

Abstract
To examine mutational pathways that lead to CXCR4 use of HIV-1, we analyzed the genotypic and phenotypic characteristics of envelope sequences from a large panel of patient virus populations and individual clones containing different V3 mutations. Basic amino acid substitutions at position 11 were strong determinants of CXCR4-mediated entry, but required multiple compensatory mutations to overcome associated reductions in infectivity. In contrast, basic amino acid substitutions at position 25, or substitutions at position 6–8 resulting in the loss of a potential N-linked glycosylation site, contributed to CXCR4-mediated entry, but required additional substitutions acting cooperatively to confer efficient CXCR4 use. Our assumptions, based upon examination of patient viruses, were largely confirmed by characterizing the coreceptor utilization of five distinct panels of isogenic envelope sequences containing V3 amino acid substitutions introduced by site-directed mutagenesis. These results further define the mutational pathways leading to CXCR4 use and their associated genetic barriers.
Keywords: HIV-1, V3, coreceptor, tropism, X4, R5, dual, CXCR4, CCR5, CCR5 antagonist
INTRODUCTION
As the principal coreceptors of human immunodeficiency virus type 1 (HIV-1) infection, chemokine receptors CCR5 and CXCR4 cooperate with the CD4 receptor to mediate virus entry (Berger, Murphy, and Farber, 1999). HIV-1 coreceptor tropism is defined as the ability of a virus to use CCR5 only (R5), CXCR4 only (X4), or both of these coreceptors (dual). R5 virus populations represent the majority of HIV-1 infections, followed by dual/mixed (R5 plus dual and/or X4) virus populations; X4 virus populations are relatively rare. R5 variants predominate during acute and early infection and generally persist throughout the entire course of HIV-1 infection (Berger, Murphy, and Farber, 1999). In contrast, CXCR4-using variants (dual and, to a lesser extent, X4 viruses) are detected infrequently in recent infections (Eshleman et al., 2007; Huang et al., 2009; Markowitz et al., 2005; Masquelier et al., 2007), but gradually increase in prevalence as chronic infection ensues (Berger, Murphy, and Farber, 1999; Connor et al., 1997; Melby et al., 2006; Schuitemaker et al., 1992; Wilkin et al., 2007). Generally, CXCR4-using variants are found in approximately 20% of antiretroviral (ARV) treatment-naïve individuals, characterized by higher CD4+ T-cell counts (Brumme et al., 2005; Moyle et al., 2005), and approximately 50% of highly treatment-experienced patients, characterized by low CD4+ T-cell counts (Coakley et al., 2006; Demarest et al., 2004; Hunt et al., 2006; Melby et al., 2006; Wilkin et al., 2007). In the absence of ARV treatment, the presence of CXCR4-using viruses is strongly associated with faster depletion of CD4+ T cells and accelerated disease progression (Daar et al., 2007; Koot et al., 1993; Richman and Bozzette, 1994; Schuitemaker et al., 1992; Shepherd et al., 2008; Tersmette et al., 1989). Whether these associations are causal or consequential has not been firmly established and is the subject of ongoing debate.
More recently, the development of CCR5 antagonists as new therapeutic agents has drawn further attention to HIV-1 coreceptor tropism. The CCR5 antagonist maraviroc (Pfizer) is approved for the treatment of HIV-1 infection in patients that have failed conventional treatment regimens comprised of nucleoside, non-nucleoside and/or protease inhibitors (Gulick et al., 2008), and more recently in the treatment naïve setting (Cooper et al., 2010). Several additional therapeutic candidates have advanced to late stage clinical evaluations, including the small molecule CCR5 antagonists vicriviroc (Gulick et al., 2007; Su et al., 2009; Suleiman et al., 2010) and INCB9471 (Erickson-Viitanen et al., 15th Conference on Retroviruses and Opportunistic Infections, Boston, MA, Feb 3–6, 2008), as well as the CCR5 monoclonal antibody Pro-140 (Jacobson et al., 17th Conference on Retroviruses and Opportunistic Infections, San Francisco, CA, Feb.16–19, 2010). In each case these CCR5 inhibitors effectively suppress the replication of R5 virus populations, but not dual/mixed or X4 virus populations (Lewis et al., 2007; Saag et al., 2009; Tsibris et al., 2009; Westby et al., 2006). Although initial attempts to develop therapeutic agents that block CXCR4-mediated entry have met with less success (Hendrix et al., 2004; Moyle et al., 2009), several new candidates have recently entered early-stage clinical evaluations (Jenkinson et al., 2010; Murakami et al., 2009). Since CXCR4 inhibitors effectively suppress the replication of X4 virus populations, but not R5 or dual/mixed virus populations (Fransen et al., 2008; Hendrix et al., 2004), their use in the clinic may be linked to co-administration with CCR5 inhibitors.
Numerous studies have firmly established the third variable region (V3) of the HIV-1 envelope glycoprotein (Env) as a primary determinant for coreceptor tropism (Cocchi et al., 1996; Deng et al., 1996; Hoffman and Doms, 1999; Hung, Vander Heyden, and Ratner, 1999; Hwang et al., 1991; O'Brien et al., 1990; Shioda, Levy, and Cheng-Mayer, 1991; Speck et al., 1997; Trkola et al., 1996; Trujillo et al., 1996; Westervelt et al., 1992; Wu et al., 1996). The substitution of as few as one or two amino acids in the V3 region of R5 viruses has been reported to confer CXCR4 use (Chesebro et al., 1996; De Jong et al., 1992; Mosier et al., 1999). However, more complex patterns of amino acid substitutions in the V3 region are typically observed among the CXCR4-using variants within patient virus populations that also contain an R5 subpopulation for comparison (Huang et al., 2007; Jensen et al., 2003; Lewis et al., 2007; Tsibris et al., 2009; Westby et al., 2006). Previous studies have defined specific determinants of coreceptor tropism within the V3 region, including the acquisition of positively-charged amino acids, particularly at positions 11 and 25 (De Jong et al., 1992; Fouchier et al., 1992; Resch, Hoffman, and Swanstrom, 2001), and, more recently, position 24 (Cardozo et al., 2007), as well as the loss of a potential N-linked glycosylation site (PNGS) at position 6–8 (Pollakis et al., 2001). A recent study has demonstrated that the ability to accurately predict CXCR4 use is CD4 independent for viruses with a positively-charged amino acid at position 11, but is CD4 dependent for viruses with positively-charged amino acids at position 25, or that lack a PNGS at position 6–8 (Low et al., 2008). Precisely how substitutions at position 11, 25 and 6–8 impact CXCR4 use remains to be defined.
To advance our understanding of how HIV-1 populations broaden or switch host range, i.e. coreceptor tropism, either as a result of host- or drug-imposed selective pressure, we have attempted to more clearly define the associations between specific V3 determinants for CXCR4 use and their impact on Env mediated entry by analyzing patient-derived viruses. The primary goal of the present study is to specifically define the selective advantages and disadvantages of V3 mutational pathways that lead to efficient CXCR4 use. In doing so, we have examined the effect of positively-charged amino acids at positions 11 and 25, as well as the absence of a PNGS at positions 6–8 on CXCR4 use by analyzing (a) the envelope (env) sequences of 35 patient virus populations containing any one of these V3 substitutions in isolation, (b) multiple individual env clones derived from 12 patient virus populations containing mixed amino acid sequences at these positions, and (c) five different env sequences containing specific V3 amino acid substitutions introduced by site-directed mutagenesis. We found that these specific V3 substitutions differentially influence CXCR4 mediated entry, and their effects are highly context dependent and reliant on the presence of additional env amino acid substitutions that serve to compensate for reductions in env infectivity, or act cooperatively to confer efficient CXCR4 use. The degree of complexity and fluidity of mutational pathways leading to CXCR4 use that we observed is consistent with a high genetic barrier and may, in part, explain why CXCR4 use typically emerges late in the course of HIV infection, if at all.
METHODS
Patient virus selection
To obtain viruses containing positively-charged amino acids at position 11, 25 or lacking PNGS at position 6–8 in the V3 region of env, we determined the V3 nucleotide sequences of 200 patient plasma-derived viruses (submitted for routine coreceptor tropism [Trofile] testing), to obtain the 47 viruses described in this study. Based on env sequences, 39 viruses were identified as subtype B and 8 samples as non-subtype B and recombinant (A=1, C=2, AE=1, A/G=2, B/C=1, B/D=1). Thirty five of 47 patient viruses contained unambiguous V3 sequences (no mixtures) and a basic amino acid substitution at either position 11 (N=7) or 25 (N=25), or that lacked a PNGS at position 6–8 (N=3). The remaining 12 patient viruses contained mixed V3 amino acid sequences at position 11 (N=4) or 25 (N=5), or at position 6–8 (N=3). For this subset of 12 viruses, we also determined the V3 nucleotide sequences and coreceptor tropisms for approximately 10 env clones per virus population. Since these 47 patient virus samples were submitted to Monogram for routine coreceptor tropism testing, no clinical information or longitudinal samples were available.
Site-directed mutagenesis
Single amino acid substitutions (S11K, S11R, E25K, E25R) were introduced into the V3 regions of three R5 molecular env clones of HIV-1: JRCSF and BaL (AIDS Research and Reference Reagent Program), and clone c11.2 (from subject 11 in this study) using site-directed mutagenesis (Sarkar and Sommer, 1990). The V3 PNGS was removed from each of these three R5 env sequences by introducing an N6Q mutation. In addition, 11R substitutions were replaced by 11S substitutions in two dual env clones (c2.41, c3.14) derived from subjects 2 and 3. The complete gp160 nucleotide sequence of each engineered env gene was determined to verify the presence of the desired mutations and confirm the absence of other mutations,
Coreceptor tropism determinations
The coreceptor tropisms of patient virus populations, molecular env clones derived from patient virus populations, and env clones containing site-directed mutations in V3 were determined using the Trofile coreceptor tropism assay (Whitcomb et al., 2007). Briefly, HIV-1 env genes were amplified from patient plasma samples by RT-PCR and incorporated into env expression vectors. HIV-1 pseudovirions were generated by co-transfecting HEK-293 cells with patient virus-derived env expression vectors and an HIV-1 genomic vector containing a firefly luciferase reporter gene. Coreceptor tropism was determined by measuring the ability of pseudovirions to infect U87 target cells that express CD4 and either CCR5 or CXCR4. In the Trofile assay, the production of luciferase activity in CXCR4 and/or CCR5 target cells that exceeds background levels (~102 RLU in this study) and is inhibited by a CXCR4 or CCR5 inhibitor, respectively, is considered a demonstration of env-mediated virus entry.
env V3 sequencing
V3 nucleotide sequences for patient virus populations and molecular clones were determined using conventional dideoxy chain terminator chemistry (ABI, Foster City, CA). V3 amino acid sequences were deduced from nucleotide sequences. Predictions of coreceptor tropism based on derived V3 amino acid sequences were determined using two well-established algorithms; 11/25 rule and PSSM (De Jong et al., 1992; Fouchier et al., 1992; Jensen et al., 2003).
env V3 structure modeling
The V3 sequences of paired R5 and dual clones from four subjects (2, 3, 7 and 9), along with template structures 2QAD, 1U6U and 2ESX, were aligned by hand and modeled using MODELLER (Sali and Blundell, 1993). Surface images with electrostatic forces were created using Discovery Studio 2.5 software (http://accelrys.com/products/discovery-studio/).
RESULTS
Patient-derived viruses that contain basic amino acids at V3 position 11 or 25, or that lack an N-linked glycosylation site in V3, vary in their ability to use CXCR4 and CCR5
To better define the contributions of positively-charged amino acid substitutions and the loss of PNGS in the env V3 region to CXCR4-mediated entry, we inspected the V3 sequences of 200 patient virus populations that were submitted for routine coreceptor tropism testing (Trofile). This initial inspection defined an unambiguous subset of 35 viruses that either contained a basic amino acid substitution at position 11 (N=7) or 25 (N=25), or lacked a PNGS at position 6–8 (N=3) (Table 1). The majority of these viruses (31/35) were subtype B (C=1, B/D=1, A/G=2). All seven of the basic substitutions at position 11 were arginine (11R); no lysine (11K) was observed. In contrast, basic substitutions at position 25 were equally represented by arginine (25R, N=15) and lysine (25K, N=11). The coreceptor tropism (R5, DM, X4) and infectivity (luciferase reporter gene activity expressed as relative light units, RLU) of these 35 viruses are summarized in Table 1. All seven viruses containing 11R substitutions utilized CXCR4 efficiently relative to CCR5, i.e. infectivity was comparable or higher in CXCR4+ cells than in CCR5+ cells (median ratio of CXCR4 RLU:CCR5 RLU = 4.673, range = 0.720 to 14.971), thus no 11R viruses were classified R5. In contrast, roughly one third (8/25) of the 25K/R virus populations were R5 viruses. The remaining 25K/R virus populations (17/25) were classified DM or X4 but varied (>4 log10 RLU) in there ability to utilize CXCR4 (median ratio of CXCR4 RLU:CCR5 RLU = 1.034, range = 0.001 to 129.905). Similarly, CXCR4 utilization varied (>4 log10 RLU) of 2/3 viruses that lacked the PNGS within the V3 region (median CXCR4 RLU:CCR5 RLU = 225.169, range 1.083 to 449.225), while the remaining one virus was R5.
Table 1.
Comparison of coreceptor tropism of patient virus populations containing different amino acid substitutions at position 11, 25 or 6–8 assigned by phenotype (Trofile) and genotype (PSSM and 11KR/25KR)
| Position in V3a | Infectivity (RLU)b | Coreceptor Tropismc | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Samples (subtype) |
11 | 25 | No. of PNGS |
CXCR4+ cells |
CCR5+ cells |
Ratio CXCR4 RLU to CCR5 RLU |
Trofile | PSSM | 11KR/25KR |
| 1(B) | R | Q | 1 | 65,466 | 90,981 | 0.720 | DM | X4 | X4 |
| 2(B) | R | A | 1 | 14,483 | 11,418 | 1.268 | DM | R5 | X4 |
| 3(B) | R | G | 1 | 481,460 | 115,108 | 4.183 | DM | X4 | X4 |
| 4(B) | R | S | 1 | 23,201 | 4,494 | 5.163 | DM | X4 | X4 |
| 5(B) | R | E | 1 | 73,866 | 9,646 | 7.658 | DM | X4 | X4 |
| 6(B) | R | E | 1 | 138,510 | 9,252 | 14.971 | DM | X4 | X4 |
| 7(A/G) | R | D | 1 | 30,551 | 69 | ND | X4 | X4 | X4 |
| 8(B) | G | R | 1 | 97 | 713,550 | ND | R5 | R5 | X4 |
| 9(B) | S | R | 1 | 80 | 517,002 | ND | R5 | R5 | X4 |
| 10(B) | S | R | 1 | 56 | 18,037 | ND | R5 | R5 | X4 |
| 11(B) | G | R | 1 | 55 | 97,220 | ND | R5 | R5 | X4 |
| 12(B) | G | R | 1 | 51 | 4,367 | ND | R5 | R5 | X4 |
| 13(B) | G | R | 1 | 582 | 540,398 | 0.001 | DM | R5 | X4 |
| 14(B) | G | R | 1 | 1,905 | 52,584 | 0.036 | DM | X4 | X4 |
| 15(B) | G | R | 1 | 625 | 167,263 | 0.004 | DM | R5 | X4 |
| 16(B/D) | S | R | 1 | 627 | 173,087 | 0.004 | DM | R5 | X4 |
| 17(B) | G | R | 1 | 1,243 | 24,309 | 0.051 | DM | R5 | X4 |
| 18(B) | S | R | 1 | 118,979 | 104,602 | 1.137 | DM | R5 | X4 |
| 19(B) | H | R | 1 | 42,390 | 25,953 | 1.633 | DM | R5 | X4 |
| 20(B) | S | R | 1 | 428,770 | 133,275 | 3.217 | DM | R5 | X4 |
| 21(B) | S | R | 1 | 21,419 | 57 | ND | X4 | X4 | X4 |
| 22(C) | G | K | 1 | 66 | 5,448 | ND | R5 | X4 | X4 |
| 23(B) | S | K | 1 | 79 | 9,667 | ND | R5 | R5 | X4 |
| 24(B) | S | K | 1 | 61 | 29,357 | ND | R5 | R5 | X4 |
| 25(B) | S | K | 1 | 589 | 138,904 | 0.004 | DM | R5 | X4 |
| 26(B) | G | K | 1 | 3,000 | 447,328 | 0.007 | DM | R5 | X4 |
| 27(B) | G | K | 1 | 221,510 | 238,062 | 0.930 | DM | X4 | X4 |
| 28(B) | S | K | 1 | 236,387 | 182,683 | 1.294 | DM | R5 | X4 |
| 29(B) | G | K | 1 | 232,884 | 170,395 | 1.367 | DM | X4 | X4 |
| 30(B) | G | K | 1 | 54,503 | 19,738 | 2.761 | DM | X4 | X4 |
| 31(B) | G | K | 1 | 51,865 | 2,890 | 17.946 | DM | X4 | X4 |
| 32(B) | G | K | 1 | 70,019 | 539 | 129.905 | DM | X4 | X4 |
| 33(A/G) | S | D | 0 | 75 | 13,292 | ND | R5 | X4 | R5 |
| 34(B) | I | D | 0 | 56,990 | 52,608 | 1.083 | DM | X4 | R5 |
| 35(B) | S | Q | 0 | 200,817 | 447 | 449.255 | DM | X4 | R5 |
| HXB2 (X4) | S | E | 1 | 620,340 | 98 | ND | X4 | X4 | X4 |
| BaL (R5) | R | E | 0 | 90 | 2,023,112 | ND | R5 | R5 | R5 |
The 11R and 25K/R substitutions and the absence of the potential N-linked glycosylation site (PNGS) at position 6–8 in V3 are shown in boldface (no mixtures were detected at these positions).
Infectivity was measured as luciferase activity (relative light units, RLU) using the Trofile assay. RLU <200 represents background levels of luciferase activity on CXCR5+ or CXCR+ cells. The ratios of CXCR4 RLU to CCR5 RLU are indicated for DM viruses, ND: CXCR4/CCR5 ratios were not determined because luciferase activity (RLU) was at or below background levels for either CXCR4 or CCR5 cells.
Coreceptor tropism predictions by genotypic algorithms that do not match the phenotypic determination by the Trofile assay are highlighted in gray.
All but one of the seven viruses containing 11R substitutions were consistently predicted to use CXCR4 based on the V3 genotype-based algorithms PSSM and 11/25 rule. These predictions were highly concordant with phenotypic measurements of CXCR4 use (Table 1). In contrast, genotypic predictions of CXCR4 use or CCR5 use for the subset of viruses containing 25K/R substitutions were often discordant with phenotypic measurements (11 of 25 viruses misclassified by PSSM, 8 of 25 viruses misclassified by 11/25 rule). Similarly, genotypic algorithms also misclassified CXCR4 use among viruses lacking the PNGS within the V3 region (1 of 3 viruses misclassified by PSSM, 2 of 3 viruses misclassified by 11/25 rule) (Table 1).
Among the 35 viruses evaluated, six additional amino acid substitutions--glutamate (E), aspartate (D), glutamine (Q), glycine (G), alanine (A), and serine (S)--were observed at position 25 (10 viruses), in addition to K and R (25 viruses). Four amino acid substitutions--predominantly S (14/28) and G (11/28), and rarely histidine (H, 1/28) and isoleucine (I, 1/28)--were observed at position 11 in 28 viruses, in addition to R in seven viruses (Table 1). These data suggest that HIV-1 can accommodate a greater diversity of amino acid substitutions at position 25 versus position 11 in V3. Notably, in addition to the 200 viruses surveyed in the current study, 11K was also not observed upon surveying 1,148 env sequences of patient viruses compiled by our laboratory (Stawiski et al; unpublished observation).
11R substitutions in V3 strongly promote, but are not essential for efficient use of CXCR4
To further examine the contribution of the 11R substitution to CXCR4 usage, we next characterized the coreceptor tropism and V3 sequences of multiple env molecular clones derived from four patient virus populations that had mixed amino acid sequences at position 11 (env subtype B=2, B/C=1, AE=1), (Table 2). Three virus populations (subjects 1–3) were comprised of mixtures of R5 and dual variants, while the remaining virus population (subject 4) was comprised solely of dual variants. In all four subjects, 11R variants were mixed with either 11S (subjects 1, 2 and 4) or 11G (subject 3) variants (consistent with the position 11 substitutions that we observed for the 35 viruses listed Table 1). Based upon infectivity, every 11R env clone isolated from all four subjects utilized CXCR4 efficiently relative to CCR5. This phenotype of 11R env clones is consistent with the 11R virus populations described in Table 1 and with our previous description of a dual-X virus sub-classification (Huang et al., 2007).
Table 2.
env clonal analysis of four patient virus populations containing mixed amino acid substitutions at position 11 in V3
| Subject (subtype) |
Virusesa | Position 11b |
V3 amino acid sequencesc | No.of AA changesd |
Coreceptor tropisme |
Infectivity (RLU)f | |
|---|---|---|---|---|---|---|---|
| CXCR4+ cells |
CCR5+ cells |
||||||
| 1(B/C) | Population | S/R | DM | 14,188 | 523,551 | ||
| clone (n=4) | S |  |
0 | R5 | 83 | 951,905 | |
| clone (n=1) | S | 4 | R5 | 63 | 556,160 | ||
| clone (n=1) | S | 5 | R5 | 157 | 1,015,357 | ||
| clone (n=2) | R | 8 | Dual-X | 564,997 | 432,732 | ||
| 2(B) | Population | G/R | DM | 73,347 | 55,808 | ||
| clone (n=1) | G |  |
0 | R5 | 177 | 69,366 | |
| clone (n=2) | G | 0 | Dual-R | 660 | 1,456,992 | ||
| clone (n=4) | R | 5 | Dual-X | 681,919 | 350,020 | ||
| clone (n=3) | R | 6 | Dual-X | 730,141 | 174,252 | ||
| 3(AE) | Population | S/R | DM | 6,091 | 142,291 | ||
| clone (n=4) | S | 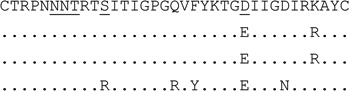 |
0 | R5 | 121 | 603,308 | |
| clone (n=1) | S | 2 | R5 | 74 | 1,211,595 | ||
| clone (n=1) | S | 2 | Dual-X | 16,534 | 13,848 | ||
| clone (n=3) | R | 5 | Dual-X | 1,026,300 | 693,476 | ||
| 4(B) | Population | S/R | DM | 560,500 | 745,641 | ||
| clone (n=6) | S |  |
0 | Dual-X | 879,283 | 531,108 | |
| clone (n=1) | R | 1 | Dual-X | 173,666 | 126,407 | ||
| clone (n=1) | R | 2 | Dual-X | 29,439 | 40,645 | ||
Clones with identical V3 sequences and the same coreceptor tropism are grouped together. One dual clone from subject 2 and one from subject 3 (indicated in bold) were chosen for reverse site directed mutagenesis.
The 11R substitutions in V3 are shown in boldface
For V3 amino acid sequences, dots represent amino acids identical to the first sequence, dashes in the first sequence indicate gaps to accommodate insertions. Positions 11, 25 and 6–8 (for PNGS) are underlined.
The number of amino acids that differ from the most prevalent V3 sequence of R5 clones in the same sample is indicated, except for subject 4. Subject 4 contained only dual clones, and the number of amino acid differences between clones with or without 11R is shown.
Coreceptor tropism was determined by the Trofile assay. Dual clones were further classified as dual-X and dual-R based on efficiently or inefficiently utilized CXCR4, as defined previously (Huang et al., 2007). R5 and dual-X clones from subject 3 that shared identical V3 amino acid sequences are highlighted (bold and italics). Dual-X clones from subject 4 that shared identical V3 amino acid sequences, except at position 11 (S or R), are highlighted (bold and italics).
Infectivity was measured as luciferase activity (relative light units, RLU) in the Trofile assay. RLU <200 on CXCR5+ or CXCR+ cells was considered background luciferase activity in this study. Infectivity of multiple clones with the same V3 sequence is expressed as the average RLU.
It is important to note that we also identified dual-X variants that lacked 11R substitutions in two of the four patient viruses evaluated (Table 2). In subject 3, we identified one dual-X variant that shared an identical V3 sequence (including 11S, 25E) with R5 clones. In this case, CXCR4 use is determined entirely by sequence changes outside of the V3 region. In subject 4, all eight clones that were evaluated displayed dual-X tropism and shared similar V3 sequences, yet only two clones contained the 11R substitution, whereas the remaining six clones had 11S. We conclude from these findings that the 11R-containing variants frequently contribute to efficient CXCR4 use, but this substitution is not a requisite determinant.
CXCR4 utilization by env sequences containing 25K or 25R substitutions in V3 is context dependent
Evaluation of the contribution of basic amino acid substitutions at position 25 to CXCR4 use was similarly conducted by characterizing the tropism and V3 sequences of multiple env molecular clones derived from five patient virus populations (env subtype B=4, C=1) that exhibited mixed amino acid sequences at position 25 (Table 3). In subjects 5–9, virus subpopulations containing either 25K or 25R co-existed with subpopulations containing 25E, 25D, or 25G. In subjects 5 and 6, all of the clones evaluated shared similar V3 sequences and did not utilize CXCR4, i.e. were R5, irrespective of the presence or absence of a basic amino acid substitution (K or R) at position 25. Conversely, in subjects 7, 8 and 9, all clones containing 25K or 25R were able to use CXCR4, i.e. were dual or X4 (105–106 RLU). Notably, the V3 sequences of these variants also contained numerous additional substitutions relative to the V3 sequence of the most predominant R5 variants.
Table 3.
env clonal analysis of five patient virus populations containing mixed amino acid substitutions at position 25 in V3
| Subject (subtype) |
Viruses a | Position 25 b |
V3 amino acid sequences c | No.of AA changes d |
Coreceptor tropism e |
Infectivity (RLU)f | |
|---|---|---|---|---|---|---|---|
| CXCR4+ cells |
CCR5+ cells |
||||||
| 5(B) | Population | E/K | R5 | 62 | 383,806 | ||
| clone (n=4) | E |  |
0 | R5 | 70 | 1,796,486 | |
| clone (n=1) | E | 1 | R5 | 92 | 1,559,819 | ||
| clone (n=1) | D | 1 | R5 | 79 | 1,799,031 | ||
| clone (n=2) | K | 1 | R5 | 51 | 552,861 | ||
| 6(B) | Population | G/R | R5 | 66 | 148,806 | ||
| clone (n=3) | G | 0 | R5 | 54 | 359,517 | ||
| clone (n=8) | R | 2 | R5 | 81 | 149,570 | ||
| 7(C) | Population | E/K | DM | 709,584 | 661,721 | ||
| clone (n=3) | E | 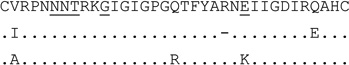 |
0 | R5 | 182 | 298,585 | |
| clone (n=1) | E | 3 | R5 | 172 | 128,256 | ||
| clone (n=4) | K | 3 | Dual-X | 630,016 | 660,567 | ||
| 8(B) | Population | D/K | DM | 2,701 | 27,905 | ||
| clone (n=5) | D | 0 | R5 | 79 | 559,320 | ||
| clone (n=3) | K | 10 | X4 | 497,805 | 133 | ||
| 9(B) | Population | E/R | DM | 62,552 | 392,667 | ||
| clone (n=4) | E |  |
0 | R5 | 85 | 503,528 | |
| clone (n=1) | E | 1 | R5 | 63 | 1,029,078 | ||
| clone (n=1) | R | 11 | Dual-X | 1,487,002 | 1,754,137 | ||
| clone (n=1) | R | 13 | X4 | 356,894 | 122 | ||
| clone (n=1) | R | 15 | X4 | 137,117 | 86 | ||
Clones with identical V3 sequences and the same coreceptor tropism are grouped together.
The 25K/R substitutions in V3 are shown in boldface.
For V3 amino acid sequences, dots represent amino acids identical to the first sequence, dashes indicate gaps to accommodate insertions. Positions 11, 25 and 6–8 (for PNGS) are underlined.
The number of amino acids that differ from the most prevalent V3 sequence of R5 clones in the same sample is indicated.
Coreceptor tropism was determined by the Trofile assay. Dual clones were further classified as dual-X and dual-R based on efficiently or inefficiently utilized CXCR4, as defined previously (Huang et al., 2007).
Infectivity was measured as luciferase activity (relative light units, RLU) in the Trofile assay. RLU <200 on CXCR5+ or CXCR+ cells was considered background luciferase activity in this study. Infectivity of multiple clones with the same V3 sequence is expressed as the average RLU.
CXCR4 utilization by env sequences lacking a PNGS at position 6–8 in V3 is context dependent
We isolated and characterized the tropism and V3 sequences of multiple env molecular clones derived from three patient virus populations (env subtype B=2, A=1) that exhibited mixed PNGS at V3 position 6–8 (Table 4). In subjects 10 and 11, none of the clones evaluated utilized CXCR4, i.e. were R5, irrespective of the presence or absence of a PNGS at position 6–8, and in the case of subject 11, the presence of 25K as well. Conversely, in subject 12, dual-X variants lacking the PNGS utilized CXCR4 efficiently (>105 RLU) when they also contained numerous additional substitutions relative to the V3 sequences of the most predominant R5 variants. Based on the clonal analyses presented in Tables 3 and 4, we conclude that the contribution of basic amino acid substitutions at position 25, or the absence of PNGS at positions 6–8, to CXCR4 use is highly dependent on the context of the env sequence, in particular, the V3 region.
Table 4.
env clonal analysis of three patient virus populations containing mixed amino acid substitutions that disrupt a PNGS at position 6–8 in V3
| Subject (subtype) |
Viruses a | No. of PNGS at position 6–8 b |
V3 amino acid sequences c | No.of AA changes d |
Coreceptor tropism e |
Infectivity (RLU) f | |
|---|---|---|---|---|---|---|---|
| CXCR4+ cells |
CCR5+ cells |
||||||
| 10(B) | Population | 0/1 | R5 | 54 | 18,687 | ||
| clone (n=8) | 0 |  |
0 | R5 | 74 | 335,296 | |
| clone (n=1) | 0 | 1 | R5 | 100 | 201,829 | ||
| clone (n=1) | 1 | 4 | R5 | 107 | 520,939 | ||
| 11(B) | Population | 0/1 | R5 | 163 | 59,137 | ||
| clone (n=5) | 1 |  |
0 | R5 | 66 | 580,001 | |
| clone (n=1) | 1 | 1 | R5 | 101 | 715,276 | ||
| clone (n=2) | 0 | 4 | R5 | 72 | 34,875 | ||
| clone (n=1) | 0 | 7 | R5 | 74 | 1,563 | ||
| clone (n=1) | 0 | 6 | R5 | 55 | 6,777 | ||
| 12(A) | Population | 0/1 | DM | 20,924 | 216,146 | ||
| clone (n=5) | 1 |  |
0 | R5 | 101 | 950,986 | |
| clone (n=2) | 1 | 0 | Dual-R | 898 | 1,148,595 | ||
| clone (n=1) | 0 | 1 | R5 | 118 | 703,962 | ||
| clone (n=2) | 0 | 12 | Dual-X | 569,619 | 1,785 | ||
Clones with identical V3 sequences and the same coreceptor tropism are grouped together. One R5 clone from subject 11 (indicated in bold) was chosen for site directed mutagenesis.
The absence of the potential N-linked glycosylation site (PNGS) at position 6–8 in V3 are shown in boldface
For V3 amino acid sequences, dots represent amino acids identical to the first sequence. Positions 11, 25 and 6–8 (for PNGS) are underlined.
The number of amino acids that differ from the most prevalent V3 sequence of R5 clones in the same sample is indicated.
Coreceptor tropism was determined by the Trofile assay. Dual clones were further classified as dual-X and dual-R based on efficiently or inefficiently utilized CXCR4, as previously described (Huang et al., 2007).
Infectivity was measured as luciferase activity (relative light units, RLU) in the Trofile assay. RLU <200 on CXCR5+ or CXCR+ cells was considered background luciferase activity in this study. Infectivity of multiple clones with the same V3 sequence is expressed as the average RLU.
An accumulation of multiple amino acid changes in V3 is required for efficient utilization of CXCR4
It should be noted that env variants that are capable of utilizing CXCR4, i.e. dual-X and X4, were characterized by multiple amino acid substitutions in V3 relative to R5 variants within the same virus population. The number of amino acid differences among env clones in each virus population is summarized in Figure 1. Furthermore, the association between the presence of basic amino acid substitutions at position 11 and 25, or the loss of a PNGS at position 6–8, and CXCR4 use appears to be reliant on the presence of additional mutations in V3 (Figure 1, Table 2–4). For example, although all 11R variants were dual-X, they also contained 5–8 additional amino acid substitutions in V3 relative to R5 clones from the same virus population (Table 2, subjects 1–3). Similarly, CXCR4-using variants containing 25K or 25R substitutions, or lacking the PNGS at position 6–8, also contained 3–15 different amino acids in V3 compared to R5 clones within the same virus population (Table 3, subjects 7, 8, 9 and Table 4, subject 12). Overall, the number of different V3 amino acid substitutions in env sequences containing 11R, 25K/R, or lacking a PNGS at position 6–8, was noticeably higher for X4 and dual-X variants (mean=9, range 3–15) compared to R5 and dual-R variants (inefficiently utilized CXCR4) within the same virus population (mean=1, range 0–5). We interpret these data as evidence that the evolution of CXCR4 use either by the acquisition of basic amino acid substitutions at position 11 or 25, or the loss of a PNGS at position 6–8, also involves the accumulation of additional V3 amino acid changes that may contribute to either efficient CXCR4 use or improvements in env infectivity.
Figure 1. The number of V3 amino acid differences among env clones from each of 12 subjects.

Each symbol represents a unique V3 sequences that differs from the most prevalent V3 sequence of R5 clones in the same sample (except for subject 4). Subject 4 contained only dual-X clones (efficiently utilize CXCR4) and the number of amino acid differences between clones with or without 11R is shown. R5 and dual-R (inefficiently utilize CXCR4) clones are indicated by open squares, dual-X and X4 clones are indicated by filled squares. The amino acid substitution or number of PNGS is shown within each symbol.
Introduction of 11K/R, 25K/R or N6Q mutations into the V3 region of R5 env sequences clarifies mutational pathways and genetic barriers to CXCR4 use
We used site-directed mutagenesis to engineer a panel of isogenic viruses that could be used to clearly establish the impact of basic amino acid substitutions at positions 11 and 25 of V3, or the loss of a PNGS site at position 6–8, on coreceptor utilization and env infectivity (Table 5). Single V3 mutations (11K, 11R, 25K, 25R, N6Q) were introduced into the env sequences of three R5 viruses: the well-characterized JRCSF and BaL strains, and molecular clone c11.2 isolated from the virus population of subject 11. Clone c11.2 (S11, E25, N6) was chosen as the third R5 env backbone sequence for these studies because all variants analyzed from the parental patient virus population were R5, irrespective the presence of a basic amino acid substation at position 25 (K) and/or the absence of a PNGS at position 6–8 (Table 4).
Table 5.
Coreceptor utilization of site-directed mutants using three R5 and two dual env backbones
| Infectivity (RLU)c | |||||
|---|---|---|---|---|---|
| Source of env backbonesa |
V3 mutationsb | Coreceptor tropism |
CXCR4+ cells | CCR5+ cells | % RLU of parental (CCR5+ cells)d |
| JRCSF (R5) | S11, E25, PNGS(+) | R5 | 193 | 1,321,140 | 100 |
| S11K | Dual | 1,929 | 299,175 | 23 | |
| S11R | Dual | 35,195 | 655,246 | 49 | |
| E25K | Dual | 22,303 | 829,696 | 63 | |
| E25R | Dual | 40,693 | 816,309 | 62 | |
| PNGS(−) | Dual | 47,206 | 3,234,479 | 245 | |
| BaL (R5) | S11, E25, PNGS(+) | R5 | 90 | 2,420,047 | 100 |
| S11K | R5 | 191 | 123,724 | 5 | |
| S11R | Dual | 11,597 | 757,409 | 31 | |
| E25K | Dual | 14,063 | 2,795,704 | 116 | |
| E25R | Dual | 37,674 | 2,079,533 | 86 | |
| PNGS(−) | Dual | 1,899 | 1,542,274 | 64 | |
| c11.2 (R5) | S11, E25, PNGS(+) | R5 | 60 | 308,852 | 100 |
| (subject 11) | S11K | R5 | 87 | 6,157 | 2 |
| S11R | R5 | 78 | 67,408 | 22 | |
| E25K | R5 | 77 | 152,441 | 49 | |
| E25R | R5 | 70 | 117,422 | 38 | |
| PNGS(−) | R5 | 78 | 62,047 | 20 | |
| c2.41 (Dual) | R11, Q25, PNGS(+) | Dual | 632,384 | 400,342 | 100 |
| (subject 2) | R11S | Dual | 586,581 | 600,991 | 150 |
| c3.14 (Dual) | R11, E25, PNGS(+) | Dual | 790,017 | 342,895 | 100 |
| (subject 3) | R11S | Dual | 525,764 | 317,581 | 93 |
Five distinct env sequences were used to generate site-directed mutants: parental JRCSF, BaL and c11.2 (isolated from subject 11) env sequences are R5; parental c2.41 and c3.14 (isolated from subjects 2 and 3) env sequences are dual.
The listed substitutions were introduced into the V3 region of three R5 env sequences (JRCSF, BaL and c11.2) and two dual env sequences (c2.41, c3.14). The presence or absence of PNGS at position 6–8 is indicated as + or −. PGNS(−) mutants of R5 env sequences were engineered by the introduction of N6Q substitutions.
Infectivity was measured as luciferase activity (relative light units, RLU) in the Trofile assay. RLU <200 on CXCR5+ or CXCR+ cells was considered background luciferase activity in this study.
CCR5-mediated entry of each mutant env sequence is represented as a percentage of the parental env sequence.
The introduction of the 11K substitution into all three R5 env sequences conferred detectable levels of CXCR4-mediated entry to JRCSF env (>103 RLU), but not for BaL and c11.2 envs (Table 5). Notably, the introduction of 11K was also associated with reductions in env-mediated infectivity on CCR5+ cells (23%, 5% and 2% relative to the parental JRCSF, BaL and c11.2 parental envs, respectively). Similarly, the introduction of the 11R substitution conferred CXCR4-mediated entry to the JRCSF and BaL envs (>104 RLU) but not to the clone c11.2 env. Again, the introduction of 11R was accompanied by reductions in infectivity on CCR5+ cells (49%, 31% and 22% relative to the JRCSF, BaL and c11.2 parental clones, respectively). Taken together, these observations suggests that the 11K and 11R substitutions are strong determinants of CXCR4 use, but when introduced as single mutations, their contributions are partially obscured by impairments in env infectivity in the absence of compensatory mutations.
The introduction of the 25K or 25R substitutions conferred CXCR4-mediated entry to JRCSF and BaL (>104 RLU) but not clone c11.2. In contrast to the 11K and 11R substitutions, the 25K and the 25R substitutions did not severely impair infectivity on CCR5 cells, suggesting that these two substitutions do not dramatically reduce overall env infectivity (Table 5). Thus, the ability of the 25K and 25R substitutions to confer CXCR4 use to some, but not all, R5 env sequences was more apparent than the 11K and 11R substitutions.
Disruption of the PNGS in the V3 regions of the three R5 env sequences was accomplished by the introduction of an N6Q mutation. Loss of the PNGS conferred detectable levels of CXCR4 use to JRCSF (>104 RLU) and BaL (>103 RLU). However, infection of CXCR4+ cells was not observed with the c11.2 env containing the N6Q mutation (Table 5). The effects of the loss of the PNGS on CCR5-mediated entry varied widely, ranging from 20% to 245% for the c11.2 and JR-CSF mutants, respectively. The reduction in CCR5 cell infectivity of the c11.2 mutant implies that compensatory mutations may be required to obtain efficient CXCR4 use by this R5 env sequence.
Reversion of R to S at position 11 does not reduce the ability of patient viruses to use CXCR4
To further test the role of V3 substitutions on CXCR4 use by patient virus isolates, we used site-directed mutagenesis to revert R substituions to S substitution at position 11. The infectivity of two dual clones (c2.41 and c3.14) containing 11R from subjects 2 and 3, and their respective mutants with a single R11S change are shown in Table 5. Interestingly, the two mutants with the R11S substitution retained the dual-tropic phenotype, and their ability to use CXCR4 and CCR5 for entry was not reduced relative to the parental clones. These data, together with experiments described above, confirm that backbone env sequences collectively contribute to CXCR4 use and can strongly influence the effects of specific V3 determinants of CXCR4 use, such as 11R, 25K/R and loss of a PNGS.
Comparative modeling of V3 structure in paired R5 and dual clones
To further examine the influence of V3 amino acid substitutions on CXCR4 use, we modeled the structure of the V3 loops of paired R5 and dual clones from two subjects with 11R variants (G11R for subject 2 and S11R for subject 3) and two subjects with 25K/R variants (E25K for subject 7 and E25R for subject 9) (Figure 2). Compared to R5 clones, each paired dual clone exhibited a large gain in positive electrostatic charge. This is in agreement with previous reports describing higher net charges in the V3 regions of CXCR4-using viruses (De Jong et al., 1992; Fouchier et al., 1992). However, our modeling indicated that a single basic amino acid substitution (R or K) at position 11 or 25 alone was not sufficient to produce the net change in positive charge observed in the paired R5 and dual clones (data not shown). This result suggests that the other V3 amino acids substitutions observed in dual clones contribute to the observed differences in electrostatic charge and are consistent with our genotypic and phenotypic characterizations described above. Furthermore, the amino acid side chains of S or G at position 11 are considerably smaller than the corresponding R and K side chains. Consequently, the S and G side chains are free to face, and perhaps form stabilizing hydrogen bonds with the opposing V3 stem, as observed in our models. In contrast, steric constraints imposed an outward positioning of the R and K side chains relative to the opposing V3 stem. At position 25, the side chains of E, R or K are more similar in size, and as such, all are sterically restricted away from the opposing V3 stem. The disparate positioning of amino acid side chains at position 11 and the consistent positioning of amino acid side chains at position 25 in R5 and dual clones may explain the impaired env infectivity that is observed for site-directed mutants containing 11R/K mutations, but not 25R/K mutations.
Figure 2. Structural modeling of the V3 regions of paired R5 and dual env clones from four patient virus populations.

Patterns of electrostatic charge across the V3 region are depicted (positive charges in blue, negative charges in red). Gains in positive charge at positions 11 and 25 are highlighted by * and ^, respectively. The V3 amino acid sequences are provided below each modeled structure, and basic amino acids are highlighted in blue. Subject 2 (G11 vs. 11R), subject 3 (S11 vs. 11R), subject 7 (E25 vs. 25K) and subject 9 (E25 vs. 25R).
DISCUSSION
In this study, we have investigated the effect of different V3 mutational pathways on CXCR4 mediated entry, including the acquisition of positively-charged amino acids or the loss of PNGS. Specifically, we characterized coreceptor usage among patient viruses that contained K or R substitutions at position 11 or 25, or that lacked a PNGS at amino acid position 6–8. We found that all patient viruses with a basic amino acid substitution at position 11 contained R substitutions and were able to utilize CXCR4. Viruses with 11K substitutions were not observed in the group of 200 patient viruses characterized in this study, or a larger survey of >1000 patient virus sequences. Conversely, CXCR4-mediated entry was variable among patient viruses containing K or R substitutions at position 25, or lacking a PNGS at position 6–8, including a subset of viruses that exhibited an R5 phenotype. Analysis of individual env clones containing these V3 changes confirmed our observations within virus populations. All env clones containing 11R substitutions utilized CXCR4 for entry, whereas only a subset of clones that contained either a 25K or 25R substitution, or that lacked a PNGS site at position 6–8 exhibited CXCR4-mediated entry. Current V3 sequence based tropism prediction methods (PSSM and 11/25 rule) are limited in their ability to accurately predict coreceptor use of viruses that lack K or R at position 11.
Our initial observations indicate that the presence of a basic amino acid at position 11 of V3 might be a stronger determinant of CXCR4 use than a basic amino acid at position 25, or the absence of a PNGS at position 6–8. To substantiate this hypothesis, we used site-directed mutagenesis to either introduce a basic amino acid substitutions at positions 11 or 25 or remove the PNGS at position 6–8 in three distinct R5 env sequences. We found that addition of either the 11K or 11R substitution alone was sufficient to confer CXCR4 use for 2/3 R5 env sequences, but that the magnitude of this affect was partially obscured by impairments in env infectivity, as noted by reductions in CCR5 mediated entry (most notably for the 11K substitution). The addition of 25K or 25R substitutions, or disruption of the PNGS was also capable of conferring CXCR4 use, however they were not consistently accompanied by reductions in CCR5-mediated infectivity. These results illustrate that the influence of individual V3 substitutions on CXCR4 use is dependent on env sequence context.
Across three distinct R5 env backbones, larger reductions in env infectivity were consistently observed with the 11K versus the 11R substitution. We speculate that 11K substitutions may be more incompatible with CCR5 utilization than 11R substitutions and consequently subject to more stringent negative selective pressure in vivo. Consistent with the observations in this study, a survey of 506 V3 sequences obtained from the Los Alamos National Laboratory database indicates that serine (S, 90.3%) and glycine (G, 7.5%) are the most prevalent substitutions at position 11 for R5 viruses. Examination of codons reveals a preferential selection for R over K at position 11: a transition from either S or G to R requires one base change while transition to K requires two base changes. This genetic disadvantage coupled with reductions in infectivity likely account for an extremely low frequency of 11K substitutions found in patient isolates.
Overall increases in V3 sequence heterogeneity among CXCR4-using virus populations have also been reported (Low et al., 2008; Troyer et al., 2005). In our study, clonal analysis of the four patient virus populations containing 11R substitutions revealed that, in three cases (subjects 1, 2, 3), all clones bearing 11R substitutions also contained additional V3 amino acid substitutions (N=5–8) relative to R5 clones that co-existed within the same virus population lacking 11R substitutions. In the remaining case (subject 4), both 11R and 11S clones utilized CXCR4 and shared similar V3 sequences, suggesting that 11R, per se, was not (or was no longer) the primary genetic determinant of CXCR4-mediated entry. Furthermore, reversion of R to S at position 11 by site-directed mutagenesis in dual env clones from subjects 2 and 3 did not alter CXCR4 utilization relative to the parental env clones. Based on the combined results of our database survey, site-directed mutagenesis experiments, and clonal analyses of 11R viruses, we invoke the following model that describes potential mutational pathways that lead to efficient CXCR4 use by 11R/K viruses: either 11R/K substitutions occur early, but require compensatory substitutions in V3 that restore obligate losses in env infectivity, or 11R/K substitutions occur late and must be preceded by V3 substitutions that provide more accommodating env contexts for 11R/K substitutions. This model is consistent with a strong association between 11R/K substitutions and efficient CXCR4 use.
Similar to 11R viruses, CXCR4-using env clones that contained 25K or 25R substitutions, or lacked a PNGS at position 6–8, also contained additional V3 amino acid substitutions (N=3–15), relative to R5 env clones that co-existed within the same virus populations (subjects 7, 8, 9 and 12). However, unlike 11R/K substitutions, 25R/K substitutions and mutations that result in loss of the PNGS at position 6–8 are not consistently associated with efficient CXCR4 utilization or reductions in env infectivity. Once again, based on the combined results of our database survey, site-directed mutagenesis experiments, and clonal analyses of 25K/R or PNGS(−) viruses, we invoke the following model to describe potential mutational pathways that lead to efficient CXCR4 use by 25R/K viruses or viruses that lack a PNGS at position 6–8: 25R/K substitutions or loss of the PNGS may occur early or late and are not associated with severe reductions in env infectivity, but require additional V3 substitutions that act cooperatively to confer efficient CXCR4 utilization. This model is consistent with a less stringent association between the presence of 25R/K substitutions or the absence of a PNGS at position 6–8 and efficient CXCR4 use.
The results of our V3 structural modeling exercises are consistent with our experimental observations. The acquisition of efficient CXCR4 use is associated with notable increases in positive electrostatic charges along the V3 stem-loop structure relative to the V3 stem-loop structures of paired R5 viruses from the same virus population. The models indicate that such increases in electrostatic charge are not acquired through single basic amino acid substitutions at position 11 or 25, but rather require multiple amino acid substitutions in the V3 sequence.
It is important to note that previous studies have demonstrated that not all determinants of CXCR4 use reside within the V3 region. Pastore et al., reported that substitutions in the V1/V2 regions of gp120 restore losses in entry infectivity resulting from V3 mutations and play an important role during coreceptor switching from CCR5 to CXCR4 of viruses selected in vitro (Pastore et al., 2006). Previously, our laboratory has reported that substitutions in gp41 of patient-derived envs impacted CXCR4 utilization (Huang et al., 2008). Furthermore, the replacement of V3 regions of X4 or dual env sequences with V3 regions of R5 env sequence, and vice versa, provides compelling evidence for the existence of determinants of CXCR4 utilization outside of the V3 region (Huang et al., 2008). Consistent with these prior observations, in this study we describe two env sequences from subject 3 that share identical V3 sequences, but differ notably in their ability to utilize CXCR4 (R5 vs. dual-X). In addition, clones that inefficiently use CXCR4 (dual-R) and contain the same V3 sequence with R5 clones were also identified.
The detailed characterization of 11R/K viruses that utilize CXCR4 in this study provides a plausible explanation for the observation that basic amino acid substitutions at position 11 but not 25, or losses in PNGS at positions 6–8, are associated with more accurate predictions of CXCR4 use. We postulate that 11R/K viruses likely escape from CCR5 inhibitor treatment by their ability to utilize CXCR4 once compensatory mutations accumulate to restore reductions in infectivity. The phenotypic impact of V3 amino acid substitutions on CXCR4 utilization by patient viruses presented in this study provides further insight into the different mutational pathways and genetic barriers for acquisition of CXCR4 use by clinical isolates.
ACKNOWLEDGEMENTS
We thank the many members of the Monogram Biosciences Clinical Reference Laboratory for the performance of tropism assays, Dr. Sunny Choe for critical review of the manuscript, and Cynthia Sedik for editorial assistance. This study was supported in part by SBIR-AT grant R44-AI-048990 (NIAID, NIH).
REFFERENCES
- Berger EA, Murphy PM, Farber JM. Chemokine receptors as HIV-1 coreceptors: roles in viral entry, tropism, and disease. Annu Rev Immunol. 1999;17:657–700. doi: 10.1146/annurev.immunol.17.1.657. [DOI] [PubMed] [Google Scholar]
- Brumme ZL, Goodrich J, Mayer HB, Brumme CJ, Henrick BM, Wynhoven B, Asselin JJ, Cheung PK, Hogg RS, Montaner JS, Harrigan PR. Molecular and clinical epidemiology of CXCR4-using HIV-1 in a large population of antiretroviral-naive individuals. J Infect Dis. 2005;192(3):466–474. doi: 10.1086/431519. [DOI] [PubMed] [Google Scholar]
- Cardozo T, Kimura T, Philpott S, Weiser B, Burger H, Zolla-Pazner S. Structural basis for coreceptor selectivity by the HIV type 1 V3 loop. AIDS Res Hum Retroviruses. 2007;23(3):415–426. doi: 10.1089/aid.2006.0130. [DOI] [PubMed] [Google Scholar]
- Chesebro B, Wehrly K, Nishio J, Perryman S. Mapping of independent V3 envelope determinants of human immunodeficiency virus type 1 macrophage tropism and syncytium formation in lymphocytes. J Virol. 1996;70(12):9055–9059. doi: 10.1128/jvi.70.12.9055-9059.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coakley E, Benhamida J, Chappey C, Whitcomb J, Goodrich J, van der Ryst E, Westby M, James I, Tressler R, Harrigan PR, Mayer H. 2nd International Workshop on Targeting HIV Entry. Boston, MA: 2006. [Google Scholar]
- Cocchi F, DeVico AL, Garzino-Demo A, Cara A, Gallo RC, Lusso P. The V3 domain of the HIV-1 gp120 envelope glycoprotein is critical for chemokine-mediated blockade of infection. Nat Med. 1996;2(11):1244–1247. doi: 10.1038/nm1196-1244. [DOI] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med. 1997;185(4):621–628. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DA, Heera J, Goodrich J, Tawadrous M, Saag M, Dejesus E, Clumeck N, Walmsley S, Ting N, Coakley E, Reeves JD, Reyes-Teran G, Westby M, Van Der Ryst E, Ive P, Mohapi L, Mingrone H, Horban A, Hackman F, Sullivan J, Mayer H. Maraviroc versus efavirenz, both in combination with zidovudine-lamivudine, for the treatment of antiretroviral-naive subjects with CCR5-tropic HIV-1 infection. J Infect Dis. 2010;201(6):803–813. doi: 10.1086/650697. [DOI] [PubMed] [Google Scholar]
- Daar ES, Kesler KL, Petropoulos CJ, Huang W, Bates M, Lail AE, Coakley EP, Gomperts ED, Donfield SM. Baseline HIV type 1 coreceptor tropism predicts disease progression. Clin Infect Dis. 2007;45(5):643–649. doi: 10.1086/520650. [DOI] [PubMed] [Google Scholar]
- De Jong JJ, De Ronde A, Keulen W, Tersmette M, Goudsmit J. Minimal requirements for the human immunodeficiency virus type 1 V3 domain to support the syncytium-inducing phenotype: analysis by single amino acid substitution. J Virol. 1992;66(11):6777–6780. doi: 10.1128/jvi.66.11.6777-6780.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarest J, Bonny T, Vavaro C, LaBranche C, Kitrinos K, McDanal C, Sparks S, Chavers S, Castillo S, Elrick D, McCarty D, Whitcomb J, Huang W, Petropoulos C, Piscitelli S. 44th Interscience Conference on Antimicrobial Agents and Chemotherapeutics; Washington, DC. 2004. [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, Davis CB, Peiper SC, Schall TJ, Littman DR, Landau NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661–666. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Eshleman SH, Husnik M, Hudelson S, Donnell D, Huang Y, Huang W, Hart S, Jackson B, Coates T, Chesney M, Koblin B. Antiretroviral drug resistance, HIV-1 tropism, and HIV-1 subtype among men who have sex with men with recent HIV-1 infection. Aids. 2007;21(9):1165–1174. doi: 10.1097/QAD.0b013e32810fd72e. [DOI] [PubMed] [Google Scholar]
- Fouchier RA, Groenink M, Kootstra NA, Tersmette M, Huisman HG, Miedema F, Schuitemaker H. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol. 1992;66(5):3183–3187. doi: 10.1128/jvi.66.5.3183-3187.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransen S, Bridger G, Whitcomb JM, Toma J, Stawiski E, Parkin N, Petropoulos CJ, Huang W. Suppression of dualtropic human immunodeficiency virus type 1 by the CXCR4 antagonist AMD3100 is associated with efficiency of CXCR4 use and baseline virus composition. Antimicrob Agents Chemother. 2008;52(7):2608–2615. doi: 10.1128/AAC.01226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick RM, Lalezari J, Goodrich J, Clumeck N, DeJesus E, Horban A, Nadler J, Clotet B, Karlsson A, Wohlfeiler M, Montana JB, McHale M, Sullivan J, Ridgway C, Felstead S, Dunne MW, van der Ryst E, Mayer H. Maraviroc for previously treated patients with R5 HIV-1 infection. N Engl J Med. 2008;359(14):1429–1441. doi: 10.1056/NEJMoa0803152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick RM, Su Z, Flexner C, Hughes MD, Skolnik PR, Wilkin TJ, Gross R, Krambrink A, Coakley E, Greaves WL, Zolopa A, Reichman R, Godfrey C, Hirsch M, Kuritzkes DR. Phase 2 study of the safety and efficacy of vicriviroc, a CCR5 inhibitor, in HIV-1-Infected, treatment-experienced patients: AIDS clinical trials group 5211. J Infect Dis. 2007;196(2):304–312. doi: 10.1086/518797. [DOI] [PubMed] [Google Scholar]
- Hendrix CW, Collier AC, Lederman MM, Schols D, Pollard RB, Brown S, Jackson JB, Coombs RW, Glesby MJ, Flexner CW, Bridger GJ, Badel K, MacFarland RT, Henson GW, Calandra G. Safety, pharmacokinetics, and antiviral activity of AMD3100, a selective CXCR4 receptor inhibitor, in HIV-1 infection. J Acquir Immune Defic Syndr. 2004;37(2):1253–1262. doi: 10.1097/01.qai.0000137371.80695.ef. [DOI] [PubMed] [Google Scholar]
- Hoffman TL, Doms RW. HIV-1 envelope determinants for cell tropism and chemokine receptor use. Mol Membr Biol. 1999;16(1):57–65. doi: 10.1080/096876899294760. [DOI] [PubMed] [Google Scholar]
- Huang W, Eshleman SH, Toma J, Fransen S, Stawiski E, Paxinos EE, Whitcomb JM, Young AM, Donnell D, Mmiro F, Musoke P, Guay LA, Jackson JB, Parkin NT, Petropoulos CJ. Coreceptor tropism in human immunodeficiency virus type 1 subtype D: high prevalence of CXCR4 tropism and heterogeneous composition of viral populations. J Virol. 2007;81(15):7885–7893. doi: 10.1128/JVI.00218-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Toma J, Fransen S, Stawiski E, Reeves JD, Whitcomb JM, Parkin N, Petropoulos CJ. Coreceptor tropism can be influenced by amino acid substitutions in the gp41 transmembrane subunit of human immunodeficiency virus type 1 envelope protein. J Virol. 2008;82(11):5584–5593. doi: 10.1128/JVI.02676-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Toma J, Stawiski E, Fransen S, Wrin T, Parkin N, Whitcomb JM, Coakley E, Hecht FM, Deeks SG, Gandhi RT, Eshleman SH, Petropoulos CJ. Characterization of human immunodeficiency virus type 1 populations containing CXCR4-using variants from recently infected individuals. AIDS Res Hum Retroviruses. 2009;25(8):795–802. doi: 10.1089/aid.2008.0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CS, Vander Heyden N, Ratner L. Analysis of the critical domain in the V3 loop of human immunodeficiency virus type 1 gp120 involved in CCR5 utilization. J Virol. 1999;73(10):8216–8226. doi: 10.1128/jvi.73.10.8216-8226.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt PW, Harrigan PR, Huang W, Bates M, Williamson DW, McCune JM, Price RW, Spudich SS, Lampiris H, Hoh R, Leigler T, Martin JN, Deeks SG. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J Infect Dis. 2006;194(7):926–930. doi: 10.1086/507312. [DOI] [PubMed] [Google Scholar]
- Hwang SS, Boyle TJ, Lyerly HK, Cullen BR. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253(5015):71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- Jenkinson S, Thomson M, McCoy D, Edelstein M, Danehower S, Lawrence W, Wheelan P, Spaltenstein A, Gudmundsson K. Blockade of X4-tropic HIV-1 cellular entry by GSK812397, a potent noncompetitive CXCR4 receptor antagonist. Antimicrob Agents Chemother. 2010;54(2):817–824. doi: 10.1128/AAC.01293-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MA, Li FS, van 't Wout AB, Nickle DC, Shriner D, He HX, McLaughlin S, Shankarappa R, Margolick JB, Mullins JI. Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J Virol. 2003;77(24):13376–13388. doi: 10.1128/JVI.77.24.13376-13388.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koot M, Keet IP, Vos AH, de Goede RE, Roos MT, Coutinho RA, Miedema F, Schellekens PT, Tersmette M. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann Intern Med. 1993;118(9):681–688. doi: 10.7326/0003-4819-118-9-199305010-00004. [DOI] [PubMed] [Google Scholar]
- Lewis M, Simpson P, Fransen S, Huang W, Whitcomb J, Mosley M, Robertson DL, Mansfield R, Ciaramella G, Westby M. CXCR4-using virus detected in patients receiving maraviroc in the Phase III studies MOTIVATE 1 and 2 originates from a pre-existing minority of CXCR4-using virus. Antivir Ther. 2007;12:S65. [Google Scholar]
- Low AJ, Marchant D, Brumme CJ, Brumme ZL, Dong W, Sing T, Hogg RS, Montaner JS, Gill V, Cheung PK, Harrigan PR. CD4-dependent characteristics of coreceptor use and HIV type 1 V3 sequence in a large population of therapy-naive individuals. AIDS Res Hum Retroviruses. 2008;24(2):219–228. doi: 10.1089/aid.2007.0140. [DOI] [PubMed] [Google Scholar]
- Markowitz M, Mohri H, Mehandru S, Shet A, Berry L, Kalyanaraman R, Kim A, Chung C, Jean-Pierre P, Horowitz A, LaMar M, Wrin T, Parkin N, Poles M, Petropoulos C, Mullen M, Boden D, Ho DD. Infection with multidrug resistant, dual-tropic HIV-1 and rapid progression to AIDS: a case report. Lancet. 2005;365(9464):1031–1038. doi: 10.1016/S0140-6736(05)71139-9. [DOI] [PubMed] [Google Scholar]
- Masquelier B, Capdepont S, Neau D, Peuchant O, Taupin JL, Coakley E, Lie Y, Carpentier W, Dabis F, Fleury HJ. Virological characterization of an infection with a dual-tropic, multidrug-resistant HIV-1 and further evolution on antiretroviral therapy. Aids. 2007;21(1):103–106. doi: 10.1097/QAD.0b013e3280117053. [DOI] [PubMed] [Google Scholar]
- Melby T, Despirito M, Demasi R, Heilek-Snyder G, Greenberg ML, Graham N. HIV-1 coreceptor use in triple-class treatment-experienced patients: baseline prevalence, correlates, and relationship to enfuvirtide response. J Infect Dis. 2006;194(2):238–246. doi: 10.1086/504693. [DOI] [PubMed] [Google Scholar]
- Mosier DE, Picchio GR, Gulizia RJ, Sabbe R, Poignard P, Picard L, Offord RE, Thompson DA, Wilken J. Highly potent RANTES analogues either prevent CCR5-using human immunodeficiency virus type 1 infection in vivo or rapidly select for CXCR4-using variants. J Virol. 1999;73(5):3544–3550. doi: 10.1128/jvi.73.5.3544-3550.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle G, DeJesus E, Boffito M, Wong RS, Gibney C, Badel K, MacFarland R, Calandra G, Bridger G, Becker S. Proof of activity with AMD11070, an orally bioavailable inhibitor of CXCR4-tropic HIV type 1. Clin Infect Dis. 2009;48(6):798–805. doi: 10.1086/597097. [DOI] [PubMed] [Google Scholar]
- Moyle GJ, Wildfire A, Mandalia S, Mayer H, Goodrich J, Whitcomb J, Gazzard BG. Epidemiology and predictive factors for chemokine receptor use in HIV-1 infection. J Infect Dis. 2005;191(6):866–872. doi: 10.1086/428096. [DOI] [PubMed] [Google Scholar]
- Murakami T, Kumakura S, Yamazaki T, Tanaka R, Hamatake M, Okuma K, Huang W, Toma J, Komano J, Yanaka M, Tanaka Y, Yamamoto N. The novel CXCR4 antagonist KRH-3955 is an orally bioavailable and extremely potent inhibitor of human immunodeficiency virus type 1 infection: comparative studies with AMD3100. Antimicrob Agents Chemother. 2009;53(7):2940–2948. doi: 10.1128/AAC.01727-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien WA, Koyanagi Y, Namazie A, Zhao JQ, Diagne A, Idler K, Zack JA, Chen IS. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nature. 1990;348(6296):69–73. doi: 10.1038/348069a0. [DOI] [PubMed] [Google Scholar]
- Pastore C, Nedellec R, Ramos A, Pontow S, Ratner L, Mosier DE. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J Virol. 2006;80(2):750–758. doi: 10.1128/JVI.80.2.750-758.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollakis G, Kang S, Kliphuis A, Chalaby MI, Goudsmit J, Paxton WA. N-linked glycosylation of the HIV type-1 gp120 envelope glycoprotein as a major determinant of CCR5 and CXCR4 coreceptor utilization. J Biol Chem. 2001;276(16):13433–13441. doi: 10.1074/jbc.M009779200. [DOI] [PubMed] [Google Scholar]
- Resch W, Hoffman N, Swanstrom R. Improved success of phenotype prediction of the human immunodeficiency virus type 1 from envelope variable loop 3 sequence using neural networks. Virology. 2001;288(1):51–62. doi: 10.1006/viro.2001.1087. [DOI] [PubMed] [Google Scholar]
- Richman DD, Bozzette SA. The impact of the syncytium-inducing phenotype of human immunodeficiency virus on disease progression. J Infect Dis. 1994;169(5):968–974. doi: 10.1093/infdis/169.5.968. [DOI] [PubMed] [Google Scholar]
- Saag M, Goodrich J, Fatkenheuer G, Clotet B, Clumeck N, Sullivan J, Westby M, van der Ryst E, Mayer H. A double-blind, placebo-controlled trial of maraviroc in treatment-experienced patients infected with non-R5 HIV-1. J Infect Dis. 2009;199(11):1638–1647. doi: 10.1086/598965. [DOI] [PubMed] [Google Scholar]
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234(3):779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- Sarkar G, Sommer SS. The "megaprimer" method of site-directed mutagenesis. Biotechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- Schuitemaker H, Koot M, Kootstra NA, Dercksen MW, de Goede RE, van Steenwijk RP, Lange JM, Schattenkerk JK, Miedema F, Tersmette M. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol. 1992;66(3):1354–1360. doi: 10.1128/jvi.66.3.1354-1360.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JC, Jacobson LP, Qiao W, Jamieson BD, Phair JP, Piazza P, Quinn TC, Margolick JB. Emergence and persistence of CXCR4-tropic HIV-1 in a population of men from the multicenter AIDS cohort study. J Infect Dis. 2008;198(8):1104–1112. doi: 10.1086/591623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shioda T, Levy JA, Cheng-Mayer C. Macrophage and T cell-line tropisms of HIV-1 are determined by specific regions of the envelope gp120 gene. Nature. 1991;349(6305):167–169. doi: 10.1038/349167a0. [DOI] [PubMed] [Google Scholar]
- Speck RF, Wehrly K, Platt EJ, Atchison RE, Charo IF, Kabat D, Chesebro B, Goldsmith MA. Selective employment of chemokine receptors as human immunodeficiency virus type 1 coreceptors determined by individual amino acids within the envelope V3 loop. J Virol. 1997;71(9):7136–7139. doi: 10.1128/jvi.71.9.7136-7139.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z, Gulick RM, Krambrink A, Coakley E, Hughes MD, Han D, Flexner C, Wilkin TJ, Skolnik PR, Greaves WL, Kuritzkes DR, Reeves JD. Response to vicriviroc in treatment-experienced subjects, as determined by an enhanced-sensitivity coreceptor tropism assay: reanalysis of AIDS clinical trials group A5211. J Infect Dis. 2009;200(11):1724–1728. doi: 10.1086/648090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suleiman J, Zingman BS, Diaz RS, Madruga JV, DeJesus E, Slim J, Mak C, Lee E, McCarthy MC, Dunkle LM, Walmsley S. Vicriviroc in combination therapy with an optimized regimen for treatment-experienced subjects:48-week results of the VICTOR-E1 phase 2 trial. J Infect Dis. 2010;201(4):590–599. doi: 10.1086/650342. [DOI] [PubMed] [Google Scholar]
- Tersmette M, Gruters RA, de Wolf F, de Goede RE, Lange JM, Schellekens PT, Goudsmit J, Huisman HG, Miedema F. Evidence for a role of virulent human immunodeficiency virus (HIV) variants in the pathogenesis of acquired immunodeficiency syndrome: studies on sequential HIV isolates. J Virol. 1989;63(5):2118–2125. doi: 10.1128/jvi.63.5.2118-2125.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trkola A, Dragic T, Arthos J, Binley JM, Olson WC, Allaway GP, Cheng-Mayer C, Robinson J, Maddon PJ, Moore JP. CD4-dependent, antibody-sensitive interactions between HIV-1 and its co-receptor CCR-5. Nature. 1996;384(6605):184–187. doi: 10.1038/384184a0. [DOI] [PubMed] [Google Scholar]
- Troyer RM, Collins KR, Abraha A, Fraundorf E, Moore DM, Krizan RW, Toossi Z, Colebunders RL, Jensen MA, Mullins JI, Vanham G, Arts EJ. Changes in human immunodeficiency virus type 1 fitness and genetic diversity during disease progression. J Virol. 2005;79(14):9006–9018. doi: 10.1128/JVI.79.14.9006-9018.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trujillo JR, Wang WK, Lee TH, Essex M. Identification of the envelope V3 loop as a determinant of a CD4-negative neuronal cell tropism for HIV-1. Virology. 1996;217(2):613–617. doi: 10.1006/viro.1996.0158. [DOI] [PubMed] [Google Scholar]
- Tsibris AM, Korber B, Arnaout R, Russ C, Lo CC, Leitner T, Gaschen B, Theiler J, Paredes R, Su Z, Hughes MD, Gulick RM, Greaves W, Coakley E, Flexner C, Nusbaum C, Kuritzkes DR. Quantitative deep sequencing reveals dynamic HIV-1 escape and large population shifts during CCR5 antagonist therapy in vivo. PLoS One. 2009;4(5):e5683. doi: 10.1371/journal.pone.0005683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westby M, Lewis M, Whitcomb J, Youle M, Pozniak AL, James IT, Jenkins TM, Perros M, van der Ryst E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority of HIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J Virol. 2006;80(10):4909–4920. doi: 10.1128/JVI.80.10.4909-4920.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westervelt P, Trowbridge DB, Epstein LG, Blumberg BM, Li Y, Hahn BH, Shaw GM, Price RW, Ratner L. Macrophage tropism determinants of human immunodeficiency virus type 1 in vivo. J Virol. 1992;66(4):2577–2582. doi: 10.1128/jvi.66.4.2577-2582.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitcomb JM, Huang W, Fransen S, Limoli K, Toma J, Wrin T, Chappey C, Kiss LD, Paxinos EE, Petropoulos CJ. Development and characterization of a novel single-cycle recombinant-virus assay to determine human immunodeficiency virus type 1 coreceptor tropism. Antimicrob Agents Chemother. 2007;51(2):566–575. doi: 10.1128/AAC.00853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkin TJ, Su Z, Kuritzkes DR, Hughes M, Flexner C, Gross R, Coakley E, Greaves W, Godfrey C, Skolnik PR, Timpone J, Rodriguez B, Gulick RM. HIV Type 1 Chemokine Coreceptor Use among Antiretroviral-Experienced Patients Screened for a Clinical Trial of a CCR5 Inhibitor: AIDS Clinical Trial Group A5211. Clin Infect Dis. 2007;44(4):591–595. doi: 10.1086/511035. [DOI] [PubMed] [Google Scholar]
- Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384(6605):179–183. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]