spent decades on these drugs (which include lipodystrophies, metabolic disorders and premature aging) are indeed attributable to the inhibition of ZMPSTE24. It is also worthwhile to note that the down-regulation of ZMPSTE24 expression has been associated with normal physiological aging (61). Further work on the activity and regulation of ZMPSTE24 will be needed to fully probe these potentially important connections.
MATERIALS AND METHODS
Plasmid constructs
The experiments in this study were carried out using CEN URA3 yeast plasmid constructs expressed in the strain SM3614 (MATa ste24Δ rce1Δ). These plasmids encode an N-terminally His10HA3-tagged version of human ZMPSTE24 expressed from a PGK promoter with the following mutations: WT (pSM2677), P248L (pSM2676), H335A (pSM2673), W340R (pSM2672), N265S (pSM2671), T159_L209del (pSM2670), L362F(fsX18) (pSM2669), L94P (pSM2678), L438F (pSM2680) and W450X (pSM2679). These constructs were made as described below.
Constructs used to express human ZMPSTE24 and individual disease mutations were first created in a yeast 2µm URA3 vector backbone. The method of homologous recombination of overlapping PCR and vector products in S. cerevisiae was used to introduce mutations. Plasmids were rescued from yeast and transformed into the recombination deficient STBL2 E. coli strain (Invitrogen). The base plasmid pSM1282 (17) containing the yeast STE24 gene with an N-terminal HIS-HA tag (His10HA3) under the control of the PGK promoter was gapped by removal of the ySTE24 ORF and used as the vector backbone for all homologous recombination reactions. PCR products containing the entire ORF of wild-type or mutant ZMPSTE24 alleles with overlapping homologous 5′ and 3′ ends and gapped pSM1282 were co-transformed into the yeast strain SM3103 (trp1 leu2 ura3 his4 can1 ste24::LEU2) (48). It is important to note that bacteria-containing ZMPSTE24 plasmids were propagated on selective media at 25°C. The use of STBL2 bacterial cells and low temperature was necessary to prevent toxicity and/or recombination and rearrangement events in E. coli. Such events occur frequently for ZMPSTE24 in bacteria when these conditions are not used. Plasmids were purified and sequenced to ensure that the desired mutation was obtained. The wild-type and ZMPSTE24 mutant alleles were subsequently transferred to a CEN URA3 vector (pRS316), using standard restriction digestion/ligation-mediated cloning techniques.
All plasmids and strains used in this study are freely available upon request.
Halo and mating assays
Halo assays were performed essentially as previously described (49,51,54,56). Briefly, yeast strains (SM3614, MATa ste24Δ rce1Δ) expressing wild-type or mutant ZMPSTE24 on the CEN URA3 plasmid (see above) were grown overnight in selective (SC-URA) media, concentrated by centrifugation and resuspended in sterile water. The yeast strains used were as follows: WT (SM5973), P248L (SM5978), H335A (SM5975), W340R (SM5977), N265S (SM5974), T159_L209del (SM5976), L362F(fsX18) (SM5981), L94P (SM5983), L438F (SM5985) and W450X (SM5984). Five microliters of each strain was spotted onto a lawn of the halo tester SM2375 (MATα sst2) mutant yeast cells spread on YPD plates containing 0.04% Triton X-100. Inclusion of TX-100 in the plates allows and even diffusion of a-factor and a sharper halo. Plates were incubated at 30°C for 2 days.
The qualitative spot-mating assay was performed essentially as described previously (49,51,54,56). Briefly, overnight cultures (SC-URA) of the MATa yeast strains expressing wild-type ZMPSTE24 or the mutant alleles (described above) were diluted to OD600 of 1.0 in sterile water. Ten microliters of each strain was spotted onto a lawn of MATα far1 mating tester strain (SM2882) spread on synthetic medium (SD) to select for diploids. Plates were incubated at 30°C for 2 days.
Quantitative mating assays were performed essentially as described previously (49,51,54,56), except that a far1 strain was used as a mating partner (55). Briefly, overnight cultures of the yeast strains expressing wild-type ZMPSTE24 or mutant alleles (described above) were diluted to OD600 1 in sterile water. Equal volumes (0.5 ml) of the tester strain (MATα far1) and each of the strains expressing ZMPSTE24 alleles were mixed and applied to a nitrocellulose filter, excess liquid was removed by applying a vacuum and the filters were placed on a YPD plate. Plates were incubated at 30°C for 4 h to allow mating to occur. Cells were washed off of the filters by vortexing in sterile water, and serial 10-fold dilutions were prepared and plated on diploid-selective plates (SD) or non-selective plates (YPD). The mating efficiency for each of the strains tested was calculated as the ratio of diploids to total cells, and graphed as a percentage of the mating efficiency of the yeast strain expressing wild-type ZMPSTE24. Mating tests were performed in triplicate and the error bars indicate standard deviation.
ZMPSTE24 in vitro assay
Human ZMPSTE24 activity was determined using a radioactive in vitro-coupled proteolysis and methyltransferase vapor diffusion assay as described previously (57), with minor modifications. Briefly, the human wild-type and mutant ZMPSTE24 variants used for the mating and halo tests above, or the yeast Ste14p-isoprenylcysteine carboxyl methyltransferase (ICMT), were expressed in the SM3614 (Δste24Δrce1) yeast strain and the membrane fractions were isolated as described previously (59). It should be noted that mutant L362F(fsX18) was expressed from a high copy (2μ) yeast plasmid, rather than a low copy CEN plasmid for the in vitro assay and immunoblot (Figs 4 and 5, and Table 1). Reaction mixtures contained 5 µg of membranes expressing ZMPSTE24 or its variants, an excess (8 µg) of membranes expressing Ste14p, 25 µm farnesylated a-factor peptide (YIIKGVFWDPA-farnCVIA) (EZ Biolabs), and 20 µm S-adenosyl [14C-methyl]-L-methionine in 100 mm Tris–HCl (pH 7.5) in a total volume of 60 μl. The reaction mixtures were incubated at 30°C for 30 min and quenched by the addition of 50 μl 1 m NaOH/1% SDS. The reaction mixture (100 μl) was spotted onto pleated filter and placed in the neck of a scintillation vial containing 10 ml of Biosafe II scintillation fluid (RPI, Mount Prospect, IL, USA) and capped. The [14C]methanol released from the cleaved and methylated substrate as a result of quenching the reaction was allowed to diffuse into the scintillation fluid for 2.5 h at room temperature. The filter paper was then removed and the radioactivity was quantified by scintillation counting. Specific activity was reported as pmol of VIA groups removed/min/mg ZMPSTE24, as measured by the methyltransferase assay. Each value was derived from three assays performed in triplicate.
Immunoblotting and antibodies
Crude membranes prepared from yeast strains expressing wild-type and mutant His10HA3-N-terminally tagged ZMPSTE24 were heated at 65°C for 30 min in 2× SDS. The samples were subjected to SDS–PAGE (10% gel) and transferred to a 0.2 µm nitrocellulose membrane. The nitrocellulose membrane was blocked overnight in 20% non-fat dry milk. ZMPSTE24 was detected with the α-HA primary antibody (1:15,000) and goat α-mouse horseradish peroxidase-conjugated secondary antibody (1:4,000), using enhanced chemiluminescence (ECL) to visualize the signal (Pierce SuperSignal West Pico Chemiluminescent Substrate).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the National Institutes of Health (grant number RO1 GM41223 to S.M.).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Megan Kane for critical comments on this manuscript
Conflict of Interest: The authors declare no conflict of interest.
REFERENCES
- 1.Barrowman J., Michaelis S. ZMPSTE24, an integral membrane zinc metalloprotease with a connection to progeroid disorders. Biol. Chem. 2009;390:761–773. doi: 10.1515/BC.2009.080. [DOI] [PubMed] [Google Scholar]
- 2.Bergo M.O., Gavino B., Ross J., Schmidt W.K., Hong C., Kendall L.V., Mohr A., Meta M., Genant H., Jiang Y., et al. Zmpste24 deficiency in mice causes spontaneous bone fractures, muscle weakness, and a prelamin A processing defect. Proc. Natl Acad. Sci. USA. 2002;99:13049–13054. doi: 10.1073/pnas.192460799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pendas A.M., Zhou Z., Cadinanos J., Freije J.M., Wang J., Hultenby K., Astudillo A., Wernerson A., Rodriguez F., Tryggvason K., et al. Defective prelamin A processing and muscular and adipocyte alterations in Zmpste24 metalloproteinase-deficient mice. Nat. Genet. 2002;31:94–99. doi: 10.1038/ng871. [DOI] [PubMed] [Google Scholar]
- 4.Young S.G., Fong L.G., Michaelis S. Prelamin A, Zmpste24, misshapen cell nuclei, and progeria–new evidence suggesting that protein farnesylation could be important for disease pathogenesis. J. Lipid Res. 2005;46:2531–2558. doi: 10.1194/jlr.R500011-JLR200. [DOI] [PubMed] [Google Scholar]
- 5.Dittmer T.A., Misteli T. The lamin protein family. Genome Biol. 2011;12:222. doi: 10.1186/gb-2011-12-5-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gruenbaum Y., Margalit A., Goldman R.D., Shumaker D.K., Wilson K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005;6:21–31. doi: 10.1038/nrm1550. [DOI] [PubMed] [Google Scholar]
- 7.Mattout A., Dechat T., Adam S.A., Goldman R.D., Gruenbaum Y. Nuclear lamins, diseases and aging. Curr. Opin. Cell Biol. 2006;18:335–341. doi: 10.1016/j.ceb.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 8.Worman H.J., Bonne G. ‘Laminopathies’: a wide spectrum of human diseases. Exp. Cell Res. 2007;313:2121–2133. doi: 10.1016/j.yexcr.2007.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wright L.P., Philips M.R. Thematic review series: lipid posttranslational modifications. CAAX modification and membrane targeting of Ras. J. Lipid Res. 2006;47:883–891. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Beck L.A., Hosick T.J., Sinensky M. Isoprenylation is required for the processing of the lamin A precursor. J. Cell Biol. 1990;110:1489–1499. doi: 10.1083/jcb.110.5.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weber K., Plessmann U., Traub P. Maturation of nuclear lamin A involves a specific carboxy-terminal trimming, which removes the polyisoprenylation site from the precursor; implications for the structure of the nuclear lamina. FEBS Lett. 1989;257:411–414. doi: 10.1016/0014-5793(89)81584-4. [DOI] [PubMed] [Google Scholar]
- 12.Davies B.S., Fong L.G., Yang S.H., Coffinier C., Young S.G. The posttranslational processing of prelamin A and disease. Annu. Rev. Genomics Hum. Genet. 2009;10:153–174. doi: 10.1146/annurev-genom-082908-150150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barrowman J., Hamblet C., Kane M.S., Michaelis S. Requirements for efficient proteolytic cleavage of prelamin A by ZMPSTE24. PloS ONE. 2012;7:e32120. doi: 10.1371/journal.pone.0032120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barrowman J., Hamblet C., George C.M., Michaelis S. Analysis of prelamin A biogenesis reveals the nucleus to be a CaaX processing compartment. Mol. Biol. Cell. 2008;19:5398–5408. doi: 10.1091/mbc.E08-07-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmidt W.K., Tam A., Fujimura-Kamada K., Michaelis S. Endoplasmic reticulum membrane localization of Rce1p and Ste24p, yeast proteases involved in carboxyl-terminal CAAX protein processing and amino-terminal a-factor cleavage. Proc. Natl Acad. Sci. USA. 1998;95:11175–11180. doi: 10.1073/pnas.95.19.11175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tam A., Nouvet F.J., Fujimura-Kamada K., Slunt H., Sisodia S.S., Michaelis S. Dual roles for Ste24p in yeast a-factor maturation: NH2-terminal proteolysis and COOH-terminal CAAX processing. J. Cell Biol. 1998;142:635–649. doi: 10.1083/jcb.142.3.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tam A., Schmidt W.K., Michaelis S. The multispanning membrane protein Ste24p catalyzes CAAX proteolysis and NH2-terminal processing of the yeast a-factor precursor. J. Biol. Chem. 2001;276:46798–46806. doi: 10.1074/jbc.M106150200. [DOI] [PubMed] [Google Scholar]
- 18.De Sandre-Giovannoli A., Levy N. Altered splicing in prelamin A-associated premature aging phenotypes. Prog. Mol. Subcell. Biol. 2006;44:199–232. doi: 10.1007/978-3-540-34449-0_9. [DOI] [PubMed] [Google Scholar]
- 19.Eriksson M., Brown W.T., Gordon L.B., Glynn M.W., Singer J., Scott L., Erdos M.R., Robbins C.M., Moses T.Y., Berglund P., et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–298. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merideth M.A., Gordon L.B., Clauss S., Sachdev V., Smith A.C., Perry M.B., Brewer C.C., Zalewski C., Kim H.J., Solomon B., et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 2008;358:592–604. doi: 10.1056/NEJMoa0706898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scaffidi P., Gordon L., Misteli T. The cell nucleus and aging: tantalizing clues and hopeful promises. PLoS Biol. 2005;3:e395. doi: 10.1371/journal.pbio.0030395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moulson C.L., Fong L.G., Gardner J.M., Farber E.A., Go G., Passariello A., Grange D.K., Young S.G., Miner J.H. Increased progerin expression associated with unusual LMNA mutations causes severe progeroid syndromes. Hum. Mutat. 2007;28:882–889. doi: 10.1002/humu.20536. [DOI] [PubMed] [Google Scholar]
- 23.Fong L.G., Frost D., Meta M., Qiao X., Yang S.H., Coffinier C., Young S.G. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science. 2006;311:1621–1623. doi: 10.1126/science.1124875. [DOI] [PubMed] [Google Scholar]
- 24.Yang S.H., Chang S.Y., Ren S., Wang Y., Andres D.A., Spielmann H.P., Fong L.G., Young S.G. Absence of progeria-like disease phenotypes in knock-in mice expressing a non-farnesylated version of progerin. Hum. Mol. Genet. 2011;20:436–444. doi: 10.1093/hmg/ddq490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Capell B.C., Olive M., Erdos M.R., Cao K., Faddah D.A., Tavarez U.L., Conneely K.N., Qu X., San H., Ganesh S.K., et al. A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model. Proc. Natl Acad. Sci. USA. 2008;105:15902–15907. doi: 10.1073/pnas.0807840105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fong L.G., Ng J.K., Meta M., Cote N., Yang S.H., Stewart C.L., Sullivan T., Burghardt A., Majumdar S., Reue K., et al. Heterozygosity for Lmna deficiency eliminates the progeria-like phenotypes in Zmpste24-deficient mice. Proc. Natl Acad. Sci. USA. 2004;101:18111–18116. doi: 10.1073/pnas.0408558102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Agarwal A.K., Fryns J.P., Auchus R.J., Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in mandibuloacral dysplasia. Hum. Mol. Genet. 2003;12:1995–2001. doi: 10.1093/hmg/ddg213. [DOI] [PubMed] [Google Scholar]
- 28.Miyoshi Y., Akagi M., Agarwal A.K., Namba N., Kato-Nishimura K., Mohri I., Yamagata M., Nakajima S., Mushiake S., Shima M., et al. Severe mandibuloacral dysplasia caused by novel compound heterozygous ZMPSTE24 mutations in two Japanese siblings. Clin. Genet. 2008;73:535–544. doi: 10.1111/j.1399-0004.2008.00992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moulson C.L., Go G., Gardner J.M., van der Wal A.C., Smitt J.H., van Hagen J.M., Miner J.H. Homozygous and compound heterozygous mutations in ZMPSTE24 cause the laminopathy restrictive dermopathy. J. Invest. Dermatol. 2005;125:913–919. doi: 10.1111/j.0022-202X.2005.23846.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smigiel R., Jakubiak A., Esteves-Vieira V., Szela K., Halon A., Jurek T., Levy N., De Sandre-Giovannoli A. Novel frameshifting mutations of the ZMPSTE24 gene in two siblings affected with restrictive dermopathy and review of the mutations described in the literature. Am. J. Med. Genet. A. 2010;152A:447–452. doi: 10.1002/ajmg.a.33221. [DOI] [PubMed] [Google Scholar]
- 31.Varela I., Cadinanos J., Pendas A.M., Gutierrez-Fernandez A., Folgueras A.R., Sanchez L.M., Zhou Z., Rodriguez F.J., Stewart C.L., Vega J.A., et al. Accelerated ageing in mice deficient in Zmpste24 protease is linked to p53 signalling activation. Nature. 2005;437:564–568. doi: 10.1038/nature04019. [DOI] [PubMed] [Google Scholar]
- 32.Ahmad Z., Zackai E., Medne L., Garg A. Early onset mandibuloacral dysplasia due to compound heterozygous mutations in ZMPSTE24. Am. J. Med. Genet. A. 2010;152A:2703–2710. doi: 10.1002/ajmg.a.33664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shackleton S., Smallwood D.T., Clayton P., Wilson L.C., Agarwal A.K., Garg A., Trembath R.C. Compound heterozygous ZMPSTE24 mutations reduce prelamin A processing and result in a severe progeroid phenotype. J. Med. Genet. 2005;42:e36. doi: 10.1136/jmg.2004.029751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agarwal A.K., Zhou X.J., Hall R.K., Nicholls K., Bankier A., Van Esch H., Fryns J.P., Garg A. Focal segmental glomerulosclerosis in patients with mandibuloacral dysplasia owing to ZMPSTE24 deficiency. J. Investig. Med. 2006;54:208–213. doi: 10.2310/6650.2006.05068. [DOI] [PubMed] [Google Scholar]
- 35.Ben Yaou R., Navarro C., Quijano-Roy S., Bertrand A.T., Massart C., De Sandre-Giovannoli A., Cadinanos J., Mamchaoui K., Butler-Browne G., Estournet B., et al. Type B mandibuloacral dysplasia with congenital myopathy due to homozygous ZMPSTE24 missense mutation. Eur. J. Hum. Genet. 2011;19:647–654. doi: 10.1038/ejhg.2010.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cunningham V.J., D'Apice M.R., Licata N., Novelli G., Cundy T. Skeletal phenotype of mandibuloacral dysplasia associated with mutations in ZMPSTE24. Bone. 2010;47:591–597. doi: 10.1016/j.bone.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Thill M., Nguyen T.D., Wehnert M., Fischer D., Hausser I., Braun S., Jackisch C. Restrictive dermopathy: a rare laminopathy. Arch. Gynecol. Obstet. 2008;278:201–208. doi: 10.1007/s00404-008-0676-6. [DOI] [PubMed] [Google Scholar]
- 38.Denecke J., Brune T., Feldhaus T., Robenek H., Kranz C., Auchus R.J., Agarwal A.K., Marquardt T. A homozygous ZMPSTE24 null mutation in combination with a heterozygous mutation in the LMNA gene causes Hutchinson-Gilford progeria syndrome (HGPS): insights into the pathophysiology of HGPS. Hum. Mutat. 2006;27:524–531. doi: 10.1002/humu.20315. [DOI] [PubMed] [Google Scholar]
- 39.Chen M., Kuo H.H., Huang Y.C., Ke Y.Y., Chang S.P., Chen C.P., Lee D.J., Lee M.L., Lee M.H., Chen T.H., et al. A case of restrictive dermopathy with complete chorioamniotic membrane separation caused by a novel homozygous nonsense mutation in the ZMPSTE24 gene. Am. J. Med. Genet. A. 2009;149A:1550–1554. doi: 10.1002/ajmg.a.32768. [DOI] [PubMed] [Google Scholar]
- 40.Jagadeesh S., Bhat L., Suresh I., Muralidhar S.L. Prenatal diagnosis of restrictive dermopathy. Indian Pediatr. 2009;46:349–351. [PubMed] [Google Scholar]
- 41.Kariminejad A., Goodarzi P., Thanh Huong le T., Wehnert M.S. Restrictive dermopathy. Molecular diagnosis of restrictive dermopathy in a stillborn fetus from a consanguineous Iranian family. Saudi Med. J. 2009;30:150–153. [PubMed] [Google Scholar]
- 42.Morais P., Magina S., Ribeiro M.D., Rodrigues M., Lopes J.M., Thanh H.L., Wehnert M., Guimaraes H. Restrictive dermopathy-a lethal congenital laminopathy. Case report and review of the literature. Eur. J. Pediatr. 2008 doi: 10.1007/s00431-008-0868-x. published online doi:10.1007/s00431-00008-00868-x. [DOI] [PubMed] [Google Scholar]
- 43.Navarro C.L., Cadinanos J., De Sandre-Giovannoli A., Bernard R., Courrier S., Boccaccio I., Boyer A., Kleijer W.J., Wagner A., Giuliano F., et al. Loss of ZMPSTE24 (FACE-1) causes autosomal recessive restrictive dermopathy and accumulation of Lamin A precursors. Hum. Mol. Genet. 2005;14:1503–1513. doi: 10.1093/hmg/ddi159. [DOI] [PubMed] [Google Scholar]
- 44.Sander C.S., Salman N., van Geel M., Broers J.L., Al-Rahmani A., Chedid F., Hausser I., Oji V., Al Nuaimi K., Berger T.G., et al. A newly identified splice site mutation in ZMPSTE24 causes restrictive dermopathy in the Middle East. Br. J. Dermatol. 2008;159:961–967. doi: 10.1111/j.1365-2133.2008.08772.x. [DOI] [PubMed] [Google Scholar]
- 45.Navarro C.L., De Sandre-Giovannoli A., Bernard R., Boccaccio I., Boyer A., Genevieve D., Hadj-Rabia S., Gaudy-Marqueste C., Smitt H.S., Vabres P., et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum. Mol. Genet. 2004;13:2493–2503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 46.Agarwal A.K., Garg A. Genetic disorders of adipose tissue development, differentiation, and death. Annu. Rev. Genomics Hum. Genet. 2006;7:175–199. doi: 10.1146/annurev.genom.7.080505.115715. [DOI] [PubMed] [Google Scholar]
- 47.Dutour A., Roll P., Gaborit B., Courrier S., Alessi M.C., Tregouet D.A., Angelis F., Robaglia-Schlupp A., Lesavre N., Cau P., et al. High prevalence of laminopathies among patients with metabolic syndrome. Hum. Mol. Genet. 2011;20:3779–3786. doi: 10.1093/hmg/ddr294. [DOI] [PubMed] [Google Scholar]
- 48.Fujimura-Kamada K., Nouvet F.J., Michaelis S. A novel membrane-associated metalloprotease, Ste24p, is required for the first step of NH2-terminal processing of the yeast a-factor precursor. J. Cell Biol. 1997;136:271–285. doi: 10.1083/jcb.136.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Barrowman J., Michaelis S. In: The Enzymes. Hrycyna C.A., Bergo M.O., Tamanoi F., editors. Vol. 30. Burlington: Academic Press; 2011. pp. 13–41. [Google Scholar]
- 50.Boyartchuk V.L., Rine J. Roles of prenyl protein proteases in maturation of Saccharomyces cerevisiae a-factor. Genetics. 1998;150:95–101. doi: 10.1093/genetics/150.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huyer G., Kistler A., Nouvet F.J., George C.M., Boyle M.L., Michaelis S. Saccharomyces cerevisiae a-factor mutants reveal residues critical for processing, activity, and export. Eukaryot. Cell. 2006;5:1560–1570. doi: 10.1128/EC.00161-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt W.K., Tam A., Michaelis S. Reconstitution of the Ste24p-dependent N-terminal proteolytic step in yeast a-factor biogenesis. J. Biol. Chem. 2000;275:6227–6233. doi: 10.1074/jbc.275.9.6227. [DOI] [PubMed] [Google Scholar]
- 53.Leung G.K., Schmidt W.K., Bergo M.O., Gavino B., Wong D.H., Tam A., Ashby M.N., Michaelis S., Young S.G. Biochemical studies of Zmpste24-deficient mice. J. Biol. Chem. 2001;276:29051–29058. doi: 10.1074/jbc.M102908200. [DOI] [PubMed] [Google Scholar]
- 54.Nijbroek G.L., Michaelis S. Functional assays for analysis of yeast ste6 mutants. Methods Enzymol. 1998;292:193–212. doi: 10.1016/s0076-6879(98)92016-x. [DOI] [PubMed] [Google Scholar]
- 55.Peter M., Herskowitz I. Direct inhibition of the yeast cyclin-dependent kinase Cdc28-Cln by Far1. Science. 1994;265:1228–1231. doi: 10.1126/science.8066461. [DOI] [PubMed] [Google Scholar]
- 56.Berkower C., Michaelis S. Mutational analysis of the yeast a-factor transporter STE6, a member of the ATP binding cassette (ABC) protein superfamily. EMBO J. 1991;10:3777–3785. doi: 10.1002/j.1460-2075.1991.tb04947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Coffinier C., Hudon S.E., Farber E.A., Chang S.Y., Hrycyna C.A., Young S.G., Fong L.G. HIV protease inhibitors block the zinc metalloproteinase ZMPSTE24 and lead to an accumulation of prelamin A in cells. Proc. Natl Acad. Sci. USA. 2007;104:13432–13437. doi: 10.1073/pnas.0704212104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hudon S.E., Coffinier C., Michaelis S., Fong L.G., Young S.G., Hrycyna C.A. HIV-protease inhibitors block the enzymatic activity of purified Ste24p. Biochem. Biophys. Res. Commun. 2008;374:365–368. doi: 10.1016/j.bbrc.2008.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson J.L., Frase H., Michaelis S., Hrycyna C.A. Purification, functional reconstitution, and characterization of the Saccharomyces cerevisiae isoprenylcysteine carboxylmethyltransferase Ste14p. J. Biol. Chem. 2005;280:7336–7345. doi: 10.1074/jbc.M410292200. [DOI] [PubMed] [Google Scholar]
- 60.Agarwal A.K., Garg A. Genetic basis of lipodystrophies and management of metabolic complications. Annu. Rev. Med. 2006;57:297–311. doi: 10.1146/annurev.med.57.022605.114424. [DOI] [PubMed] [Google Scholar]
- 61.Ragnauth C.D., Warren D.T., Liu Y., McNair R., Tajsic T., Figg N., Shroff R., Skepper J., Shanahan C.M. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. 2010;121:2200–2210. doi: 10.1161/CIRCULATIONAHA.109.902056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.