

Abstract
Bipolar disorder is characterized by a cycle of mania and depression, which affects approximately 5 million people in the United States. Current treatment regimes include the so-called “mood-stabilizing drugs”, such as lithium and valproate that are relatively dated drugs with various known side effects. Glycogen synthase kinase-3β (GSK-3β) plays a central role in regulating circadian rhythms, and lithium is known to be a direct inhibitor of GSK-3β. We designed a series of second generation benzofuran-3-yl-(indol-3-yl)maleimides containing a piperidine ring that possess IC50 values in the range of 4 to 680 nm against human GSK-3β. One of these compounds exhibits reasonable kinase selectivity and promising preliminary absorption, distribution, metabolism, and excretion (ADME) data. The administration of this compound at doses of 10 to 25 mgkg−1 resulted in the attenuation of hyperactivity in amphetamine/ chlordiazepoxide-induced manic-like mice together with enhancement of prepulse inhibition, similar to the effects found for valproate (400 mgkg−1) and the antipsychotic haloperidol (1 mgkg−1). We also tested this compound in mice carrying a mutation in the central transcriptional activator of molecular rhythms, the CLOCK gene, and found that the same compound attenuates locomotor hyperactivity in response to novelty. This study further demonstrates the use of inhibitors of GSK-3β in the treatment of manic episodes of bipolar/mood disorders, thus further validating GSK-3β as a relevant therapeutic target in the identification of new therapies for bipolar patients.
Keywords: antimanic agents, bipolar disorder, circadian rhythms, CLOCK mutant mice, GSK-3 inhibitors
Introduction
Malfunctioning protein kinase signaling in the human body has been correlated with a number of disease processes. In this regard, one particular kinase that has emerged as an attractive target in pharmaceutical research is the glycogen synthase kinase-3 (GSK-3). Initially reported in the late 1970s as the kinase that phosphorylates and inactivates glycogen synthase, GSK-3 is now recognized as a multifunctional serine/ threonine kinase involved in a number of central signaling pathways, including cell development, gene transcription, metabolic homeostasis, neurogenesis, and apoptosis.[1] In mammals, GSK-3 exists as two homologues: GSK-3a (51 kDa) and GSK-3β (47 kDa), that are distributed ubiquitously and constitutively active in unstimulated tissues both peripherally and centrally. At present, GSK-3 has been shown to phosphorylate over 50 identified protein substrates and is itself regulated by the phosphorylation of specific residues Tyr 279/Tyr 216 (GSK3α/β) and Ser 21/Ser 9 (GSK-3α/β) to bring about either activation or inhibition, respectively.[2]
The groundbreaking discovery that lithium directly inhibits GSK-3 was first reported in 1996 by the groups of Klein and Woodgett, spurring an exponential increase in research interest in this area.[3] There is now an increasing amount of evidence identifying the role of GSK-3-related Wnt signal transduction in mood disorders.[4] AR-A014418 has been reported to display neuroprotective activity in neuroblastoma cells through the inhibition of GSK-3.[5] In a behavioral context, haploinsufficient GSK-3β mice showed a more active response in an antidepressant assay, the forced swim test, as well as reduced amphetamine-induced hyperactivity similar to the behavioral profile observed for lithium-treated mice.[6] Valproic acid is also reported to directly inhibit GSK-3β, although there are some conflicting results in the literature.[7] Other drugs, such as the antidepressants fluoxetine and imipramine and the antipsychotic drug haloperidol, have all been shown to reduce GSK-3β activity in the brain.[8] Lithium is also reported to cause an increase in the synthesis of brain-derived neurotrophic factor (BDNF), which is increasingly known to be involved in mood regula-tion.[9] Furthermore, GSK-3β is reported to interact directly with serotonin 5-HT1B receptors but not 5-HT1A, consistent with the previous finding of lithium regulation of 5-HT1B.[10] Increasing number of studies also pointed out to the involvement of GSK-3β in behavioral aspects induced by dopaminergic activity.[11] Taken together, these studies validate GSK-3β as an attractive therapeutic target, not only in the treatment of bipolar disorder and other mood disorders, but also for neurodegenerative diseases, such as Alzheimer’s. As lithium, valproic acid, and carbamazepine have been used in the treatment of bipolar disease for over 40 years, and suffer from side effects, the introduction of safe, bioavailable GSK-3β inhibitors may offer a means for discovering improved drug treatments.[12]
To date, there are many small, drug-like GSK-3β inhibitors that have been reported by both pharmaceutical companies and academic groups. However, many of these are yet to be thoroughly characterized in a variety of in vivo studies. Moreover, no central nervous system (CNS)-active, GSK-3β-selective inhibitor has been used clinically. Our group reported a series of benzofuran-3-yl-(indol-3-yl)maleimides as potent ATP-competitive GSK-3β inhibitors,[13] one of which (compound 1) was found to block mania-like effects in rodents (Figure 1).[13a] We recently solved the crystal structure of one of our first-generation benzofuran-3-yl-(indol-3-yl)maleimide 2 bound to the GSK-3b active site. Consequently, as part of our ongoing efforts to identify GSK-3β inhibitor that could be advanced to the clinic for the treatment of bipolar/mood disorders, we designed piperidylmaleimides 3 by exploiting the structural information gleaned from the X-ray co-crystal structure of 2 (Figure 2). Aiming to improve the water solubility, blood–brain barrier penetration and desirable pharmacokinetic profile, while maintaining the potency and selectivity at GSK-3β, herein we describe the synthesis, structure–activity relationship (SAR), in vivo pharmacology and preliminary ADME profile of these new piperidylmaleimides.
Figure 1.

Benzofuran-3-yl-(indol-3-yl)maleimides 1–3. Compound 3, second generation piperidylmaleimides, exhibits improved water solubility and metabolic stability.
Figure 2.

X-ray co-crystal structure of benzofuran-3-yl-(indol-3-yl)maleimide bound to the GSK-3β (PDB ID: 3SD0; http://www.pdb.org). a) Electron density surrounding inhibitor 2 bound to the active site of GSK-3β. The Fo–Fc Fomit map is contoured at + 3s. b) Hydrogen bonds formed between GSK-3β and 2 are shown as dashed ellipsoids. The bromine atom of 2 is colored green. c) Solvent-exposed surface map of GSK-3β (grey) showing binding pocket for 2 and solvent exposure of the hydroxypropyl group.
Results and Discussion
Design, synthesis and in Vitro SAR of the benzofuran-3-yl-(indol-3-yl)maleimides
The X-ray co-crystal structure of one of our previously identi-fied benzofuran-3-yl-(indol-3-yl)maleimide analogues 2 bound to the GSK-3β (Figure 2) was determined to 2.7 resolution. Residual electron-density difference maps (Fo–Fc) reveal that inhibitor 2 binds in the active site with well-ordered density. The position of the bromine atom of 2 is strikingly evident in Fo–Fc maps contoured at +10σ. The orientation of 2 within the active site reveals the characteristic hydrogen bonding of the maleimide core to the carbonyl group of Asp133 and amino group of Val135, as well as the interaction of the hy-droxypropyl group extension from the indole ring with the residue Arg141. In addition, a water molecule acts as a bridge between the carbonyl group of maleimide to Asp200, while another water molecule connects the oxygen atom of the benzofuran ring to Lys85. The hydroxypropyl substituent on the indole ring forms a hydrogen bond with the Arg141 but is largely solvent exposed and therefore could be substituted with the more rigid piperidine analogue. Such a substitution would not only improve water solubility when prepared in a salt form, but also acts as a versatile handle to introduce further chemical modifications when required.
With this hypothesis in hand, a series of piperidyl maleimides were synthesized by condensation of the appropriately substituted 3-indolylglyoxylic acid esters and benzofuran-3-acetamide as previously described (Scheme 1).[13a] Reductive amination of piperidones 5 with indolines 4 was carried out in the presence of NaBH(OAc)3 in acetic acid, followed by aromatization with 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in anhydrous THF to form indoles 6, which were subsequently treated with oxalyl chloride and methanol to form the substituted 3-indolylglyoxylic acid esters 7. Benzofuran-3-acetamides 10 were acquired from Wittig reaction of 8 with (carbethoxymethylene)triphenylphosphorane, followed by subsequent conversion of esters 9 to the corresponding acetamides.[13a] Final deprotection of 11 and subsequent reductive amination or amide bond formation afforded the desired final compounds.
Scheme 1.

Reagents and conditions: a) NaBH(OAc)3, HOAc, RT, 1 h; b) DDQ, THF, RT, 16 h; c) 1. (COCl)2, Et2O; 2. MeOH, 0°C → RT, 16 h; d) Ph3P=CHCO2Et, PhMe, 100°C, 16 h; e) NH (l), EtOH, 80° 3 C, 3 d; f) tBuOK, Et2O or THF, −20°C→RT, 16 h; g) TFA, CH2Cl2, RT, 2 h; h) R1CO2H, HOBt, EDC, DMAP, CH2Cl2, RT, 16 h; i) ArCHO or HCHO, NaBH(OAc)3, HOAc, THF, RT, 16 h.
The synthesized maleimides were then evaluated for their ability to inhibit phosphorylation of the primed substrate (YR RAAVPPSPSLSRHSSPHQ(pS)EDEEE; 20 μm) in the presence of 10 μm ATP by human GSK-3β. As presented in Table 1, these compounds possess IC50 values in the range of 4 to 680 nm. The unsubstituted piperidine analogue 3a showed good potency (IC50=67 nm) comparable to compound 1. Substitution of the benzofuran ring with 6- or 7-methoxybenzofuran (3b,c) resulted in two- to tenfold less potent inhibitors. On the 5-sub-stituted indole ring, the following trend is observed: F (3d) > Cl (3e) >Br (3 f), with the 5-F compound (3d) having an IC50 value of 30 nm. Further attachment of 7-methoxy group on the benzofuran ring (3g) maintained the potency. Extension of the piperidine ring with methyl (3h) or methylpyridyl (3i,j) substituent gave similar potency to the unsubstituted maleimide 3a. However, to our delight, installment of arylamide groups gave several compounds (3k–o) with higher potency than staurosporine and two other known potent, selective inhibitors of GSK-3β (SB216763 and AR-A0144183). Compound 3a was chosen as a representative compound of this class for selectivity studies against 15 kinases (Supporting Information). The piperidine maleimide 3a is reasonably selective amongst other kinases with the exception of PKA and PKC-b2, which could be attributed to the unsubstituted indole ring, where our previous studies showed that a bulky 5-substituent is required for GSK-3β selectivity.
Table 1.
In vitro GSK-3β inhibition by the piperidyl maleimides.[a]
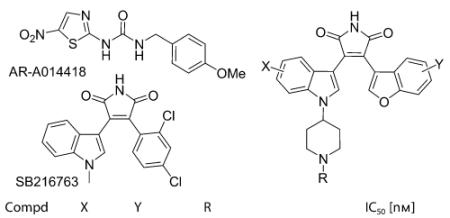
| Compd | X | Y | R | IC50 [nm] |
|---|---|---|---|---|
| 3a | H | H | H | 67±6 |
| 3b | H | 6-OMe | H | 629±32 |
| 3c | H | 7-OMe | H | 134±5 |
| 3d | 5-F | H | H | 30±4 |
| 3e | 5-Cl | H | H | 272±27 |
| 3 f | 5-Br | H | H | 684±17 |
| 3g | 5-F | 7-OMe | H | 28±4 |
| 3h | H | H | CH3 | 155±2 |
| 3i | H | H | CH2(2-pyridyl) | 110±4 |
| 3j | H | H | CH2(4-pyridyl) | 65±4 |
| 3k | H | H | CO(1-morpholinyl) | 10±2 |
| 3l | H | H | CO(2-pyridyl) | 6±1 |
| 3m | H | H | CO(3-pyridyl) | 8±2 |
| 3n | H | H | CO(4-pyridyl) | 4±1 |
| 3o | H | H | CO(2-pyrazyl) | 4±1 |
| 3p | H | H | COCH2NH2 | 55±2 |
| 3q | H | H | CO(4-methylfurazanyl) | 25±2 |
| 1 | – | – | – | 87±24[b] |
| Staurosporine | 12±8 | |||
| SB216763 | 23±1 | |||
| AR-A014418 | 229±11 | |||
The synthesized maleimides were evaluated for their ability to inhibit phosphorylation of primed substrate (YRRAAVPPSPSLSRHSSPHQ(pS)E DEEE; 20 μm) by human GSK-3β in the presence of 10 μm ATP concentration. These compounds were tested at Reaction Biology Inc. (http://www.reactionbiology.com).
An IC50 value of 7±3 nm was obtained when compound 1 was tested using the conditions reported in Ref. [13a].
In vivo behavioral studies and pharmacokinetic profile
The efficacy of GSK-3β inhibitors for the treatment of bipolar/mood disorders is best determined from animal behavioral studies. Compound 3a was therefore evaluated in an animal model of mania/hyperactivity induced by the stimulant amphetamine (Amph) and the anxiolytic chlordiazepoxide (CDP). In this assay, the combination of the Amph+CDP produces an increase in locomotor activity, which is blocked by known mood stabilizers, such as lithium, valproate and lamotrigine.[13a,14] C57BL/6J male mice received a 5 min pretreatment with either vehicle, valproate (400 mgkg−1), or compound 3a (3 or 10 mgkg−1; reductions in baseline locomotor activity precluded testing of higher doses of compound 3a). Mice were then administered a mixture of CDP (2.5 mgkg−1)+Amph (4 mgkg−1) and locomotor activity was recorded for 60 min. Data were analyzed for the second 30 min period of a 60 min test session, the time when no baseline reductions in locomotor activity were observed for compound 3a at 10 mgkg−1. Pretreatment with compound 3a (10 mgkg−1) reduced the locomotor activity induced by the Amph+CDP mixture to about 65% of the vehicle–Amph+CDP mixture, while the valproate-treated mice (400 mgkg−1) had their activity reduced to about 35%. The 3 mgkg−1 dose of compound 3a was not active in the Amph/CDP assay (Figure 3). These results demonstrate that the GSK-3β inhibitor 3a has a profile similar to known mood stabilizers like valproate in the Amph/CDP mania model.
Figure 3.
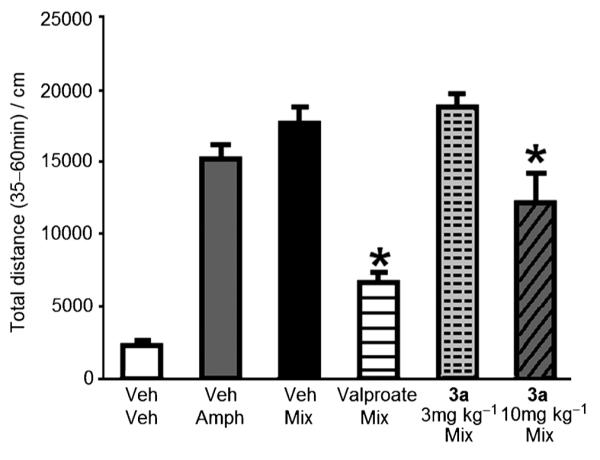
Pretreatment with 3a (10 mgkg−1) or valproate (400 mgkg−1) reduced the locomotor hyperactivity induced by the amphetamine (Amph) + chlordiazepoxide (CDP) mixture (Mix). Each bar represents mean distance traveled ( SEM) during the second 30 min of a 60 min test session. Analysis of variance showed a main effect of treatment [F(5,47=31.6; p<0.0001)] and the Fisher’s PLSD post-hoc confirmed that mice treated with compound 3a (10 mgkg−1) or valproate showed reduced locomotor activity compared to mice treated with vehicle (Veh)+ Mix (*p<0.05 vs Veh Mix; n=8–9 per group).
To further confirm the antimanic effect of 3a, prepulse inhibition (PPI) studies were performed. PPI is a test of sensory motor gating in which the auditory startle response is reduced as a result of a weaker auditory prestimulus application prior to the main startle stimulus. Patients suffering from acute mania, similar to patients with schizophrenia, have deficits in sensorimotor gating and mood stabilizers, such as lamotrigine improve PPI in mice.[15] Compound 3a at 25 mgkg−1 enhanced PPI similar to that observed for mice treated with haloperidol (1 mgkg−1) and valproate (400 mgkg−1). The 10 mgkg−1 dose of compound 3a was not active in the PPI assay (Figure 4). These results demonstrate that the GSK-3β inhibitor 3a has a profile similar to known mood stabilizers like valproate in the PPI model.
Figure 4.
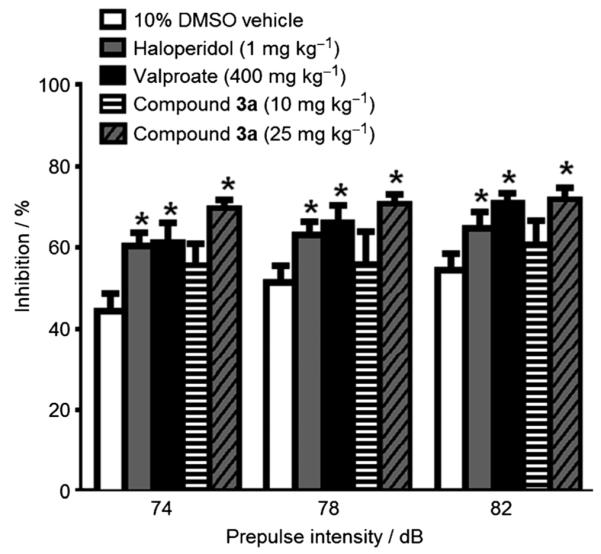
C57BL/6J male mice received a 30 min pretreatment with either vehicle, haloperidol (1 mgkg−1), valproate (400 mgkg−1), or compound 3a (10 or 25 mgkg−1). Each bar represents the mean percent inhibition of the startle response ( SEM) following the prepulse stimulus. Analysis of variance showed a main effect of treatment [F(4,54=3.6; p<0.05)] and Fisher’s PLSD post-hoc confirmed that haloperidol, valproate and compound 3a (10 mgkg−1) increase PPI (*p<0.05 vs 10% DMSO vehicle; n=8–14 per group).
On the basis of the cyclical nature of bipolar disorder, which cycles between mania and depression, disruption of the CLOCK gene that regulates the biological rhythms in mice has been used to develop a novel animal model of mania, reminiscent of humans with bipolar disorder in the manic state.[16] CLOCK mutant mice display behaviors, such as hyperactivity, decreased need for sleep, fewer depression-like characteristics, increased risk-taking behavior, and higher sensitivity to cocaine. Certain aspects of the behavioral profile of CLOCK mutant mice can be corrected back to that of wild-type mice upon chronic administration of lithium. Compound 3a was therefore administered to CLOCK mutant mice at 12.5 mgkg−1 over five days, after which the locomotor activity (determined by beam breaks) was recorded every 5 min over a 2 h period (Figure 5). Administration of compound 3a resulted in attenuation of the locomotor hyperactivity of the CLOCK mutant mice, similar to the effects achieved by the chronic administration of lithium.[16] While further behavioral studies are currently in progress, this preliminary finding is consistent with the notion of the antimanic-like effects of GSK-3β inhibition as seen in the Amph+ CDP-treated mice (Figure 3) and the prepulse inhibition studies (Figure 4).
Figure 5.
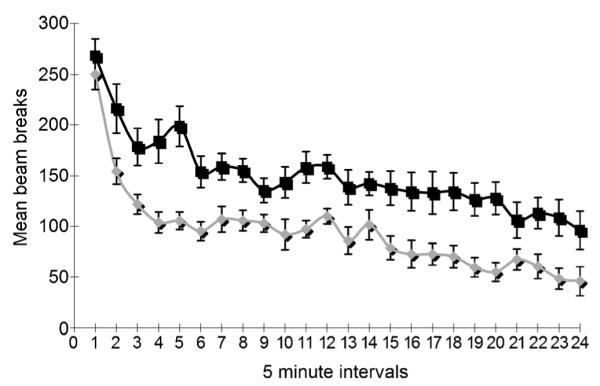
Locomotor hyperactivity in response to novelty of compound 3a is reduced after five days of 12.5 mgkg−1 dosage (*p<0.0006 vs saline; n=9–12 per group). The locomotor response to novelty was measured in adult, male, ClockΔ19 mutant mice. All mice were housed in a standard 12:12 h light/dark cycle. The experimental group (n= 9) consisted of mice treated with 12.5 mgkg−1 of 3a in 1:20 DMSO/saline, and the control group (n=12) consisted of mice treated with vehicle only. Both groups were injected once a day (i.p.) for five consecutive days prior to the test. Compound 3a:  ; saline: ∎.
; saline: ∎.
A preliminary ADME profile was obtained for compound 3a, specifically for the inhibition of the cytochrome P450s (CYPs), human liver microsomal stability, and plasma protein binding. In the presence of up to 10 μm of 3a, none of the CYP isoforms tested (CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A4) showed more than 30% inhibition, indicative of minimal adverse drug interactions. When incubated with human or mouse liver microsomes, more than 83% of compound 3a remained after 60 min at 1 and 10 μm concentrations, suggesting a reasonable stability and duration of action. Plasma protein binding of 3a was studied using equilibrium dialysis in mouse plasma, where it was found to be greater than 96% at 1 and 10 mm. Lastly, the aqueous solubility of 3a was determined to be 196 mgL−1 using an HPLC-based method.
Conclusions
Based on the X-ray co-crystal structure of our reported indolyl-benzofuranyl-maleimides, we have designed a second generation of piperidyl-bearing maleimides that show excellent inhibitory activity against GSK-3β, and one member of which shows promising mood stabilizing effects. Maleimide 3a was found to possess a reasonable kinase selectivity profile, and more importantly, was able to reduce locomotor activity in the chlor-diazepoxide/amphetamine-induced hyperactivity model in vivo at 10 mgkg−1 dose. Compound 3a also produced an enhancement of prepulse inhibition comparable to that shown by haloperidol at 1 mgkg−1. Furthermore, the study of 3a in the CLOCK mutant mice revealed its ability to counter hyperactivity to a degree similar to that achieved by the chronic administration of large doses of lithium. Preliminary ADME studies showed this compound to possess good metabolic stability and low CYP inhibition, together with an improved aqueous solubility. The present work thus further expands on the possible utility of lithium mimetics, namely small-molecule GSK-3β inhibitors, in the treatment of the manic stages of bipolar disorders.
Experimental Section
Chemistry
General information
All starting materials were purchased from Sigma–Aldrich. 1H NMR and 13C NMR spectra were recorded on a Bruker spectrometer at 400 MHz and 100 MHz, respectively, with tetramethylsilane (TMS) as an internal standard. Standard abbreviations for multiplicity were used: s=singlet, d=doublet, dd=doublet of doublets, dt=doublet of triplets, t=triplet, q=quadruplet, m=multiplet and br=broad. High-resolution mass spectrometry (HRMS) experiments were performed on a Q-TOF-2TM instrument (Micromass). Thin-layer chromatography (TLC) was performed on Merck 60 F254 silica gel plates. Preparative TLC was performed with Analtech 1000 μm silica gel GF plates. Column chromatography was performed using Merck silica gel (40–60 mesh). Preparative HPLC was carried out on an ACE 5 AQ column (150 mm × 21.2 mm), with detection at 254 and 280 nm on a Shimadzu SPD10A VP detector; flow rate 17 mLmin−1; and a 35 min gradient from 50% CH3CN in H2O to 100% CH3CN with 0.05% trifluoroacetic acid (TFA). Analytical HPLC was carried out on a Lune 5u C18 column (150 mm×4.6 mm), with detection at 254 and 280 nm on a Shimadzu SPD-10A VP detector; flow rate 1.0 mLmin−1; and a 25 min gradient from 50% CH3CN in H2O to 100% CH3CN with 0.05% TFA. All final compounds were purified by preparative HPLC.
General procedures for the synthesis of compounds 3–7 and 9–11
Method A: General method for the synthesis of N-(azacycloalkyl)indoles 6a–d, via reductive amination of indolines 4a–d and 1-alkyl-4-piperidones 5
A solution of indoline 4a–d (1.0 equiv) and 5 (1.0 equiv) in HOAc solution was treated with powdered NaBH(OAc)3 (1.5 equiv) at RT under Ar and the reaction was stirred at RT for 1 h. Upon completion of the reaction (TLC, eluent: hexanes/EtOAc 4:1), the solution was concentrated underreduced pressure. Aq concd NaOH was added dropwise to adjust the pH to 10–11, keeping the reaction temperature below 40°C, and the resulting mixture was diluted with EtOAc (200 mL) and washed thoroughly with saturated brine (20 mL), dried (Na2SO4), filtered, and concentrated to afford the crude indoline intermediate. A solution of indoline intermediate (1.0 equiv) in anhydrous THF was treated dropwise with a solution of DDQ (1.0 equiv) in anhydrous THF at 0°C for more than 60 min under Ar. The reaction mixture was allowed to warm to RT and stirred overnight. Upon completion of the reaction, the resulting mixture was diluted with EtOAc (200 mL) and washed thoroughly with satd aq NaHCO3 (50 mL) and brine (50 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography of the crude mixture afforded 6a–d.
Method B: General method for the synthesis of glyoxylyl esters 7a–d
A solution of 6a–d (1.0 equiv) in Et2O was treated dropwise with oxalyl chloride (1.1 equiv) at 0°C under Ar and the reaction was stirred at 0°C for 90 min, then cooled to −78°C. MeOH or EtOH was added in one portion and the resulting yellow slurry mixture was allowed to warm to RT and stirred overnight. Upon completion of the reaction (TLC, eluent: hexanes/EtOAc 2:1), the solution was concentrated under reduced pressure. The resulting mixture was diluted with CH2Cl2 (200 mL) and washed thoroughly with satd aq NaHCO3 (50 mL) and brine (50 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography (hexanes/EtOAc 2:1) of the crude mixture afforded 7a–d.
Method C: General method for the synthesis of acetamide 10a–c
The ester 9a–c was added to liquid NH3 at −78°C, and the reaction flask was sealed and heated at 80°C for 3 d. The reaction mixture was cooled to −78°C and then allowed to reach RT slowly so that the excess of NH3 was evaporated, and the residue was washed with hexane to afford acetamide 10a–c, which was dried in vacuo and used without further purification.
Method D: General method for the synthesis of maleimides 11a–g
A solution of glyoxylyl esters 7a–d (1.0 equiv) and acetamide 10a–c (1.0 equiv) in Et2O was treated dropwise with 1 m tBuOK in THF (3.0 equiv) at −20°C under Ar, and the reaction was allowed to warm to RT immediately and then stirred at RT overnight. Upon completion of the reaction (TLC, eluent: hexanes/ EtOAc 2:1), the reaction was concentrated under reduced pressure. The resulting mixture was diluted with EtOAc (200 mL) and washed thoroughly with 1 m HCl (20 mL), satd aq NaHCO3 (50 mL) >and brine (50 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography of the crude mixture afforded 11a–g.
Method E: General method for the synthesis of amine 3a–g
A solution of 11a–g in CH2Cl2/TFA (10:1) was stirred at RT for 2 h. Upon completion of the reaction (TLC, eluent: hexanes/EtOAc 2:1), the resulting mixture was concentrated under reduced pressure. The final product 3a–g was then purified by HPLC.
Method F: General method for the synthesis of amine 3h–j
A solution of 3a (1.0 equiv) and appropriate RCHO (1.5 equiv) in anhydrous THF was treated with HOAc (3.0 equiv) and NaBH(OAc)3 (3.0 equiv) at RT under Ar, and the reaction was stirred at RT overnight. Upon completion of the reaction (TLC, eluent: EtOAc/MeOH 19:1), the solution was concentrated under reduced pressure. The residue was diluted with EtOAc (200 mL) and washed thoroughly with satd aq K2CO3 (20 mL) and brine (20 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography of the crude mixture afforded 3h–j using the conditions specified. The solution of 3h–j in CH2Cl2/TFA (10:1) was stirred at RT for 2 h. The resulting mixture was concentrated and then purified by HPLC.
Method G: General method for the synthesis of amide 3k–q
A solution of 3a (1.0 equiv) and appropriate RCOOH (1.5 equiv) in anhydrous CH2Cl2 was treated with HOBt (2.0 equiv), EDC (2.0 equiv) and DMAP (2.0 equiv) at RT under Ar, and the reaction was stirred at RT overnight. Upon completion of the reaction (TLC, eluent: EtOAc/MeOH 19:1), the solution was concentrated under reduced pressure. The residue was diluted with EtOAc (200 mL) and washed thoroughly with satd K2CO3 (20 mL) and brine (20 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography of the crude mixture afforded 3k–q using the conditions specified. The solution of 3k–q in CH2Cl2/TFA (10:1) was stirred at RT for 2 h. The resulting mixture was concentrated and then purified by HPLC.
4-Indol-1-yl-piperidine-1-carboxylic acid tert-butyl ester (6a)
Synthesized by method A using indoline and 4-oxo-piperidine-1-carboxylic acid tert-butyl ester as starting materials. Yield 80% (yellow-brown solid); 1H NMR (CDCl3): δ=1.51 (s, 9H), 1.85–2.00 (m, 2H), 2.05–2.15 (m, 2H), 2.85–3.05 (m, 2H), 4.20–4.50 (m, 3H), 6.54 (d, J=2.8 Hz, 1H), 7.08–7.16 (m, 1H), 7.16–7.26 (m, 2H), 7.39 (d, J=8.0 Hz, 1H), 7.65 ppm (d, J=8.0 Hz, 1H).
4-(5-Fluoro-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (6b)
Synthesized by method A using 5-fluoro-2,3-dihydro-1H-indole[17] and 4-oxo-piperidine-1-carboxylic acid tert-butyl ester as starting materials. Yield 78% (yellow solid); 1H NMR (CDCl3): δ= 1.50 (s, 9H), 1.79–1.99 (m, 2H), 2.00–2.15 (m, 2H), 2.84–3.00 (m, 2H), 4.24–4.40 (m, 3H), 6.48 (d, J=2.8 Hz, 1H), 6.96 (dt, J=9.2, 2.4 Hz, 1H), 7.21 (d, J=3.2 Hz, 1H), 7.27–7.29 ppm (m, 2H).
4-(5-Chloro-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (6c)
Synthesized by method A using 5-chloro-2,3-dihydro-1H-indole and 4-oxo-piperidine-1-carboxylic acid tert-butyl ester as starting materials. Yield 83% (yellow-brown solid); 1H NMR (CDCl3): δ=1.51 (s, 9H), 1.80–2.00 (m, 2H), 2.05–2.15 (m, 2H), 2.80–3.00 (m, 2H), 4.25–4.45 (m, 3H), 6.48 (d, J=3.2 Hz, 1H), 7.17 (dd, J=2.0 Hz, J=8.8 Hz, 1H), 7.21 (d, J=3.2 Hz, 1H), 7.30 (d, J=8.8 Hz, 1H), 7.61 ppm (d, J=2.0 Hz, 1H).
4-(5-Bromo-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (6d)
Synthesized by method A using 5-bromo-2,3-dihydro-1H-indole and 4-oxo-piperidine-1-carboxylic acid tert-butyl ester as starting materials. Yield 32% (yellow-brown solid); 1H NMR (CDCl3): δ=1.52 (s, 9H), 1.80–1.95 (m, 2H), 2.00–2.10 (m, 2H), 2.80–3.00 (m, 2H), 4.25–4.45 (m, 3H), 6.46 (d, J=3.6 Hz, 1H), 7.17 (d, J=3.6 Hz, 1H), 7.20–7.35 (m, 2H), 7.76 ppm (d, J=1.2 Hz, 1H).
4-(3-Methoxyoxalyl-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (7a)
Synthesized by method B. Yield 76% (beige solid); 1H NMR (CDCl3): δ=1.52 (s, 9H), 1.90–2.08 (m, 2H), 2.15–2.27 (m, 2H), 2.85–3.05 (m, 2H), 3.97 (s, 3H), 4.30–4.55 (m, 3H), 7.35–7.42 (m, 2H), 7.42–7.50 (m, 1H), 8.47 (s, 1H), 8.47–8.52 ppm (m, 1H).
4-(5-Fluoro-3-ethoxyoxalyl-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (7b)
Synthesized by method B. Yield 77% (beige solid); 1H NMR (CDCl3): δ=1.44 (t, J=7.2 Hz, 3H), 1.51 (s, 9H), 1.86–2.05 (m, 2H), 2.11–2.23 (m, 2H), 2.86–3.06 (m, 2H), 4.30–4.49 (m, 5H), 7.09 (dt, J=8.8, 2.4 Hz, 1H), 7.31–7.40 (m, 1H), 8.14 (d, J=9.2, 2.4 Hz, 1H), 8.47 ppm (s, 1H).
4-(5-Chloro-3-methoxyoxalyl-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (7c)
Synthesized by method B. Yield 98% (beige solid); 1H NMR (CDCl3): δ=1.52 (s, 9H), 1.90–2.05 (m, 2H), 2.10–2.25 (m, 2H), 2.85–3.05 (m, 2H), 3.97 (s, 3H), 4.30–4.50 (m, 3H), 7.32 (dd, J=1.6 Hz, J=8.8 Hz, 1H), 7.36 (d, J=8.8 Hz, 1H), 8.46–8.48 (m, 1H), 8.48 ppm (s, 1H).
4-(5-Bromo-3-methoxyoxalyl-indol-1-yl)-piperidine-1-carboxylic acid tert-butyl ester (7d)
Synthesized by method B. Yield 63% (beige solid); 1H NMR (CDCl3): δ=1.52 (s, 9H), 1.90–2.05 (m, 2H), 2.10–2.25 (m, 2H), 2.85–3.05 (m, 2H), 3.97 (s, 3H), 4.30–4.50 (m, 3H), 7.31 (d, J=8.8 Hz, 1H), 7.46 (dd, J=1.6 Hz, J=8.8 Hz 1H), 8.46 (s, 1H), 8.64 ppm (d, J=1.6 Hz, 1H).
Benzofuran-3-yl-acetamide (10a)
Synthesized by method C using benzofuran-3-yl-acetic acid ethyl ester as a starting material. Yield 87% (brown powder); 1H NMR ([D6]DMSO): δ=3.49 (s, 2H), 6.90–7.10 (br s, 1H), 7.23–7.31 (m, 2H), 7.55 (d, J=8.0 Hz, 1H), 7.57–7.61 (br s, 1H), 7.63 (d, J=7.6 Hz, 1H), 7.82 ppm (s, 1H).
7-Methoxy-benzofuran-3-yl-acetamide (10b)
Synthesized by method C using (7-methoxy-benzofuran-3-yl)acetic acid ethyl ester as a starting material. Yield 85% (brown powder); 1H NMR ([D6]DMSO): δ=3.45 (s, 2H), 3.92 (s, 3H), 6.85–6.95 (m, 1H), 6.90– 7.05 (br s, 1H), 7.10–7.25 (m, 2H), 7.50–7.60 (br s, 1H), 7.79 ppm (s, 1H).
6-Methoxy-benzofuran-3-yl-acetamide (10c)
Synthesized by method C using (6-methoxy-benzofuran-3-yl)acetic acid ethyl ester as a starting material. Yield 75% (brown powder); 1H NMR ([D6]DMSO): δ=3.43 (s, 2H), 3.79 (s, 3H), 6.89 (dd, J=2.0 Hz, J= 8.4 Hz, 1H), 6.90–7.10 (br s 1H), 7.15 (d, J=2.0 Hz, 1H), 7.49 (d, J= 8.4 Hz, 1H), 7.50–7.60 (br s, 1H), 7.69 ppm (s, 1H).
4-[3-(4-Benzofuran-3-yl-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)-indol-1-yl]-piperidine-1-carboxylic acid tert-butyl ester (11a)
Synthesized by method D. Yield 63% (orange solid); 1H NMR (CDCl3): δ=1.51 (s, 9H), 1.75–1.90 (m, 2H), 2.00–2.12 (m, 2H), 2.80– 3.00 (m, 2H), 4.25–4.45 (m, 3H), 6.80 (d, J=8.0 Hz, 1H), 6.87 (t, J= 7.6 Hz, 1H), 6.92 (t, J=7.6 Hz, 1H), 7.14–7.24 (m, 3H), 7.38 (d, J= 8.4 Hz, 1H), 7.44–7.49 (br s, 1H), 7.50 (d, J=8.4 Hz, 1H), 7.70 (s, 1H), 8.14 ppm (s, 1H); 13C NMR (CDCl3): δ=28.4, 32.1, 43.1 (br), 54.0, 80.4, 105.7, 109.6, 111.5, 111.6, 121.2, 122.3, 122.4, 122.7, 122.8, 123.7, 124.7, 124.8, 126.1, 129.2, 132.4, 135.9, 147.4, 154.6, 155.0, 170.8, 171.1 ppm; HRMS (ESI): m/z [M H] calcd for C30H29N3O5: 510.2034, found: 510.2013; HPLC purity : 99.9%.
3-Benzofuran-3-yl-4-(1-piperidin-4-yl-1H-indol-3-yl)-pyrrole-2,5-dione TFA salt (3a)
Synthesized by method E. Yield 71% (orange powder); 1H NMR (CDCl3/CD3OD): δ=2.15–2.29 (m, 4H), 3.08–3.22 (m, 2H), 3.51–3.60 (m, 2H), 4.48–4.60 (m, 1H), 6.74 (d, J=7.6 Hz, 1H), 6.81 (t, J=7.6 Hz, 1H), 6.86 (t, J=7.6 Hz, 1H), 7.05–7.18 (m, 3H), 7.36 (d, J=8.0 Hz, 1H), 7.44 (d, J=8.4 Hz, 1H), 7.67 (s, 1H), 8.08 ppm (s, 1H); 13C NMR (CDCl3/CD3OD): δ=28.9, 43.3, 51.0, 106.2, 109.4, 111.4, 111.5, 121.3, 122.1, 122.2, 122.6, 123.1, 124.6, 124.7, 124.8, 126.0, 128.4, 131.8, 135.6, 147.4, 155.0, 171.7, 172.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C25H21N3O3: 412.1656, found: 412.1680; HPLC purity: 99.2%.
4-{3-[4-(6-Methoxy-benzofuran-3-yl)-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl]-indol-1-yl}-piperidine-1-carboxylic acid tert-butyl ester (11b)
Synthesized by method D. Yield 49% (orange solid); 1H NMR (CDCl3): δ=1.51 (s, 9H), 1.75–1.90 (m, 2H), 2.00–2.15 (m, 2H), 2.80–3.00 (m, 2H), 3.78 (s, 3H), 4.20–4.50 (m, 3H), 6.48 (dd, J= 2.4 Hz, J=8.8 Hz, 1H), 6.63 (d, J=8.8 Hz, 1H), 6.92 (t, J=7.6 Hz, 1H), 7.00 (d, J=2.0 Hz, 1H), 7.18 (t, J=7.6 Hz, 1H), 7.22 (d, J= 8.0 Hz, 1H), 7.37 (d, J=8.4 Hz, 1H), 7.70 (s, 1H), 8.05 (s, 1H), 8.16 ppm (s, 1H).
3-(6-Methoxy-benzofuran-3-yl)-4-(1-piperidin-4-yl-1H-indol-3-yl)-pyrrole-2,5-dione TFA salt (3b)
Synthesized by method E. Yield 25% (orange powder); 1H NMR ([D6]DMSO): δ=2.00–2.20 (m, 4H), 3.05–3.25 (m, 2H), 3.35–3.55 (m, 2H), 3.73 (s, 3H), 4.70–4.90 (m, 1H), 6.50 (d, J=8.0 Hz, 1H), 6.59 (d, J=8.0 Hz, 1H), 6.84 (t, J= 8.0 Hz, 1H), 7.03 (d, J=8.0 Hz, 1H), 7.14 (t, J=8.0 Hz, 1H), 7.20 (d, J=8.0 Hz, 1H), 7.65 (d, J=8.0 Hz, 1H), 7.79 (s, 1H), 8.18 (s, 1H), 8.30–8.50 (br s, 1H), 8.70–8.90 (br s, 1H), 11.20 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=28.6, 43.1, 50.3, 55.6, 96.0, 105.2, 110.6, 111.2, 111.9, 118.1, 120.6, 121.3, 122.0, 122.4, 123.8, 125.7, 129.0, 131.9, 135.5, 146.4, 155.5, 157.8, 171.8, 172.2 ppm; HRMS (ESI): m/z [M+H]+ calcd for C26H23N3O4: 442.1761, found: 442.1796; HPLC purity: 99.5%.
4-{3-[4-(7-Methoxy-benzofuran-3-yl)-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl]-indol-1-yl}-piperidine-1-carboxylic acid tert-butyl ester (11c)
Synthesized by method D. Yield 36% (orange solid); 1H NMR (CDCl3): δ=1.51 (s, 9H), 1.75–1.90 (m, 2H), 2.00–2.15 (m, 2H), 2.80–3.00 (m, 2H), 4.00 (s, 3H), 4.25–4.45 (m, 3H), 6.38 (d, J= 8.0 Hz, 1H), 6.70 (d, J=7.6 Hz, 1H), 6.78 (t, J=8.0 Hz, 1H), 6.93 (t, J=7.6 Hz, 1H), 7.18 (t, J=7.6 Hz, 1H), 7.23 (d, J=8.0 Hz, 1H), 7.38 (d, J=8.0 Hz, 1H), 7.62 (s. 1H), 7.70 (s, 1H), 8.13 ppm (s, 1H).
3-(7-Methoxy-benzofuran-3-yl)-4-(1-piperidin-4-yl-1H-indol-3-yl)-pyrrole-2,5-dione TFA salt (3c)
Synthesized by method E. Yield 30% (orange powder); 1H NMR (CDCl3/CD3OD): δ=2.15–2.35 (m, 4H), 3.10–3.25 (m, 2H), 3.50–3.60 (m, 2H), 3.94 (s, 3H), 4.45–4.63 (m, 1H), 6.32 (d, J=8.0 Hz, 1H), 6.65 (d, J=8.0 Hz, 1H), 6.72 (t, J= 8.0 Hz, 1H), 6.86 (t, J=7.6 Hz, 1H), 7.10 (d, J=8.0 Hz, 1H), 7.15 (d, J=7.6 Hz, 1H), 7.36 (d, J=8.4 Hz, 1H), 7.68 (s, 1H), 8.07 ppm (s, 1H); 13C NMR (CDCl3/CD3OD): δ=28.4, 42.9, 50.6, 55.6, 105.8, 106.4, 109.1, 111.5, 113.9, 120.9, 121.7, 122.6, 122.9, 123.9, 125.7, 126.3, 128.0, 131.6, 135.2, 144.0, 144.9, 146.8, 171.4, 171.9 ppm; HRMS (ESI): m/z [M+H]+ calcd for C26H23N3O4: 442.1761, found: 442.1783; HPLC purity: 99.7%.
4-[3-(4-Benzofuran-3-yl-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)-5-fluoro-indol-1-yl]-piperidine-1-carboxylic acid tert-butyl ester (11d)
Synthesized by method D. Yield 19% (orange solid); 1H NMR (CDCl3): δ=1.48 (s, 9H), 1.69–1.86 (m, 2H), 1.96–2.10 (m, 2H), 2.83– 2.95 (m, 2H), 4.24–4.40 (m, 3H), 6.70 (d, J=7.6 Hz, 1H), 6.85 (t, J= 7.6 Hz, 1H), 6.87–6.97 (m, 2H), 7.20 (t, J=7.2 Hz, 1H), 7.24–7.33 (m, 1H), 7.45–7.54 (m, 2H), 7.66 (s, 1H), 8.17 ppm (s, 1H).
3-Benzofuran-3-yl-4-(5-fluoro-1-piperidin-4-yl-1H-indol-3-yl)-pyr-role-2,5-dione TFA salt (3d)
Synthesized by method E. Yield 50% (orange powder); 1H NMR ([D6]DMSO): δ=1.96–2.13 (m, 4H), 3.07–3.17 (m, 2H), 3.38–3.51 (m, 2H), 4.76–4.86 (m, 1H), 6.74 (t, J= 12.0 Hz, 2H), 6.90 (t, J=8.0 Hz, 1H), 7.02 (t, J=8.0 Hz, 1H), 7.24 (t, J=8.0 Hz, 1H), 7.59–7.72 (m, 2H), 7.81 (s, 1H), 8.34 (s, 1H), 8.41 (br s, 1H), 8.81 (br s, 1H), 11.26 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=28.6, 43.0, 50.6, 105.2, 105.3, 106.1, 106.4, 110.4, 110.7, 111.1, 111.6, 111.8, 111.9, 121.7, 122.9, 123.8, 124.9, 125.0, 126.1, 126.2, 130.6, 132.0, 132.2, 147.5, 154.3, 156.2, 158.5, 171.7, 172.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C25H20FN3O3: 430.1561, found: 430.1555; HPLC purity: 96.9%.
4-[3-(4-Benzofuran-3-yl-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)-5-chloro-indol-1-yl]-piperidine-1-carboxylic acid tert-butyl ester (11e)
Synthesized by method D using DMF as the solvent. Yield 22% (orange solid); 1H NMR (CDCl3): δ=1.53 (s, 9H), 1.80–2.00 (m, 2H), 2.00–2.20 (m, 2H), 2.85–3.05 (m, 2H), 4.30–4.50 (m, 3H), 6.80 (t, J=8.0 Hz, 1H), 6.84 (s, 1H), 6.88 (d, J=11.2 Hz, 1H), 7.04 (d, J= 7.6 Hz, 1H), 7.15 (t, J=8.0 Hz, 1H), 7.21 (d, J=9.2 Hz, 1H), 7.33 (d, J=8.8 Hz, 1H), 7.36 (s, 1H), 7.84 (s, 1H), 7.88 ppm (s, 1H).
3-Benzofuran-3-yl-4-(5-chloro-1-piperidin-4-yl-1H-indol-3-yl)-pyr-role-2,5-dione TFA salt (3e)
Synthesized by method E. Yield 20% (orange powder); 1H NMR ([D6]DMSO): δ=1.85–2.15 (m, 4H), 3.00–3.15 (m, 2H), 3.35–3.50 (m, 2H), 4.70–4.90 (m, 1H), 6.69 (d, J= 7.6 Hz, 1H), 6.90 (t, J=7.2 Hz, 1H), 7.11 (s, 1H), 7.18 (d, J=8.4 Hz, 1H), 7.25 (t, J=7.6 Hz, 1H), 7.64 (d, J=8.4 Hz, 1H), 7.68 (d, J= 8.8 Hz, 1H), 7.77 (s, 1H), 8.36 (s, 1H), 8.50 (br s, 2H), 11.27 ppm (br s, 1H); 13C NMR ([D6]DMSO): δ=28.6, 43.0, 50.5, 104.9, 111.1, 111.6, 112.2, 120.8, 121.6, 122.3, 122.9, 124.4, 124.7, 125.0, 125.2, 126.7, 130.4, 131.8, 134.1, 147.6, 154.4, 171.7, 172.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C25H20ClN3O3: 446.1266, found: 446.1257; HPLC purity: 99.8%.
4-[3-(4-Benzofuran-3-yl-2,5-dioxo-2,5-dihydro-1H-pyrrol-3-yl)-5-bromo-indol-1-yl]-piperidine-1-carboxylic acid tert-butyl ester (11 f)
A solution of appropriate glyoxylyl ester (1.0 equiv) and acet-amide (1.0 equiv) in DMF was treated dropwise with 1.6 m nBuLi in THF (4.0 equiv) at −78°C under Ar, and the reaction was allowed to warm to RT immediately and then stirred at RT overnight. Upon completion of the reaction (TLC, eluent: hexanes/EtOAc 2:1), the solution was concentrated under reduced pressure. The resulting mixture was diluted with EtOAc (200 mL) and washed thoroughly with 1 m HCl (20 mL), satd aq K2CO3 (50 mL) and brine (50 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography of the crude mixture afforded 11 f in 30% yield (orange solid); 1H NMR (CDCl3): δ=1.50 (s, 9H), 1.75–1.90 (m, 2H), 1.90–2.10 (m, 2H), 2.80–3.00 (m, 2H), 4.19–4.45 (m, 3H), 6.66 (d, J=7.6 Hz, 1H), 6.86 (t, J=7.6 Hz, 1H), 7.15–7.30 (m, 3H), 7.42 (s, 1H), 7.53 (d, J=8.4 Hz, 1H), 7.61 (s, 1H), 7.85 (s, 1H), 8.19 ppm (s, 1H).
3-Benzofuran-3-yl-4-(5-bromo-1-piperidin-4-yl-1H-indol-3-yl)-pyr-role-2,5-dione TFA salt (3 f)
Synthesized by method E. Yield 20% (orange powder); 1H NMR ([D6]acetone): δ=2.20–2.35 (m, 2H), 2.40–2.56 (m, 2H), 3.30–3.45 (m, 2H), 3.60–3.75 (m, 2H), 4.90–5.05 (m, 1H), 6.77 (d, J=8.4 Hz, 1H), 6.89 (t, J=7.6 Hz, 1H), 7.18–7.30 (m, 2H), 7.43 (s, 1H), 7.56 (d, J=8.0 Hz, 1H), 7.70 (d, J=8.4 Hz, 1H), 7.91 (s, 1H), 8.30 (s, 1H), 10.1 ppm (br s, 1H); 13C NMR ([D6]DMSO): δ=28.5, 43.0, 50.5, 104.8, 111.1, 111.6, 112.6, 113.2, 121.6, 122.9, 123.8, 124.3, 124.7, 124.8, 125.0, 127.3, 130.2, 131.8, 134.3, 147.5, 154.4, 171.7, 172.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C25H20BrN3O3: 490.0761, found: 490.0813; HPLC purity: 99.3%.
3-(5-Fluoro-1-piperidin-4-yl-1H-indol-3-yl)-4-(7-methoxy-benzo-furan-3-yl)-pyrrole-2,5-dione TFA salt (3g)
Synthesized by method E. Yield 20% (orange powder); 1H NMR ([D6]DMSO): δ=1.92–2.24 (m, 4H), 3.04–3.27 (m, 2H), 3.39–3.49 (m, 2H), 3.91 (s, 3H), 4.74–4.86 (m, 1H), 6.27 (d, J=8.0 Hz, 1H), 6.74–6.91 (m, 3H), 7.04 (dt, J=8.0, 4.0 Hz, 1H), 7.62–7.72 (m, 1H), 7.79 (s, 1H), 8.28– 8.43 (m, 2H), 8.72 (br d, 1H), 11.24 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=28.6, 43.0, 50.5, 55.9, 105.2, 105.3, 106.2, 106.4, 107.2, 110.5, 110.7, 111.4, 111.8, 111.9, 113.6, 123.7, 123.8, 126.2, 126.3, 126.5, 130.6, 132.0, 132.2, 143.7, 145.1, 147.4, 156.2, 158.6, 171.7, 172.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C26H22FN3O4: 460.1667, found: 460.1662; HPLC purity: 98.2%.
3-Benzofuran-3-yl-4-[1-(1-methyl-piperidin-4-yl)-1H-indol-3-yl]-pyrrole-2,5-dione TFA salt (3h)
Synthesized by method F using 3a and HCHO as starting materials. Yield 29% (orange powder); 1H NMR ([D6]DMSO): δ=2.00–2.30 (m, 4H), 2.84 (s, 3H), 3.10–3.30 (m, 2H), 3.50–3.65 (m, 2H), 4.70–4.85 (m, 1H), 6.74 (d, J=8.0 Hz, 1H), 6.83 (t, J=8.0 Hz, 1H), 6.88 (t, J=8.0 Hz, 1H), 7.04 (d, J= 8.0 Hz, 1H), 7.14 (t, J=8.0 Hz, 1H), 7.22 (t, J=8.0 Hz, 1H), 7.57–7.65 (m, 2H), 7.78 (s, 1H), 8.31 (s, 1H), 9.50–9.70 (br s, 1H), 11.23 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=29.1, 42.7, 49.9, 53.0, 105.2, 110.6, 111.3, 111.6, 120.6, 121.3, 121.8, 122.4, 122.8, 123.7, 124.9, 125.0, 125.7, 129.1, 132.3, 135.6, 147.4, 154.3, 171.8, 172.2 ppm; HRMS (ESI): m/z [M+H]+ calcd for C26H23N3O3: 426.1812, found: 426.1841; HPLC purity: 96.0%.
3-Benzofuran-3-yl-4-[1-(1-pyridin-2-ylmethyl-piperidin-4-yl)-1H-indol-3-yl]-pyrrole-2,5-dione TFA salt (3i)
Synthesized by method F using 3a and pyridine-2-carboxaldehyde as starting materials. Yield 67% (orange powder); 1H NMR ([D6]DMSO): δ=2.05–2.25 (m, 2H), 2.25–2.45 (m, 2H), 3.25–3.45 (m, 4H), 4.59 (s, 2H), 4.80–4.95 (m, 1H), 6.77 (t, J=8.0 Hz, 2H), 6.81 (d, J=7.6 Hz, 1H), 6.88 (t, J= 7.2 Hz, 1H), 7.00 (d, J=8.0 Hz, 1H), 7.12 (t, J=7.6 Hz, 1H), 7.21 (t, J=8.0 Hz, 1H), 7.52 (t, J=6.4 Hz, 1H), 7.60 (t, J=6.4 Hz, 2H), 7.66 (d, J=8.0 Hz, 1H), 7.86 (s, 1H), 7.97 (t, J=8.8 Hz, 1H), 8.30 (s, 1H), 8.72 (d, J=4.4 Hz, 1H), 11.24 ppm (s, 1H); 13C NMR ([D6]DMSO): δ= 28.2, 50.1, 51.5, 55.0, 105.2, 110.7, 111.3, 111.6, 120.6, 121.2, 121.7, 122.3, 122.8, 123.5, 124.3, 124.8, 124.9, 125.1, 125.7, 129.4, 132.3, 135.6, 137.9, 147.4, 149.7, 150.5, 154.3, 171.8, 172.2 ppm; HRMS (ESI): m/z [M+H]+ calcd for C31H26N4O3: 503.2078, found: 503.2076; HPLC purity: 99.8%.
3-Benzofuran-3-yl-4-[1-(1-pyridin-4-ylmethyl-piperidin-4-yl)-1H-indol-3-yl]-pyrrole-2,5-dione TFA salt (3j)
Synthesized by method F using 3a and pyridine-4-carboxaldehyde as starting materials. Yield 50% (orange powder); 1H NMR ([D6]DMSO): δ=2.10–2.25 (m, 4H), 3.10–3.30 (m, 2H), 3.40–3.60 (m, 2H), 4.42 (s, 2H), 4.70–4.90 (br s, 1H), 6.75 (d, J=7.6 Hz, 1H), 6.81 (t, J=7.2 Hz, 1H), 6.88 (t, J= 7.2 Hz, 1H), 7.01 (d, J=8.0 Hz, 1H), 7.12 (t, J=8.0 Hz, 1H), 7.21 (t, J=8.0 Hz, 1H), 7.55–7.70 (m, 4H), 7.79 (br s, 1H), 8.30 (s, 1H), 8.75 (d, J=6.0 Hz, 2H), 11.23 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=29.0, 50.1, 51.3, 57.8, 105.2, 110.7, 111.3, 111.6, 115.2, 118.1, 120.6, 121.3, 121.8, 122.4, 122.8, 123.6, 124.9, 125.1, 125.7, 126.1, 129.2, 132.3, 135.6, 147.4, 149.8, 154.3, 158.1, 171.8, 172.2 ppm; HRMS (ESI): m/z [M+H]+ calcd for C31H26N4O3: 503.2078, found: 503.2074; HPLC purity: 99.5%.
3-Benzofuran-3-yl-4-{1-[1-(morpholine-4-carbonyl)-piperidin-4-yl]-1H-indol-3-yl}-pyrrole-2,5-dione TFA salt (3k)
A solution of 3a (1.0 equiv) and Et3N (3.0 equiv) in CH3CN was treated dropwise with morpholine-4-carbonyl chloride (1.5 equiv) at 0°C (ice-water bath) under Ar, and the reaction was allowed to warm to RT and then stirred at RT for 3 h. Upon completion of the reaction (TLC, eluent: EtOAc/MeOH 98:2), the solution was concentrated under reduced pressure. The resulting mixture was diluted with EtOAc (50 mL) and washed thoroughly with 1 m HCl (20 mL), satd aq K2CO3 (20 mL) and brine (20 mL), dried (Na2SO4), filtered, and concentrated under reduced pressure. Silica gel chromatography of the crude mixture afforded 3k iny 57% yield (orange powder); 1H NMR ([D6]DMSO): δ=1.70–1.85 (m, 2H), 1.85–2.00 (m, 2H), 2.98 (t, J=12.0 Hz, 2H), 3.10–3.30 (m, 4H), 3.50–3.65 (m, 4H), 3.72 (d, J=12.0 Hz, 2H), 4.55–4.75 (m, 1H), 6.72 (d, J=8.0 Hz, 1H), 6.81 (t, J=8.0 Hz, 1H), 6.88 (t, J=8.0 Hz, 1H), 7.05 (d, J=8.0 Hz, 1H), 7.10 (t, J=8.0 Hz, 1H), 7.21 (t, J=8.0 Hz, 1H), 7.59 (d, J=8.0 Hz, 1H), 7.63 (d, J=8.0 Hz, 1H), 7.85 (s, 1H), 8.29 (s, 1H), 11.19 ppm (s, 1H); 13C NMR (CDCl3): δ=17.5, 24.4, 34.0, 35.5, 49.8, 50.8, 57.3, 64.1, 70.1, 109.2, 113.1, 114.9, 115.1, 124.5, 125.7, 125.8, 126.1, 126.2, 127.2, 128.1, 128.5, 129.6, 132.5, 135.9, 139.3, 150.7, 158.5, 175.3, 175.7, 175.9 ppm; HRMS (ESI): m/z [M+H]+ calcd for C30H28N4O5: 525.2132, found: 525.2138; HPLC purity: 97.5%.
3-Benzofuran-3-yl-4-{1-[1-(pyridine-2-carbonyl)-piperidin-4-yl]-1H-indol-3-yl}-pyrrole-2,5-dione TFA salt (3l)
Synthesized by method G using 3a and pyridine-2-carboxylic acid as starting materials. Yield 70% (orange powder); 1H NMR ([D6]DMSO): δ=1.75–1.95 (m, 3H), 2.00–2.10 (m, 1H), 2.90–3.10 (m, 1H), 3.25–3.45 (m,1H), 3.70–3.90 (m, 1H), 4.60–4.75 (m, 1H), 4.75–4.90 (m, 1H), 6.72 (d, J=8.0 Hz, 1H), 6.82 (t, J=8.0 Hz, 1H), 6.87 (t, J=8.0 Hz, 1H), 7.06 (d, J=8.0 Hz, 1H), 7.11 (t, J=8.0 Hz, 1H), 7.20 (t, J=8.0 Hz, 1H), 7.50 (t, J=8.0 Hz, 1H), 7.61 (t, J=8.0 Hz, 2H), 7.65 (d, J= 8.0 Hz, 1H), 7.90 (s, 1H), 7.95 (t, J=8.0 Hz, 1H), 8.30 (s, 1H), 8.61 (d, J=8.0 Hz, 1H), 11.19 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=31.5, 32.4, 41.6, 46.2, 53.5, 105.4, 109.2, 111.2, 111.2, 120.8, 121.9, 122.0, 122.3, 122.4, 123.5, 123.7, 124.3, 124.4, 124.5, 125.7, 128.8, 131.9, 135.5, 136.9, 147.0, 147.9, 153.3, 154.6, 167.3, 170.6, 171.0 ppm; HRMS (ESI): m/z [M+H]+ calcd for C31H24N4O4: 517.1870, found: 517.1888; HPLC purity: 99.7%.
3-Benzofuran-3-yl-4-{1-[1-(pyridine-3-carbonyl)-piperidin-4-yl]-1H-indol-3-yl}-pyrrole-2,5-dione TFA salt (3m)
Synthesized by method G using 3a and pyridine-3-carboxylic acid as starting materials. Yield 76% (orange powder); 1H NMR ([D6]DMSO): δ=1.80–2.10 (m, 4H), 2.90–3.10 (m, 2H), 3.50–3.75 (m, 1H), 4.50–4.75 (m, 1H), 4.75–4.90 (m, 1H), 6.70 (d, J=8.0 Hz, 1H), 6.82 (t, J=8.0 Hz, 1H), 6.87 (t, J=8.0 Hz, 1H), 7.07 (d, J=8.0 Hz, 1H), 7.12 (d, J= 8.0 Hz, 1H), 7.20 (t, J=8.0 Hz, 1H), 7.45–7.55 (m, 1H), 7.60 (d, J= 8.0 Hz, 1H), 7.67 (d, J=8.0 Hz, 1H), 7.89 (d, J=8.0 Hz, 1H), 7.95 (s, 1H), 8.31 (s, 1H), 8.60–8.75 (m, 2H), 11.21 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=8.7, 45.8, 52.5, 104.9, 110.6, 111.3, 111.5, 120.4, 121.2, 121.8, 122.2, 122.8, 123.4, 123.6, 124.8, 125.0, 125.6, 129.9, 132.0, 132.6, 134.7, 135.7, 147.4, 147.6, 150.4, 154.3, 166.8, 171.8, 172.1 ppm; HRMS (ESI): m/z [M+H]+ calcd for C31H24N4O4: 517.1870, found: 517.1868; HPLC purity: 99.9%.
3-Benzofuran-3-yl-4-{1-[1-(pyridine-4-carbonyl)-piperidin-4-yl]-1H-indol-3-yl}-pyrrole-2,5-dione TFA salt (3n)
Synthesized by method G using 3a and pyridine-4-carboxylic acid as starting materials. Yield 63% (orange powder); 1H NMR ([D6]DMSO): δ=1.80–2.10 (m, 4H), 2.90–3.10 (m, 1H), 3.20–3.40 (m, 1H), 3.40–3.60 (m, 1H), 4.75–4.85 (m, 1H), 4.85–4.95 (m, 1H), 6.70 (d, J=8.0 Hz, 1H), 6.82 (t, J=8.0 Hz, 1H), 6.87 (t, J=8.0 Hz, 1H), 7.07 (d, J=8.0 Hz, 1H), 7.11 (t, J=8.0 Hz, 1H), 7.21 (t, J=8.0 Hz, 1H), 7.62 (d, J= 8.0 Hz, 1H), 7.65–7.75 (m, 3H), 7.95 (s, 1H), 8.32 (s, 1H), 8.82 (d, J= 4.0 Hz, 2H), 11.22 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=31.7, 32.4, 41.1, 46.5, 52.8, 105.3, 111.0, 111.7, 111.9, 120.8, 121.5, 122.2, 122.5, 122.6, 123.1, 123.8, 125.2, 125.3, 126.0, 130.3, 132.9, 136.1, 146.7, 147.8, 148.5, 154.7, 166.4, 172.2, 172.5 ppm; HRMS (ESI): m/z [M+ H]+ calcd for C31H24N4O4: 517.1870, found: 517.1881; HPLC purity: 99.9%.
3-Benzofuran-3-yl-4-{1-[1-(pyrazine-2-carbonyl)-piperidin-4-yl]-1H-indol-3-yl}-pyrrole-2,5-dione TFA salt (3o)
Synthesized by method G using 3a and pyrazine-2-carboxylic acid as starting materials. Yield 30% (orange powder); 1H NMR (CDCl3): δ=1.94–2.19 (m, 3H), 2.18–2.32 (m, 1H), 3.02 (t, J=12.0 Hz, 1H), 3.32 (t, J= 12.0 Hz, 1H), 4.23–4.33 (m, 1H), 4.50–4.62 (m, 1H), 4.93–5.02 (m, 1H), 6.78 (d, J=8.0 Hz, 1H), 6.85 (t, J=8.0 Hz, 1H), 6.91 (t, J= 8.0 Hz, 1H), 7.11–7.26 (m, 3H), 7.39 (d, J=8.0 Hz, 1H), 7.42–7.56 (m, 2H), 7.72 (s, 1H), 8.13 (s, 1H), 8.57 (s, 1H), 8.68 (s, 1H), 9.00 ppm (s, 1H); 13C NMR (CDCl3): δ=31.5, 32.3, 41.8, 46.2, 53.4, 105.5, 109.2, 111.1, 111.2, 120.9, 121.9, 122.0, 122.3, 122.5, 123.7, 124.4, 124.5, 125.7, 128.7, 131.8, 135.4, 142.1, 145.2, 145.5, 147.1, 148.6, 154.6, 164.8, 170.2, 170.6 ppm; HRMS (ESI): m/z [M+H]+ calcd for C30H23N5O4: 518.1823, found: 518.1825; HPLC purity: 99.6%.
3-{1-[1-(2-Amino-acetyl)-piperidin-4-yl]-1H-indol-3-yl}-4-benzofuran-3-yl-pyrrole-2,5-dione TFA salt (3p)
Synthesized by method G using 3a and N-Boc glycine as starting materials. Yield 40% (orange powder); 1H NMR ([D6]DMSO): δ=1.57–1.75 (m, 1H), 1.87–2.06 (m, 3H), 2.91 (t, J=12.0 Hz, 1H), 3.29 (t, J=12.0 Hz, 1H), 3.78–3.91 (m, 1H), 3.94 (br s, 2H), 4.47–4.60 (m, 1H), 4.73–4.89 (m, 1H), 6.74 (d, J=8.0 Hz, 1H), 6.80 (t, J=8.0 Hz, 1H), 6.88 (t, J= 8.0 Hz, 1H), 7.02 (d, J=8.0 Hz, 1H), 7.12 (t, J=8.0 Hz, 1H), 7.22 (t, J=8.0 Hz, 1H), 7.60 (d, J=12.0 Hz, 1H), 7.65 (d, J=8.0 Hz, 1H), 7.82 (s, 1H), 8.06 (br s, 3H), 8.30 (s, 1H), 11.21 ppm (s, 1H); 13C NMR ([D6]DMSO): δ=30.7, 31.6, 31.8, 41.0, 43.5, 52.4, 104.9, 110.7, 111.4, 111.5, 120.5, 121.2, 121.8, 122.3, 122.8, 123.3, 124.8, 125.1, 125.7, 129.4, 132.5, 135.7, 147.3, 154.3, 164.5, 171.8, 172.2 ppm; HRMS (ESI): m/z [M+H]+ calcd for C27H24N4O4: 469.1870, found: 469.1860; HPLC purity: 99.6%.
3-Benzofuran-3-yl-4-{1-[1-(4-methyl-furazan-3-carbonyl)-piperidin-4-yl]-1H-indol-3-yl}-pyrrole-2,5-dione (3q)
Synthesized by method G using 3a and 4-methyl-furazan-3-carboxylic acid as starting materials. Yield 30% (orange powder); 1H NMR (CDCl3): δ=1.86–2.09 (m, 2H), 2.15–2.33 (m, 2H), 2.56 (s, 3H), 3.02 (t, J= 12.0 Hz, 1H), 3.38 (t, J=12.0 Hz, 1H), 4.44–4.65 (m, 2H), 4.88–5.02 (m, 1H), 6.77 (d, J=8.0 Hz, 1H), 6.85 (t, J=8.0 Hz, 1H), 6.92 (t, J= 8.0 Hz, 1H), 7.12–7.28 (m, 3H), 7.38 (d, J=8.0 Hz, 1H), 7.48 (d, J= 8.0 Hz, 1H), 7.68 (s, 2H), 8.13 ppm (s, 1H); 13C NMR (CDCl3): δ=8.9, 31.8, 32.8, 42.0, 46.5, 53.5, 106.0, 109.5, 111.5, 111.6, 121.3, 122.3, 122.4, 122.7, 123.0, 124.2, 124.7, 124.8, 126.1, 128.8, 132.1, 135.8, 147.5, 148.1, 152.2, 155.0, 157.3, 170.6, 171.0 ppm; HRMS (ESI): m/z [M H] calcd for C29H23N5O5: 520.1626, found: 520.1622; HPLC purity: 99.3%.
Biology
Amphetamine–chlordiazepoxide locomotor hyperactivity procedure
Locomotor activity was measured in a photocell apparatus (Med Associates Inc., St Albans, VT, USA). The apparatus consisted of plexiglas square chambers (27.3 27.3 20.3 cm) surrounded by 16 infrared photobeam sources. Distance traveled (cm) was measured as the index for activity. Mice were administered either water vehicle, 3a (3 or 10 mgkg−1), or valproate (400 mgkg−1), and placed into individual holding cages for 5 min. Mice were then injected with amphetamine, amphetamine+chlordiazepoxide (CDP) (amphetamine: 4 mgkg−1; CDP: 2.5 mgkg−1), or vehicle (water) and placed in the open field for a period of 60 min.
Prepulse inhibition of the acoustic startle response procedure
Prepulse inhibition (PPI) of the acoustic startle response was measured in the startle reflex system (Med Associates Inc.). Mice were injected with 10% DMSO vehicle, haloperidol (1 mgkg−1), valoprate (400 mgkg−1), or 3a (10 or 25 mgkg−1) and placed into individual holding cages for 30 min. Mice were then placed into the PPI chambers and received a 5 min habituation to the background white noise (70 dB) followed by a habituation block of six presentations of the startle stimulus alone. PPI trials consisted of ten blocks of six different types of trials: null (no stimuli), startle (120 dB), startle plus prepulse (4, 8 and 12 dB over background noise) and prepulse alone (12 dB over background noise). Trial types were presented at random within each block. Each trial started with a 50 ms null period during which baseline movements were recorded. During a subsequent 20 ms period, prepulse stimuli were presented and responses to the prepulse were measured. After an interval of 100 ms, the startle stimuli were presented for 40 ms and responses recorded for 100 ms from startle onset. Responses were sampled every 1 ms. The inter-trial interval was variable with an average of 15 s (range: 10–20 s). In startle alone trials, the basic auditory startle was measured and in prepulse plus startle trials the amount of inhibition of the normal startle was measured. PPI was calculated as [1-(startle amplitude on prepulse plus startle trial)/(startle amplitude on startle trial)] 100%. Startle re-sponses from the initial habituation trials were not used to calculate PPI.
Analyses
Locomotor activity data were analyzed using one-way analysis of variance (ANOVA) with drug treatment as the betweengroup variable. PPI data were analyzed with ANOVA with drug treatment as a between group variable and prepulse intensity as the repeated measure. All significant main effects and interactions were followed up by Fisher’s PLSD post-hoc test when appropriate. An effect was considered significant if p <0.05.
Drugs
All compounds were administered intraperitoneally (i.p.) in a volume of 10 mLkg−1. Valproate, d-amphetamine, chlordiazepoxide (Sigma, St.Louis, MO, USA) and 3a were dissolved in water for the Amph/CDP hyperactivity study. Valproate, haloperidol (Sigma) and 3a were dissolved in 10% DMSO for the PPI study.
Animals
Male C57BL/6J mice (Jackson Laboratory, Bar Harbor, ME, USA) were housed in groups of four in OptiMICE cages in a colony room on a 12 h light/dark cycle. Mice were approximately 7–14 weeks of age at the time of testing, and all behavioral tests were carried out during the light portion of the cycle. The temperature was maintained between 20 and 23°C with a relative humidity between 30% and 70%. Chow and water were provided ad libitum for the duration of the study. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of PsychoGenics (Tarrytown, NY, USA).
Locomotor activity in CLOCK mutant mice
Each mouse was placed in a clean activity chamber for 2 h, during which their gross activity was recorded by the number of times they crossed five parallel photobeams in the chamber (San Diego Instruments, San Diego, CA, USA). Beam breaks were recorded in 5 min bins. Significance between the activity of the drug-treated group and saline-treated group was determined by the Student’s t-test. These procedures were approved by the IACUC of the University of Texas Southwestern Medical Center (Dallas, TX, USA).
Supplementary Material
Acknowledgements
This work was supported in part by the US National Institutes of Health (1R01 MH072940–01 to APK). ADM is funded by the US National Cancer Institute (CA48112) and the Walther Cancer Foundation. CAM is funded by the US National Institute of Mental Health and the US National Institute on Drug Abuse. X-ray data collection was performed by the Life Sciences Collaborative Access Team (LS-CAT) at the Advanced Photon Source (Argonne National Laboratory, Argonne, USA). Use of the advanced photon source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. W-31–109-Eng-38.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/cmdc.201100188.
References
- [1]a).Embi N, Rylatt DB, Cohen P. Eur. J. Biochem. 1980;107:519. [PubMed] [Google Scholar]; b) Doble BW, Woodgett JR. J. Cell Sci. 2003;116:1175. doi: 10.1242/jcs.00384. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Eldar-Finkelman H. Trends Mol. Med. 2002;8:126. doi: 10.1016/s1471-4914(01)02266-3. [DOI] [PubMed] [Google Scholar]
- [2]a).Bhat RV, Shanley J, Correll MP, Fieles WE, Keith RA, Scott CW, Lee C-M. Proc. Natl. Acad. Sci. USA. 2000;97:11074. doi: 10.1073/pnas.190297597. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sayas CL, Ariaens A, Ponsioen B, Moolenaar WH. Mol. Biol. Cell. 2006;17:1834. doi: 10.1091/mbc.E05-07-0688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]a).Klein PS, Melton DA. Proc. Natl. Acad. Sci. USA. 1996;93:8455. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Stambolic V, Ruel L, Woodgett JR. Curr. Biol. 1996;6:1664. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- [4].Gould TD, Manji HK. Neuroscientist. 2002;8:497. doi: 10.1177/107385802237176. [DOI] [PubMed] [Google Scholar]
- [5].Bhat R, Xue Y, Berg S, Hellberg S, Ormoe M, Nilsson Y, Radesaeter A-C, Jerning E, Markgren P-O, Borgegard T, Nyloef M, Gimenez-Cassina A, Hernandez F, Lucas JJ, Diaz-Nido J, Avila J. J. Biol. Chem. 2003;278:45937. doi: 10.1074/jbc.M306268200. [DOI] [PubMed] [Google Scholar]
- [6]a).Hoeflich KP, Luo J, Ruble EA, Tsao M-S, Jin O, Woodgett JR. Nature. 2000;406:86. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]; b) O’Brien WT, Harper AD, Jov F, Woodgett JR, Maretto S, Piccolo S, Klein PS. J. Neurosci. 2004;24:6791. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Beaulieu J-M, Sotnikova TD, Yao W-D, Kockeritz L, Woodgett JR, Gainetdinov RR, Caron MG. Proc. Natl. Acad. Sci. USA. 2004;101:5099. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7]a).Chen G, Huang L-D, Jiang Y-M, Manji HK. J. Neurochem. 1999;72:1327. doi: 10.1046/j.1471-4159.2000.0721327.x. [DOI] [PubMed] [Google Scholar]; b) Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS. J. Biol. Chem. 2001;276:36734. doi: 10.1074/jbc.M101287200. [DOI] [PubMed] [Google Scholar]
- [8].Li X, Zhu W, Roh M-S, Friedman AB, Rosborough K, Jope RS. Neuropsychopharmacology. 2004;29:1426. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Fukumoto T, Morinobu S, Okamoto Y, Kagaya A, Yamawaki S. Psychopharmacology. 2001;158:100. doi: 10.1007/s002130100871. [DOI] [PubMed] [Google Scholar]
- [10].Chen L, Salinas GD, Li X. Mol. Pharmacol. 2009;76:1150. doi: 10.1124/mol.109.056994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Miller JS, Tallarida RJ, Unterwald EM. Brain Res. Bull. 2010;82:184. doi: 10.1016/j.brainresbull.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12]a).Cohen P, Goedert M. Nat. Rev. Drug Discovery. 2004;3:479. doi: 10.1038/nrd1415. [DOI] [PubMed] [Google Scholar]; b) Hur E-M, Zhou F-Q. Nat. Rev. Neurosci. 2010;11:539. doi: 10.1038/nrn2870. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Kypta RM. Expert Opin. Ther. Pat. 2005;15:1315. [Google Scholar]
- [13]a).Kozikowski AP, Gaisina IN, Yuan H, Petukhov PA, Blond SY, Fedolak A, Caldarone B, McGonigle P. J. Am. Chem. Soc. 2007;129:8328. doi: 10.1021/ja068969w. [DOI] [PubMed] [Google Scholar]; b) Gaisina IN, Gallier F, Ougolkov AV, Kim KH, Kurome T, Guo S, Holzle D, Luchini DN, Blond SY, Billadeau DD, Kozikowski AP. J. Med. Chem. 2009;52:1853. doi: 10.1021/jm801317h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]a).Vale AL, Ratcliffe F. Psychopharmacology. 1987;91:352. doi: 10.1007/BF00518190. [DOI] [PubMed] [Google Scholar]; b) Hanania T, Dillon GM, Malekiani SA, Manzano ML, Leahy E. Soc. Neurosci. 35th Annual Meeting; Washington DC, USA. 2005. abstract 108.9. [Google Scholar]
- [15]a).Perry W, Minassian A, Feifel D, Braff DL. Biol. Psychiatry. 2001;50:418. doi: 10.1016/s0006-3223(01)01184-2. [DOI] [PubMed] [Google Scholar]; b) Brody SA, Geyer MA, Large CH. Psychopharmacology. 2003;169:240. doi: 10.1007/s00213-003-1421-2. [DOI] [PubMed] [Google Scholar]
- [16].Roybal K, Theobold D, Graham A, DiNieri JA, Russo SJ, Krishnan V, Chakravarty S, Peevey J, Oehrlein N, Birnbaum S, Vitaterna MH, Orsulak P, Takahashi JS, Nestler EJ, Carlezon WA, Jr., McClung CA. Proc. Natl. Acad. Sci. USA. 2007;104:6406. doi: 10.1073/pnas.0609625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aubry C, Wilson AJ, Emmerson D, Murphy E, Yam Chan Y, Dickens MP, Garc MD, Jenkins PR, Mahale S, Chaudhuri B. Bioorg. Med. Chem. 2009;17:6073. doi: 10.1016/j.bmc.2009.06.070. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.