

Abstract
BACKGROUND & AIMS
Bilirubin is a natural and potent antioxidant that accumulates in the blood of newborn children and leads to physiological jaundice. Breast-fed infants have higher serum levels of bilirubin than formula-fed infants and are at risk for bilirubin-induced neurological dysfunction (BIND). Clearance of bilirubin requires the expression of uridine diphosphate glucuronosyltransferase (UGT) 1A1; we investigated its role in the association between breast feeding with jaundice in mice.
METHODS
We studied mice in which the original Ugt1 locus was disrupted and replaced with the human UGT1 locus (hUGT1 mice); these mice spontaneously develop neonatal hyperbilirubinemia and BIND. We fed human breast milk or formula to neonatal hUGT1 mice and examined activation of the intestinal xenobiotic receptors pregnane X receptor and constitutive androstane receptor. We also examined inflammatory signaling pathways in mice with disruptions in IκB-kinase–α and IκB kinase–β in the intestinal epithelium.
RESULTS
hUGT1 mice that were fed breast milk developed severe hyperbilirubinemia because of suppression of UGT1A1 in the gastrointestinal tract. Formula-fed hUGT1 mice had lower serum levels of bilirubin, which resulted from induction of UGT1A1 in the gastrointestinal tract. hUGT1/Pxr-null mice did not develop severe hyperbilirubinemia, whereas hUGT1/Car-null mice were susceptible to BIND when they were fed breast milk. Breast milk appeared to suppress intestinal IκB kinase α and β, resulting in inactivation of nuclear factor–κB and loss of expression of UGT1A1, leading to hyperbilirubinemia.
CONCLUSIONS
Breast milk reduces expression of intestinal UGT1A1, which leads to hyperbilirubinemia and BIND; suppression of this gene appears to involve inactivation of nuclear factor–κB. Hyperbilirubinemia can be reduced by activation of pregnane X receptor, constitutive androstane receptor, or nuclear factor–κB.
Keywords: Nursing, Mouse Model, Neonatal Jaundice, Gene Regulation
Overabundance of serum bilirubin leads to neonatal jaundice (hyperbilirubinemia), a common malady in 50%– 60% of newborn children and to a greater extent in premature infants.1–3 Newborn children can accumulate more bilirubin than adults because of the increase in red blood cell turnover coupled with reduced expression of bilirubin uridine diphosphate– glucuronosyltransferase (UGT) activity.4,5 Clearance of bilirubin is solely dependent upon expression of UGT1A1,6 which metabolizes bilirubin through glucuronidation to the bilirubin mono/di-glucuronide, followed by transport of the glucuronide into the bile.7 Thus, neonatal hyperbilirubinemia is characterized by production rate of bilirubin that cannot be matched by glucuronidation and elimination of bilirubin.8 Exaggerated physiological jaundice can lead to brain toxicity, which is characterized by acute bilirubin enceph-alopathy9 or the more severe chronic encephalopathy that results from the permanent clinical sequelae of bilirubin-induced neurologic dysfunction (BIND).10 Permanent brain damage by BIND results from irreversible accumulation of unconjugated bilirubin in brain tissue, termed kernicterus.11 Thus, understanding the normal and pathological events leading to hyperbilirubinemia is important in clinical practice to prevent the adverse effects of accumulating serum bilirubin.
Although it can cause brain damage at abnormal physiological concentrations, bilirubin is also known as a potent antioxidant.12,13 Increased serum bilirubin elevates the antioxidant threshold, resulting in a number of clinical benefits, such as lower risks for free radical–producing diseases,14 oxygen-radical diseases,15 cancer,16 coronary artery disease,17 and peripheral vascular disease.18 It has been recognized for more than 50 years that newborns who are breastfed have a 3- to 6-fold greater probability of developing elevated total serum bilirubin (TSB) levels than formula-fed newborns.19 –21 Investigations have led to a number of clues linking the components of breast milk to neonatal jaundice, such as steroids,20 fats,20 cytokines,22 and epidermal growth factor.23 However, experimental evidence that any of these mechanisms underlie the onset of breast milk jaundice remains elusive. A significant obstacle in defining those events that lead to breast milk jaundice has been the lack of an appropriate animal model displaying neonatal hyperbilirubinemia.
The selective deletion of the Ugt1 locus in Ugt1−/− mice leads to neonatal lethality,24 resulting from the inability to metabolize bilirubin by UGT1A1-dependent glucuronidation. By introducing the human UGT1 locus and the UGT1A1 gene as a transgene25 into Ugt1−/− mice,24 we have rescued neonatal lethality and created humanized UGT1 (hUGT1) mice. Interestingly, the human UGT1A1 gene is regulated in a developmental fashion both in liver and the gastrointestinal (GI) tract, resulting in extreme neonatal hyperbilirubinemia.26 In addition, onset of hyperbilirubinemia in hUGT1 mice culminates in seizures and eventually death in 7%–10% of neonatal mice. Development of seizures is one of the conditions classically observed when humans develop BIND.10 The neurological sequelae associated with severe hyperbilirubinemia in normal infants has also been attributed to breast milk.27
With breast milk contributing to the natural antioxidative properties of the blood by promoting physiological jaundice, we sought to define the underlying mechanism leading to breast milk–induced hyperbilirubinemia in hUGT1 mice. The model developed for these studies is based upon findings that normal instant formula fed to neonatal hUGT1 mice in place of normal breast milk induces intestinal UGT1A1 and reduces TSB levels. Using mouse genetics, new findings have confirmed that the xenobiotic receptors pregnane X receptor (PXR) and constitutive androstane receptor (CAR) play crucial roles in contributing to breast milk–induced hyperbilirubinemia and the onset of BIND and seizures, while the antioxidant protection induced by breast milk is linked to early development through suppression of the intestinal IκB kinase (IKK)/nuclear factor–κB (NF-κB) signaling pathway.
Materials and Methods
Chemicals and Reagents
A mouse anti-human UGT1A1 antibody was a gift of Dr Joseph K. Ritter (Virginia Commonwealth University, Medical College of Virginia, Richmond, VA). Anti– cytochrome P450 (Cyp)2b9/10 antibody and anti-Cyp3a antibody were generously provided by Dr Masahiko Negishi at the National Institute of Environmental Health Sciences and Dr Frank Gonzalez at the National Institutes of Health. Anti–p-glycoprotein (Mdr1) antibody and anti– glyceraldehyde-3 -phosphate dehydrogenase antibody were purchased from Novus Biologicals (Littleton, CO) and Santa Cruz Biotech (Santa Cruz, CA). Primers for quantitative real-time polymerase chain reaction (PCR) were commercially synthesized at Integrated DNA Technologies, Inc (San Diego, CA). Infant formula was purchased from local stores. Human breast milk was kindly donated by Deirdre La Placa, a research assistant in our laboratory. All other chemicals and solvents were of analytical grade or the highest grade commercially available.
Animals and Treatments
Tg(UGT1A1*28)Ugt1−/− (hUGT1) mice were developed previously in a C57BL/6 background.26 To generate hUGT1/Car−/− and hUGT1/Pxr−/− mice, hUGT1 mice were crossed with Car-null mice provided to our laboratory by Dr Masahiko Negishi at the National Institute of Environmental Health Sciences28 and Pxr-null mice obtained from Dr Ronald Evans at the Salk Institute.29 Both Car and Pxr null mice were crossed into the C57BL/6 background before breeding with hUGT1 mice. IKK-αF/F/IKK-βF/F and Vil-Cre/IKK-αF/F/IKK-βF/F mice were previously generated on a C57BL/6 background.30 For the feeding experiments, newborn pups were separated from their nursing mothers at 9 days after birth. Pups were fed with infant formula or breast milk 8 times per day from 8 am to 11 pm until 14 days after birth. The milk was warmed in an incubator at 43°C before feeding. Pups were housed in a temperature-controlled and humidified cage at 33°–34°C. For tissue collections, mice were anesthetized by isoflurane inhalation, and the liver was perfused with ice-cold 1.15% KCl. The small intestine was opened and rinsed in cold 1.15% KCl. Tissues were stored at −80°C. All animal experiments were carried out following University of California San Diego Institutional Animal Care and Use guidelines.
Bilirubin Measurements
Blood was obtained from the submandibular vein and centrifuged at 2000×g for 5 minutes. Serum samples (20 μL) were measured for total serum bilirubin using a Unistat Bilirubinometer (Reichert, Inc., Depew, NY). For each serum sample, the bilirubin value was measured 3 times and the mean value was used for data analysis. To avoid hemolysis of the serum and photolysis of the bilirubin, the serum samples were analyzed promptly after collection of blood.
Quantitative Real-Time Reverse Transcription PCR Analysis
One microgram RNA was reverse-transcribed into complementary DNA using iScript cDNA Synthesis Kit (BioRad, Hercules, CA). Quantitative real-time PCR was performed with qPCR MasterMix Plus for SYBR (Eurogentec, Seraing, Belgium), and the reactions were run in a Mx4000 Multiplex QPCR System (Stratagene, La Jolla, CA). Forward and reverse primers used were: UGT1A1-S, 5′-CCT TGC CTC AGA ATT CCT TC-3′ and UGT1A1 AS, 5′-ATT GAT CCC AAA GAG AAA ACC AC-3′; mouse Cyp2b10-S, 5-AAA GTC CCG TGG CAA CTT CC-3′ and Cyp2b10-AS, 5′-CAT CCC AAA GTC TCT CAT GG-3′; mouse Cyp3a11-S, 5-CTC AAT GGT GTG TAT ATC CCC-3′, and Cyp3a11-AS, 5′-CCG ATG TTC TTA GAC ACT GCC-3′, mouse cyclophilin-S, 5′-CAG ACG CCA CTG TCG CTT T-3′ and mouse cyclophilin-AS, 5′-TGT CTT TGG AAC TTT GTC TGC AA-3′. Each reaction contained 1 μL complementary DNA and 0.2 μM of the primers in a total volume of 20 μL. PCR conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds, 60°C for 20 seconds, and 72°C for 40 seconds.
Western Blot Analysis
Microsomal protein (20 μg) was separated on 4%–12% NuPAGE Bis-Tris polyacrylamide gels (Invitrogen, Carlsbad, CA). After electrophoresis at 200 V, the protein was transferred onto a nitrocellulose membrane (Millipore, Billerica, MA), and the membrane was blocked with 5% nonfat dry milk in Tris-buffered saline solution (10 mM Tris [pH 8.0], 150 mM NaCl, 0.05% Tween 20) for 1 hour. Membranes were then incubated with primary antibody at a dilution of 1:3000 overnight at 4°C, followed by incubation with horseradish peroxidase– conjugated secondary antibody for 1 hour at room temperature. After application of chemiluminescence reagents (Western Lightning, PerkinElmer Life Sciences, Waltham, MA), images were obtained in a Bio-Rad Universal Hood II equipped with a ChemiDoc XRS imaging system (Bio-Rad, Hercules, CA).
Results
Feeding Suppresses GI UGT1A1 Shortly After Birth
At 5 days after birth, hUGT1 mice are accumulating serum bilirubin and show no detectable human UGT1A1 expression in liver tissue.26 However, UGT1A1 is detected in small intestinal microsomes. With the GI tract playing an important role in bilirubin homeostasis in hUGT1 mice, we examined the levels of UGT1A1 gene expression in the GI tract during embryonic development (E20) just before birth and 8 –12 hours after birth. By comparing expression levels before and after birth, it would allow us to compare the impact of early breast feeding on gene expression. In comparing UGT1A1 gene expression by quantitative real-time reverse transcription PCR, the levels of UGT1A1 RNA gene transcript are nearly 5-fold greater in GI fetal tissue on embryonic day 20 (E20) when compared to levels observed after birth when the newborn mice have been nursing for 12 hours (Figure 1A). These findings suggest that exposure to breast milk leads to a reduction or suppression of intestinal UGT1A1 gene expression.
Figure 1.

Effects of breast milk and formula on serum bilirubin levels and gene expression in hUGT1 mice. (A) RNA was isolated from intestinal tissue from hUGT1 mice at embryonic day 20 (E20) and 12 hours after birth (12 hr). Quantitative real-time reverse transcription PCR was performed to measure relative expression for UGT1A1. Fold induction of the genes is expressed as compared to E20 mice. (B) Newborn pups were nursed (N), fed with formula (F) or human breast milk (HBM) for 5 days. At 14 days, serum bilirubin levels were measured. (C) Gene expression by quantitative real-time reverse transcription PCR of UGT1A1, Cyp2b10, and Cyp3a11 were determined from small intestine RNA isolated after 5 days of formula and HBM treatment. Fold induction of the genes in the formula-fed mice is expressed as compared to nursing mice. (D) Immunoblot of microsomal human UGT1A1, mouse CYP2B10, and CYP3A11 from small intestinal tissue is shown after formula and HBM treatment. The density of the bands was also quantified. (E) Body weight of mice was determined. (F) Nine-day-old hUGT1 mice were orally treated with 50 mg/kg triglyceride (TG) mix (1:1:1:1:1 of tricaprin, tricaprylin, trilaurin, trimyristin, and tripalmitin, dissolved in corn oil) for 5 days. At 14 days, serum bilirubin levels were measured. NT indicates no treatment (control). Data are expressed as mean ± SD, n = 6; *P < .01.
Formula Reduces Serum Bilirubin and Prevents BIND
To examine the effects of breast milk and formula feeding on serum bilirubin levels in neonatal hUGT1 mice, 9-day-old pups with average TSB levels of 10 mg/dL (171 umol/L) were treated orally with formula for 5 days. Mice that were allowed to nurse continued to accumulate serum bilirubin through 14 days after birth, while mice fed Enfamil Infant Formula (Mead Johnson & Company, Glenview, IL) exhibited dramatically decreased TSB values (Figure 1B). Similar reductions in TSB were achieved in hUGT1 mice that were fed Earth’s Best Organic Soy Formula (The Hain Celestial Group, Inc, Boulder, CO) or Baby’s Only Organic Dairy Formula (Nature’s One, Inc, Lewis Center, OH) (Supplementary Figure 1). Because isolation of the pups from the nursing dams might have caused stress and a resulting decrease in TSB values, we compared the effect of feeding hUGT1 mice human breast milk with that of formula-fed mice. Neonatal 9-day-old hUGT1 mice fed human breast milk for 5 days exhibited TSB levels that were comparable to 14-day-old hUGT1 mice that were nursing (Figure 1B). There was no increase in liver UGT1A1 gene expression in hUGT1 mice fed human breast milk. The decrease in TSB after formula treatment correlated with a 200 –300-fold induction of intestinal UGT1A1 gene expression (Figure 1C) and induction of UGT1A1 protein (Figure 1D). When we treated mice orally, the body weight of the formula- and human breast milk–fed mice was lower than that of the nursing pups (Figure 1E). Although body weight gain was not observed in either formula- or human breast milk–fed mice, reduction of serum bilirubin and induction of UGT1A1 were only observed in formula-fed mice, indicating that weight gain is not associated with bilirubin reduction or UGT1A1 induction. To directly understand the role of fat and bilirubin metabolism and excretion, neonatal hUGT1 mice were treated orally with 50 mg/kg of a triglyceride mix (1:1:1:1:1 of tricaprin, tricaprylin, trilaurin, trimyristin, and tripalmitin) for 5 days. This treatment did not result in a reduction of serum bilirubin (Figure 1F), indicating that fat content in formula is not the cause of the reduced bilirubin and induced UGT1A1 in the formula-fed mice.
Role of Xenobiotic Receptors CAR and PXR and Control of Hyperbilirubinemia
Previous work in our laboratory and others has confirmed that nuclear xenobiotic receptors CAR, PXR, and the peroxisome proliferator-activated receptor–α (ppar-α), in addition to the environmental sensing aryl hydrocarbon receptor (AhR), regulate UGT1A1 gene expression.25,31–33 Data obtained from formula feeding led us to speculate that formula might regulate expression of the intestinal UGT1A1 gene through activation of these receptors. To determine the role of these receptors in UGT1A1 gene induction in the small intestine, we examined Cyp1a1 expression as a marker of AhR34 activation, Cyp2b10 induction for CAR35 activation, Cyp3a11 induction for PXR activation,36 and Cyp4a10 gene expression, which is regulated by ppar–α.37 There was no induction of intestinal Cyp1a1 or Cyp4a10 gene expression, eliminating involvement of the AhR and ppar–α in response to formula (data not shown). However, there was a statistically significant induction of Cyp3a11 gene expression (>3-fold) and a dramatic induction of Cyp2b10 gene expression (>200-fold) (Figure 1C), as well as increases in protein expression levels (Figure 1D), implicating a potential role for both PXR and CAR in regulation of the UGT1A1 gene in the GI tract after formula treatment.
When we determined gene expression levels of Cyp2b10, Cyp3a11, Car, and Pxr in the developing small intestine, dramatically lowered Cyp2b10 and Cyp3a11 were observed in the mice nursed with breast milk compared to the levels at 21 days (Supplementary Figure 2), while expression of Car and Pxr was not changed. To directly determine the role of CAR, we crossed Car−/− mice with Tg(UGT1A1*28)Ugt1−/− mice generating Tg(UGT1A1*28)Ugt1−/−Car−/− (hUGT1/Car−/−) mice. Absence of Car expression in hUGT1/Car−/− mice was confirmed by quantitative real-time PCR analysis (Supplementary Figure 3). Administration of phenobarbital (Pb) orally to 12-day-old hUGT1 mice leads to induction of intestinal and liver Cyp2b10, in addition to intestinal and liver UGT1A1 gene expression (Figure 2A). Total serum bilirubin levels after 2 days of Pb exposure were <2 mg/dL (34.2 umol/L). When Pb was orally administered to hUGT1/Car−/− mice (Figure 2B), it had no impact on TSB levels and did not induce Cyp2b10 or UGT1A1 gene expression in either the liver or GI tract. The deletion of CAR in hUGT1/Car−/− mice confirmed that Pb exposure and reduction of TSB in hUGT1 mice was dependent upon CAR activation and induction of the UGT1A1 gene.
Figure 2.

UGT1A1 gene expression in breast- and formula-fed hUGT1 (top), hUGT1/Car−/− (middle), and hUGT1/Pxr−/− mice (bottom). (A) At 12 days after birth, hUGT1 mice were orally treated with phenobarbital (50 mg/kg). After treatment, quantitative real-time reverse transcription PCR was carried out for UGT1A1 and Cyp2b10. TSB levels were analyzed. For both hUGT1/Car−/− (B) and hUGT1/Pxr−/− mice (C), litters were divided, allowing part of the litter to nurse while the other littermates were fed formula. With hUGT1/Car−/− mice, additional litters were treated orally with phenobarbital (50 mg/kg) at 12 days and RNA isolated on day 14. With hUGT1/Pxr−/− mice, additional mice were treated orally with pregnenalone-16α carbonitrile (PCN). Quantitative real-time reverse transcription PCR was conducted to quantitate UGT1A1, Cyp2b10, and Cyp3a11 gene expression. At 14 days, serum bilirubin levels were measured. Data of gene expression indicate mean ± SD, n = 3. *P < .05; **P < .01; ***P < .001.
To examine if CAR underlined the reduction in TSB levels after formula treatment, 9-day-old hUGT1 and hUGT1/Car−/− mice were treated with formula for 5 days. Total serum bilirubin levels were reduced in hUGT1/Car−/− mice (Figure 2B). The reduction in TSB levels correlated with induction of intestinal UGT1A1. There was no induction of liver UGT1A1 by formula in hUGT1 and hUGT1/Car−/− mice. Although CAR is rendered nonfunctional, formula treatment led to robust induction of Cyp2b10 in the GI tract of hUGT1/Car−/− mice (Figure 2B). These results indicate that induction of intestinal UGT1A1 gene expression and reduction in TSB levels in hUGT1 mice are independent of CAR expression.
A similar approach was developed to examine if neonatal expression of PXR plays a role in formula-initiated control of TSB levels. Absence of Pxr expression in hUGT1/Pxr−/− mice was confirmed by quantitative real-time PCR analysis (Supplementary Figure 3). When hUGT1/Pxr−/− mice were treated with formula for 5 days, TSB levels were reduced below 1 mg/dL (17.1 umol/L) (Figure 2C). The reduction in TSB levels correlated with induction of intestinal UGT1A1 (Figure 2C). Although PXR is rendered nonfunctional, formula treatment led to induction of Cyp3a11 in the GI tract of hUGT1/Pxr−/− mice (Figure 2C). The reduction in TSB levels in hUGT1/Pxr−/− mice after formula treatment indicates that activation of PXR is not a key pathway in formula-driven clearance of TSB in hUGT1 mice.
CAR and PXR Are Linked to BIND
During the period of neonatal development and normal breast feeding, up to 10% of hUGT1 mice progress into bilirubin-induced seizures (Supplementary Video 1), which eventually result in death. Interestingly, TSB levels are lower in hUGT1/Pxr−/− mice compared to hUGT1 mice (Figure 3A). None of the hUGT1/Pxr−/− mice developed seizures (Figure 3B). The mechanism behind the absence of PXR and bilirubin metabolism is currently unknown. In contrast, >50% of the hUGT1/Car−/− mice developed seizures and died (Figure 3B), indicating a protective role for CAR in preventing development of BIND. When hUGT1, hUGT1/Car−/−, and hUGT1/Pxr−/− mice were treated with formula for 5 days, TSB levels were reduced (Figure 3A), with none of the mice on the formula diet progressing into seizures (Figure 3B).
Figure 3.

Serum bilirubin levels and survival curves of breast- and formula-fed hUGT1, hUGT1/Car−/−, and hUGT1/Pxr−/− mice. (A) In newborn hUGT1, hUGT1/Car−/−, and hUGT1/Pxr−/− mice, serum bilirubin levels were measured at 3, 7, 10, 14, 16, 18, and 21 days in addition to 14-day-old mice after 5 days of formula treatment. (B) Survival curves of breast- and formula-fed hUGT1, hUGT1/Car−/−, and hUGT1/Pxr−/− mice during the neonatal developmental period were determined. Error bars show SD, n = 20. *P < .01; **P < .001.
During neonatal development, TSB levels in hUGT1/Car−/− mice are statistically equivalent to those levels in hUGT1 mice. We rationalized that the increased incidence of BIND in hUGT1/Car−/− mice may result from the inability to adequately clear bilirubin from brain tissue by efflux transporters that are regulated by CAR. Bilirubin is a substrate for the Mdr1a isoform of P-glycoprotein, which is a membrane efflux pump associated with the microvasculature at the blood-facing luminal surfaces of the endothelium.38,39 In hUGT1 mice, expression of the Mdr1a gene in brain is shown to be regulated during neonatal development (Figure 4), although its protein expression is low (Figure 4A and B). In contrast, when we carried out a quantitative real-time reverse transcription PCR analysis for Mrp1, a developmental change in Mrp1 expression was not observed (Figure 4C). Mdr1a gene expression is lower in hUGT1/Car−/− mice (Figure 4D), indicating that CAR expression is important for maturing Mdr1a in brain tissue. The reduction of Mdr1a gene expression in brain tissue of hUGT1/Car−/− mice and the heightened sensitivity of these mice toward development of BIND allows us to speculate that reduced expression of Mdr1a results in accumulation of toxic levels of bilirubin in brain tissue.
Figure 4.

Mdr1a gene expression. (A) Mdr1a expression was analyzed in brain tissue of 3, 5, 7, 10, 14, 16, 18, and 21-day-old hUGT1 mice by quantitative real-time reverse transcription PCR. (B) Total tissue homogenates were prepared and immunoblot analysis was carried out for Mdr1a in the brain of 7-, 14-, and 21-day-old hUGT1 mice as well as liver of 21-day-old mice. (C) Mrp1 expression was analyzed in brain tissue of 3, 5, 7, 10, 14, 16, 18, and 21-day-old hUGT1 mice by quantitative real-time reverse transcription PCR. (D) Mdr1a expression was analyzed in brain tissue of 14-day-old hUGT1 and hUGT1/Car−/− mice. Error bars show SD, n = 3. *P < .01.
Role of NF-κB in Breast Milk–Induced Hyperbilirubinemia
It has been reported that breast milk can inhibit NF-κB– dependent target gene expression, causing suppression of interleukin-8 production in intestinal Caco-2 cells.40 We analyzed expression of mouse macrophage inflammatory protein–2 (Mip-2), the homologue of inter-leukin-8 in mice, and Cox-2, a classical NF-κB target gene. Formula-fed mice led to induction of Mip-2 and Cox-2 in the GI tract (Figure 5A). Because dramatic developmental changes in expression were not observed (Supplementary Figure 4), these data suggest an involvement of NF-κB in regulation of the UGT1A1 gene via breast and formula feeding.
Figure 5.

Involvement of transcription factor NF-κB in the regulation of UGT1A1 expression with breast milk. (A) Macrophage inflammatory protein–2 and cox-2 expressions in the GI of nursing, formula-, and HBM-fed hUGT1 mice were analyzed by quantitative real-time reverse transcription PCR. (B) Neonatal IKK-αF/F/IKK-βF/F and Vil-Cre/IKK-αF/F/IKK-βF/F mice were fed formula for 5 days, and Cyp2b10 expression was determined in the intestinal epithelial cells. (C) 10 mg/kg cadmium was treated orally to the IKK-αF/F/IKK-βF/F and Vil-Cre/IKK-αF/F/IKK-βF/F mice at 12 days after birth, and Cyp2b10 expression in the intestinal epithelial cells was analyzed by real-time reverse transcription PCR at 14 days. (D) Fold induction of UGT1A1 and Cyp2b10 gene expression in hUGT1 mice. At 12 days after birth, hUGT1 mice were treated orally with LPS (100 mg/kg) or cadmium (Cd) (10 mg/kg). At 14 days, RNA was isolated from the small intestine and quantitative real-time reverse transcription PCR was carried out for UGT1A1 and Cyp2b10 expression. Fold induction of the gene in the LPS- or Cd-treated mice was shown as compared to nontreated mice. (E) Serum bilirubin levels were measured at 14 days in the untreated (control), LPS- or Cd-treated hUGT1 mice. Error bars show SD, n = 3. *P < .01; **P < .001.
To investigate the role of NF-κB in the formula-induced gene expression, formula was given orally to mice in which NF-κB signaling through IκB-kinase (IKK)-α and IKK-β are selectively ablated in the intestinal epithelium through conditional knockout of the genes.41 Although these mice do not carry the human UGT1A1 transgene, we evaluated Cyp2b10 expression, which is regulated in a similar fashion to that of human UGT1A1 in hUGT1 mice. Neonatal IKK-αF/F/IKK-βF/F and Vil-Cre/IKK-αF/F/IKK-βF/F mice were fed formula for 5 days, and Cyp2b10 expression in the intestinal epithelial cells was determined. The Cyp2b10 gene was still dramatically induced with formula feeding in Vil-Cre/IKK-αF/F/IKK-βF/F mice as well as in IKK-αF/F/IKK-βF/F mice (Figure 5B).
Cadmium is an NF-κB activator.42,43 To further investigate the role of NF-κB in the regulation of those genes, cadmium was given orally to Vil-Cre/IKK-αF/F/IKK-βF/F mice. When cadmium was given to IKK-αF/F/IKK-βF/F mice, Cyp2b10 gene expression levels were increased in intestinal epithelial cells (Figure 5C). In contrast, the inducibility of Cyp2b10 with cadmium treatment was completely abolished in the intestinal epithelial cells of Vil-Cre/IKK-αF/F/IKK-βF/F mice (Figure 5C). This indicates that the IKK/NF-κB signaling pathway is linked to induction of Cyp2b10 in the small intestine.
To investigate the role of NF-κB in control of UGT1A1 expression, neonatal hUGT1 mice were treated orally with lipopolysaccharide (LPS), a known activator of NF-κB.44 After 48 hours’ treatment with LPS, UGT1A1 gene expression was markedly induced (>10-fold) along with induction of Cyp2b10 gene expression (>15-fold) in the GI tract (Figure 5D). Because UGT1A1 was induced, this also led to a reduction in TSB levels following LPS treatment (Figure 5E). When neonatal hUGT1 mice were treated orally with cadmium, UGT1A1 and Cyp2b10 were induced in the GI tract (Figure 5D). The induction of UGT1A1 in the GI tract led to the decrease of TSB levels (Figure 5E). Data obtained from the treatment of hUGT1 mice with LPS and cadmium indicates that NF-κB plays an important role in regulating UGT1A1 and Cyp2b10 in the GI tract. Taking these findings together, we can speculate that breast milk promotes antioxidant protection in neonates by limiting intestinal UGT1A1 gene expression in a manner that is directly linked to inhibition of NF-κB expression (Figure 6).
Figure 6.
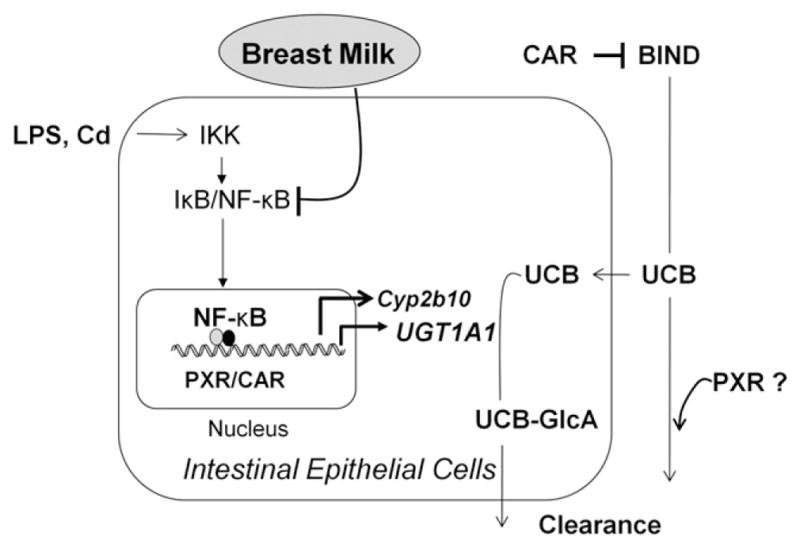
Schematic representation of pathways. In neonatal hUGT1 mice, UGT1A1 metabolizes unconjugated bilirubin (UCB) in the GI tract, suppressing the onset of BIND. Intestinal UGT1A1 is under control by IKK/NF-κB signaling but can be induced following activation of NF-κB, PXR, or CAR. In addition, CAR plays a protective role against BIND by controlling expression of Mdr1a in the brain, while PXR contributes to bilirubin homeostasis by mechanisms not yet identified.
Discussion
Humanized UGT1 mice show little expression of UGT1A1 during neonatal development in the liver, concordant to the reduced expression of bilirubin UGT activity in humans during early development.5,45 Because control of bilirubin clearance in hUGT1 mice is regulated by intestinal UGT1A1 expression,26 we rationalized that nutritional components originating from breast milk played a key role in controlling the steady-state levels of TSB by regulating intestinal UGT1A1. Although intestinal UGT1A1 gene expression is prominent at the latter stages of embryonic development, it drops quickly after birth, indicating that early effects of breast milk lead to suppression of intestinal gene expression. Proof that breast milk is suppressing intestinal UGT1A1 expression was demonstrated by feeding mice formula, resulting in a precipitous drop in TSB and an induction of intestinal UGT1A1. Because expression of human UGT1A1 is regulated in part by xenobiotic receptors as well as environmental sensors, such as the AhR, we examined the actions of formula on activation of these receptors. Concordant with induction of UGT1A1 was the simultaneous induction of intestinal Cyp3a11 and Cyp2b10 gene expression, with Cyp2b10 being induced >200-fold. These findings indicated that both PXR and CAR activation may be involved in regulation of UGT1A1. To examine this possibility, hUGT1 mice were crossed into either a Pxr-null or Car-null background to examine the impact of formula on regulating TSB levels. In both hUGT1/Pxr−/− and hUGT1/Car−/− mice, formula treatment effectively induced UGT1A1 along with Cyp3a11 and Cyp2b10 gene expression, indicating that induction of intestinal UGT1A1, Cyp2b10, and Cyp3a11 gene expression and reduction in TSB levels in formula-fed hUGT1 mice are not associated with either CAR or PXR expression.
Our findings indicate that breast milk contributes to development of hyperbilirubinemia by suppressing expression of UGT1A1 gene expression in the small intestine. Regulation of UGT1A1 gene expression in intestinal tissue is tied to control of the IKK/NF-κB signaling pathway. Indirect evidence supports this conclusion. Breast-fed hUGT1 mice have reduced expression of macrophage inflammatory protein–2, an NF-κB target gene,40 compared to formula-fed hUGT1 mice. Also, induction of the Cyp2b10 gene by cadmium was completely abolished in Vil-Cre/IKK-αF/F/IKK-βF/F mice, in which NF-κB is rendered nonfunctional. Treatment of hUGT1 mice with NF-κB activators, LPS and cadmium, resulted in the induction of UGT1A1 and Cyp2b10 expression in intestinal tissue followed by a decrease in serum bilirubin levels. These findings indicate that breast milk controls the IKK/NF-κB signaling pathway, resulting in suppression of the UGT1A1 gene in the GI tract, culminating in hyperbilirubinemia and antioxidant protection.
Conclusions
Breast feeding has been implicated in short- and long-term health benefits to growing children, which have included reduced risks of infectious diarrhea, necrotizing enterocolitis,46 type 1 and type 2 diabetes47,48; reduced frequency of food allergies49; and a protective effect on the development of early-onset inflammatory bowel disease.50 With recent findings that bilirubin is a potent and natural antioxidant,12 the beneficial actions of breast milk can also include its ability to promote mild hyperbilirubinemia. It is unclear why breast milk would promote hyperbilirubinemia, but it might be a natural defense mechanism against the potential toxicity associated with the sudden and rapid exposure to oxygen at birth. Although this is usually a benign condition, which can now be considered to be beneficial, excessive accumulation of TSB can lead to rare but serious toxicity resulting in kernicterus formation, permanent brain damage, and even death. With delayed expression of UGT1A1 in liver tissue, control of hyperbilirubinemia has been shown to be regulated by intestinal UGT1A1. Importantly, we have demonstrated that activation of intestinal xenobiotic receptors PXR and CAR can lead to induction of UGT1A1 and lowering of TSB. In cases of extreme hyperbilirubinemia, this strategy might prove beneficial with simple oral supplements designed to activate these receptors and induce intestinal UGT1A1 gene expression. Alternatively, mild induction of oxidative stress and activation of the NF-κB pathway by oral supplements or with formula will also facilitate a lowering of TSB levels.
Supplementary Material
Acknowledgments
Funding
Funding for this work was provided by US Public Health Service Grants P42ES010337 (R.H.T. and M.K.) and GM086713 (R.H.T.).
Abbreviations used in this paper
- AhR
aryl hydrocarbon receptor
- BIND
bilirubin-induced neurological dysfunction
- CAR
constitutive androstane receptor
- Cyp
cytochrome P450
- GI
gastrointestinal
- IKK
IκB kinase
- LPS
lipopolysaccharide
- Mip-2
macrophage inflammatory protein-2
- NF-κB
nuclear factor-κB
- Pb
phenobarbital
- PCR
polymerase chain reaction
- PPAR-α
peroxisome proliferator-activated receptor-α
- PXR
pregnane X receptor
- TSB
total serum bilirubin
- UGT
uridine diphosphate– glucuronosyltransferase
Footnotes
Note: To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at doi: 10.1053/j.gastro.2011.09.045.
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Watchko JF. Hyperbilirubinemia and bilirubin toxicity in the late preterm infant. Clin Perinatol. 2006;33:839– 852. doi: 10.1016/j.clp.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Bhutani VK, Maisels MJ, Stark AR, et al. Management of jaundice and prevention of severe neonatal hyperbilirubinemia in infants > or = 35 weeks gestation. Neonatology. 2008;94:63– 67. doi: 10.1159/000113463. [DOI] [PubMed] [Google Scholar]
- 3.Keren R, Luan X, Friedman S, et al. A comparison of alternative risk-assessment strategies for predicting significant neonatal hyperbilirubinemia in term and near-term infants. Pediatrics. 2008;121:e170– e179. doi: 10.1542/peds.2006-3499. [DOI] [PubMed] [Google Scholar]
- 4.Grimmer I, Moller R, Gmyrek D, et al. [Bilirubin UDP-glucuronyltransferase activity in human fetal liver homogenates] Acta Biol Med Ger. 1978;37:131–135. [PubMed] [Google Scholar]
- 5.Kawade N, Onishi S. The prenatal and postnatal development of UDP-glucuronyltransferase activity towards bilirubin and the effect of premature birth on this activity in the human liver. Biochem J. 1981;196:257–260. doi: 10.1042/bj1960257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bosma PJ, Seppen J, Goldhoorn B, et al. Bilirubin UDP-glucuronosyltransferase 1 is the only relevant bilirubin glucuronidating isoform in man. J Biol Chem. 1994;269:17960–17964. [PubMed] [Google Scholar]
- 7.Jansen PLM, Chowdhury JR, Fischberg EB, et al. Enzymatic conversion of bilirubin monoglucuronide to diglucuronide by rat liver plasma membranes. J Biol Chem. 1977;252:2710–2716. [PubMed] [Google Scholar]
- 8.Kaplan M, Muraca M, Hammerman C, et al. Imbalance between production and conjugation of bilirubin: a fundamental concept in the mechanism of neonatal jaundice. Pediatrics. 2002;110:e47. doi: 10.1542/peds.110.4.e47. [DOI] [PubMed] [Google Scholar]
- 9.Shapiro SM. Bilirubin toxicity in the developing nervous system. Pediatr Neurol. 2003;29:410– 421. doi: 10.1016/j.pediatrneurol.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 10.Shapiro SM. Definition of the clinical spectrum of kernicterus and bilirubin-induced neurologic dysfunction (BIND) J Perinatol. 2005;25:54–59. doi: 10.1038/sj.jp.7211157. [DOI] [PubMed] [Google Scholar]
- 11.Shapiro SM. Chronic bilirubin encephalopathy: diagnosis and outcome. Semin Fetal Neonatal Med. 2010;15:157–163. doi: 10.1016/j.siny.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 12.Stocker R, Glazer AN, Ames BN. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci U S A. 1987;84:5918–5922. doi: 10.1073/pnas.84.16.5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van PR. Diagnosis of kernicterus in the neonatal period. Pediatrics. 1961;28:870– 876. [PubMed] [Google Scholar]
- 14.Benaron DA, Bowen FW. Variation of initial serum bilirubin rise in newborn infants with type of illness. Lancet. 1991;338:78– 81. doi: 10.1016/0140-6736(91)90074-y. [DOI] [PubMed] [Google Scholar]
- 15.Hegyi T, Goldie E, Hiatt M. The protective role of bilirubin in oxygen-radical diseases of the preterm infant. J Perinatol. 1994;14:296–300. [PubMed] [Google Scholar]
- 16.Temme EH, Zhang J, Schouten EG, et al. Serum bilirubin and 10-year mortality risk in a Belgian population. Cancer Causes Control. 2001;12:887– 894. doi: 10.1023/a:1013794407325. [DOI] [PubMed] [Google Scholar]
- 17.Vitek L, Jirsa M, Brodanova M, et al. Gilbert syndrome and ischemic heart disease: a protective effect of elevated bilirubin levels. Atherosclerosis. 2002;160:449– 456. doi: 10.1016/s0021-9150(01)00601-3. [DOI] [PubMed] [Google Scholar]
- 18.Breimer LH, Spyropolous KA, Winder AF, et al. Is bilirubin protective against coronary artery disease? Clin Chem. 1994;40:1987–1988. [PubMed] [Google Scholar]
- 19.Newman AJ, Gross S. Hyperbilirubinemia in breast-fed infants. Pediatrics. 1963;32:995–1001. [PubMed] [Google Scholar]
- 20.Arias IM, Gartner LM, Seifter S, et al. Prolonged neonatal unconjugated hyperbilirubinemia associated with breast feeding and a steriod, pregnane-3(alpha), 20(beta)-diol, in maternal milk that inhibits glucuronide formation in vitro. J Clin Invest. 1964;43:2037–2047. doi: 10.1172/JCI105078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider AP. Breast milk jaundice in the newborn. A real entity. JAMA. 1986;255:3270–3274. [PubMed] [Google Scholar]
- 22.Zanardo V, Golin R, Amato M, et al. Cytokines in human colostrum and neonatal jaundice. Pediatr Res. 2007;62:191–194. doi: 10.1203/PDR.0b013e31809871c9. [DOI] [PubMed] [Google Scholar]
- 23.Kumral A, Ozkan H, Duman N, et al. Breast milk jaundice correlates with high levels of epidermal growth factor. Pediatr Res. 2009;66:218–221. doi: 10.1203/PDR.0b013e3181ac4a30. [DOI] [PubMed] [Google Scholar]
- 24.Nguyen N, Bonzo JA, Chen S, et al. Disruption of the Ugt1 locus in mice resembles human Crigler-Najjar type I disease. J Biol Chem. 2008;283:7901–7911. doi: 10.1074/jbc.M709244200. [DOI] [PubMed] [Google Scholar]
- 25.Chen S, Beaton D, Nguyen N, et al. Tissue-specific, inducible, and hormonal control of the human UDP-glucuronosyltransferase-1 (UGT1) locus. J Biol Chem. 2005;280:37547–37557. doi: 10.1074/jbc.M506683200. [DOI] [PubMed] [Google Scholar]
- 26.Fujiwara R, Nguyen N, Chen S, et al. Developmental hyperbilirubinemia and CNS toxicity in mice humanized with the UDP glucuronosyltransferase 1 (UGT1) locus. Proc Natl Acad Sci U S A. 2010;107:5024–5029. doi: 10.1073/pnas.0913290107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maisels MJ, Newman TB. Kernicterus in otherwise healthy, breast-fed term newborns. Pediatrics. 1995;96:730–733. [PubMed] [Google Scholar]
- 28.Ueda A, Kakizaki S, Negishi M, et al. Residue threonine 350 confers steroid hormone responsiveness to the mouse nuclear orphan receptor CAR. Mol Pharmacol. 2002;61:1284–1288. doi: 10.1124/mol.61.6.1284. [DOI] [PubMed] [Google Scholar]
- 29.Xie W, Barwick JL, Downes M, et al. Humanized xenobiotic response in mice expressing nuclear receptor SXR. Nature. 2000;406:435– 439. doi: 10.1038/35019116. [DOI] [PubMed] [Google Scholar]
- 30.Chen LW, Egan L, Li ZW, et al. The two faces of IKK and NF-kappaB inhibition: prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nat Med. 2003;9:575–581. doi: 10.1038/nm849. [DOI] [PubMed] [Google Scholar]
- 31.Yueh MF, Huang YH, Hiller A, et al. Involvement of the xenobiotic response element (XRE) in a receptor-mediated induction of human UDP-glucuronosyltransferase 1A1. J Biol Chem. 2003;278:15001–15006. doi: 10.1074/jbc.M300645200. [DOI] [PubMed] [Google Scholar]
- 32.Sugatani J, Nishitani S, Yamakawa K, et al. Transcriptional regulation of human UGT1A1 gene expression: activated glucocorticoid receptor enhances constitutive androstane receptor/pregnane X receptor-mediated UDP-glucuronosyltransferase 1A1 regulation with glucocorticoid receptor-interacting protein 1. Mol Pharmacol. 2005;67:845– 855. doi: 10.1124/mol.104.007161. [DOI] [PubMed] [Google Scholar]
- 33.Senekeo-Effenberger K, Chen S, Brace-Sinnokrak E, et al. Expression of the human UGT1 locus in transgenic mice by 4-Chloro-6-(2,3-xylidino)-2-pyrimidinylthioacetic acid (WY-14643) and implications on drug metabolism through peroxisome proliferator-activated receptor {alpha} activation. Drug Metab Dispos. 2007;35:419–427. doi: 10.1124/dmd.106.013243. [DOI] [PubMed] [Google Scholar]
- 34.Beebe L, Park SS, Anderson LM. Differential enzyme induction of mouse liver and lung following a single low or high dose of 2,3,7,8-tetrachlorodibenzo- p-dioxin (TCDD) J Biochem Toxicol. 1990;5:211–219. doi: 10.1002/jbt.2570050403. [DOI] [PubMed] [Google Scholar]
- 35.Wei P, Zhang J, Egan-Hafley M, et al. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 36.Staudinger JL, Goodwin B, Jones SA, et al. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barclay TB, Peters JM, Sewer MB, et al. Modulation of cytochrome P-450 gene expression in endotoxemic mice is tissue specific and peroxisome proliferator-activated receptor-alpha dependent. J Pharmacol Exp Ther. 1999;290:1250–1257. [PubMed] [Google Scholar]
- 38.Hanko E, Tommarello S, Watchko JF, et al. Administration of drugs known to inhibit P-glycoprotein increases brain bilirubin and alters the regional distribution of bilirubin in rat brain. Pediatr Res. 2003;54:441– 445. doi: 10.1203/01.PDR.0000085169.87948.B6. [DOI] [PubMed] [Google Scholar]
- 39.Klaassen CD, Aleksunes LM. Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev. 2010;62:1–96. doi: 10.1124/pr.109.002014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Minekawa R, Takeda T, Sakata M, et al. Human breast milk suppresses the transcriptional regulation of IL-1beta-induced NF-kappaB signaling in human intestinal cells. Am J Physiol Cell Physiol. 2004;287:C1404–C1411. doi: 10.1152/ajpcell.00471.2003. [DOI] [PubMed] [Google Scholar]
- 41.Egan LJ, Eckmann L, Greten FR, et al. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proc Natl Acad Sci U S A. 2004;101:2452–2457. doi: 10.1073/pnas.0306734101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hyun JS, Satsu H, Shimizu M. Cadmium induces interleukin-8 production via NF-kappaB activation in the human intestinal epithelial cell, Caco-2. Cytokine. 2007;37:26–34. doi: 10.1016/j.cyto.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 43.Jeong EM, Moon CH, Kim CS, et al. Cadmium stimulates the expression of ICAM-1 via NF-kappaB activation in cerebrovascular endothelial cells. Biochem Biophys Res Commun. 2004;320:887–892. doi: 10.1016/j.bbrc.2004.05.218. [DOI] [PubMed] [Google Scholar]
- 44.Luo JL, Maeda S, Hsu LC, et al. Inhibition of NF-kappaB in cancer cells converts inflammation-induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell. 2004;6:297–305. doi: 10.1016/j.ccr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 45.Onishi S, Kawade N, Itoh S, et al. Postnatal development of uridine diphosphate glucuronyltransferase activity towards bilirubin and 2-aminophenol in human liver. Biochem J. 1979;184:705–707. doi: 10.1042/bj1840705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Agostoni C, Braegger C, Decsi T, et al. Breast-feeding: a commentary by the ESPGHAN Committee on Nutrition. J Pediatr Gastroenterol Nutr. 2009;49:112–125. doi: 10.1097/MPG.0b013e31819f1e05. [DOI] [PubMed] [Google Scholar]
- 47.Mayer EJ, Hamman RF, Gay EC, et al. Reduced risk of IDDM among breast-fed children. The Colorado IDDM Registry Diabetes. 1988;37:1625–1632. doi: 10.2337/diab.37.12.1625. [DOI] [PubMed] [Google Scholar]
- 48.Pettitt DJ, Forman MR, Hanson RL, et al. Breastfeeding and incidence of non-insulin-dependent diabetes mellitus in Pima Indians. Lancet. 1997;350:166–168. doi: 10.1016/S0140-6736(96)12103-6. [DOI] [PubMed] [Google Scholar]
- 49.Saarinen UM, Kajosaari M. Breastfeeding as prophylaxis against atopic disease: prospective follow-up study until 17 years old. Lancet. 1995;346:1065–1069. doi: 10.1016/s0140-6736(95)91742-x. [DOI] [PubMed] [Google Scholar]
- 50.Barclay AR, Russell RK, Wilson ML, et al. Systematic review: the role of breastfeeding in the development of pediatric inflammatory bowel disease. J Pediatr. 2009;155:421– 426. doi: 10.1016/j.jpeds.2009.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.