

Abstract
Some apicomplexan parasites have evolved distinct protein kinase families to modulate host cell structure and function. Toxoplasma gondii rhoptry protein kinases and pseudokinases are involved in virulence and modulation of host cell signalling. The proteome of Plasmodium falciparum contains a family of putative kinases called FIKKs, some of which are exported to the host red blood cell and might play a role in erythrocyte remodelling. In this review we will discuss kinases known to be critical for host cell invasion, intracellular growth and egress, focusing on (i) calcium-dependent protein kinases and (ii) the secreted kinases that are unique to Toxoplasma (rhoptry protein kinases and pseudokinases) and Plasmodium (FIKKs).
Keywords: Protein kinase, Toxoplasma, Plasmodium, Rhoptry, Calcium-dependent protein kinase, Malaria, Red blood cell
1. Introduction
Protein kinases mediate the transfer of phosphate groups from ATP to specific residues on their target proteins, resulting in changes in the activity, stability, interactions with ligands or localisation of the phosphorylated substrates. Kinases themselves are often similarly regulated, and many of them function as signalling mediators that integrate upstream signals in the form of second messengers, post translational modifications or binding of regulatory proteins. It is therefore not surprising that many parasites have evolved distinct protein kinase families with novel domain structures and biochemical features to regulate key parasite-specific physiological processes that must be executed in a timely fashion during development or in response to external stimuli and physiological cues from the host. As signalling mediators, protein kinases are also well suited to function at the host–parasite interface, where they can perturb host signalling pathways, activate dormant mechanisms in the host cell, or disrupt the proper functioning of host proteins.
Both Toxoplasma and Plasmodium contain a family of calcium-dependent protein kinases (CDPKs), whose occurrence is restricted to plants and Alveolates (the superphylum that comprises the ciliates and apicomplexans), although trypanosomatids also possess kinases with an EF-hand calcium binding domain thought to be phylogenetically distinct from plant and Alveolate CDPKs (Parsons et al., 2005). CDPKs have a domain structure consisting of a calcium-binding domain fused to the kinase domain, such that kinase activity is stimulated upon calcium binding. Studies of apicomplexan CDPKs have revealed a conserved regulatory mechanism and highlighted the importance of calcium in regulating a number of key physiological processes including host cell attachment and invasion, gliding motility and parasite egress.
Toxoplasma gondii, a highly prevalent obligate intracellular protozoan parasite, seems to rely for a large part on protein kinases and pseudo kinases to modify the host cell. Toxoplasma is capable of infecting all nucleated cells of most warm-blooded animals. This ability to establish a chronic infection in such a wide range of host species and cell types is likely associated with its ability to modify many aspects of the host cell’s normal physiology. It does this by secreting proteins from the rhoptry, a specialised secretory organelle that is found only in apicomplexan parasites, directly into the host cytosol where they can exert their function. Many rhoptry proteins have homology to kinases (Bradley et al., 2005) and a large proportion of them are predicted to be pseudokinases (Peixoto et al., 2010).
Malaria parasites (genus Plasmodium) also belong to the Apicomplexa, however, unlike Toxoplasma, they have a very restricted range of host cells which they can invade and in which they can replicate. After inoculation into the vertebrate host through the bite of an infected mosquito, Plasmodium sporozoites must first invade hepatocytes, where they undergo asexual proliferation (exoerythrocytic schizogony), generating several thousand merozoites in the process. This strict dependence on hepatocytes for the pre-erythrocytic stage of infection has, however, recently been challenged by in vivo observations of Plasmodium berghei parasites developing in dermal and epidermal cells at the site of inoculation and generating infective merozoites (Gueirard et al., 2010).
The only cell type permissive for invasion by Plasmodium merozoites is the erythrocyte (or the reticulocyte in some instances). Invasion involves release of the contents of rhoptries and micronemes (another type of specialised secretory organelle) and formation of a parasitophorous vacuole (PV). The parasite causes considerable modifications to its host red blood cell (RBC) that include the establishment of a complex trafficking system that mediates translocation of parasite-encoded proteins to the RBC membrane skeleton and surface (Cooke et al., 2004). In the case of Plasmodium falciparum, the most lethal of the five species of malaria parasites that can infect humans, such proteins include components of the so-called “knob” structures, through which parasite-infected RBCs adhere to vascular endothelial cells, thereby significantly contributing to pathogenesis (Cooke et al., 2001). There are no orthologs of the Toxoplasma rhoptry kinases in malaria parasites, however, the P. falciparum kinome includes a family (20 members) of related kinase-like sequences called FIKKs, due to the presence of a Phe-Ile-Lys-Lys motif they share in their kinase domain (Ward et al., 2004). Most notably, 18 of the 20 fikk genes in P. falciparum are predicted to encode fully functional kinases, 16 of which are predicted to be exported into the host RBC (Schneider and Mercereau-Puijalon, 2005), and at least some of these enzymes have been shown to be exported to the host RBC and to be associated with kinase activity (Nunes et al., 2007, 2010).
Many of the Toxoplasma and Plasmodium kinases are related to kinases with known functions in other eukaryotes and these will not be discussed in this review. Instead, we will discuss in more detail, kinases from these parasites that are known to be critical for host cell invasion, intracellular growth and parasite egress, focusing on (i) calcium-dependent protein kinases and (ii) the secreted kinases that are unique to Toxoplasma (ROPKs) and Plasmodium (FIKKs).
2. Calcium-dependent regulation of invasion and egress
Calcium levels in T. gondii play key roles in regulating micronemal secretion-dependent processes including host cell invasion, gliding motility and parasite egress (Nagamune et al., 2008). Chelation of parasite intracellular calcium strongly inhibited both microneme release and invasion of host cells, and this effect was partially reversed by raising intracellular calcium using the ionophore A23187 (Carruthers et al., 1999). Additionally, evidence was provided in this study for the requirement of a staurosporine-sensitive kinase activity in regulating microneme discharge and for parasite invasion of host cells.
In Toxoplasma parasites, CDPKs were shown to be involved in host cell invasion through the use of KT5926, an inhibitor of CDPKs in other systems (Kieschnick et al., 2001). KT5926 blocks the motility of Toxoplasma tachyzoites and their attachment to host cells. In vivo, the phosphorylation of only three parasite proteins was found to be blocked by KT5926 (although there may be more), and in parasite extracts, a single KT5926-sensitive protein kinase activity was detected. This activity was calcium-dependent but did not require calmodulin. In a search for CDPKs in Toxoplasma, two members of the class of calcium-dependent protein kinases (CDPKs) were detected. TgCDPK2 was only expressed at the mRNA level in tachyzoites, with no detectable protein. TgCDPK1 protein present in Toxoplasma tachyzoites cofractionated precisely with the peak of KT5926-sensitive protein kinase activity. TgCDPK1 kinase activity was calcium-dependent but did not require calmodulin or phospholipids. TgCDPK1 was found to be inhibited effectively by KT5926 at concentrations that block parasite attachment to host cells. In vitro, TgCDPK1 phosphorylated three parasite proteins that migrated identically to the three KT5926-sensitive phosphoproteins detected in vivo. These observations suggested a central role for TgCDPK1 in regulating Toxoplasma motility and host cell invasion.
Subsequent chemical genetic and conditional knockout studies have shown that TgCDPK1 is the key transducer downstream of calcium fluxes in regulating these processes. Down-regulation of TgCDPK1 expression severely impaired the secretion of micronemal proteins, including adhesins needed for gliding motility (Lourido et al., 2010). The resulting defect in gliding motility led to a significant decrease in host cell attachment and invasion. Conditional knockout of TgCDPK1 also impaired parasite egress from host cells upon induction with a calcium ionophore, with immotile parasites remaining within vacuoles. This coincided with a defect in PV membrane (PVM) permeabilisation upon calcium ionophore treatment, which likely resulted from impaired microneme secretion of the perforin-like TgPLP1 protein (Lourido et al., 2010).
The canonical CDPK domain structure consists of an N-terminal protein kinase domain, that is highly homologous to calmodulin-dependent kinases (CaMKs), and a C-terminal CDPK activation domain (CAD) with four calcium-binding EF-hands (EF1 to EF4). A recent biochemical and structural study of three apicomplexan CDPKs (TgCDPK1 and TgCDPK3 from T. gondii, and CpCDPK1 from Cryptosporidium parvum) has provided novel insights into how these CDPKs are activated upon calcium binding by the CAD (Wernimont et al., 2010). In in vitro assays, the kinase domain was activated by low micromolar calcium concentrations. Calcium ‘in-out’ experiments suggested a partial irreversibility of this calcium-dependent activation for TgCDPK1 and TgCDPK3, while CpCDPK1 activity was reduced to basal level upon calcium removal. Actual mapping of potential auto-phosphorylation sites may provide further insights into the reversibility of calcium-induced activation of these CDPKs.
Crystal structures have been solved for auto-inhibited (calcium-free) TgCDPK1 and TgCDPK3 and for calcium-bound TgCDPK1 and CpCDPK1 (Wernimont et al., 2010). The kinase domain adopts the bi-lobal structure characteristic of eukaryotic protein kinases (ePKs), with a smaller N-terminal lobe (consisting mainly of β-strands) and a larger C-terminal lobe (predominantly α-helical) connected together via a single stretch of polypeptide (hinge region). A deep cleft formed between the two lobes contains the ATP binding pocket and active site (Dar et al., 2005). In the calcium-free state, the CAD adopts an overall dumbbell shape (resembling calmodulin in the absence of calcium) with two EF-hands at each end of the dumbbell. The CAD packs against the front face of the kinase domain, making contacts with both the N- and C-terminal lobes. The N-terminal helix of EF1 extends into a long helix (CH1) that spans the length of the CAD and packs anti-parallel along a second long helix (CH2). The N-terminus of CH1 connects directly with the C-terminus of the kinase domain and packs against the kinase C-terminal lobe to block substrate binding, analogous to the pseudo-substrate segment of CAMKII (Rosenberg et al., 2005) (Fig. 1). Additional hydrophobic contacts with the N-and C-terminal lobes of the kinase domain are made by EF2 and EF3, respectively. Calcium binding to all four EF hands within the CAD causes conformational changes that expose hydrophobic surfaces and partial unwinding and bending of the CH1 and CH2 helices. This leads to a dramatic rearrangement of the CAD into a more compact structure. The calcium-bound CAD is also repositioned and binds to the back face of the kinase domain, which induces a widening of the active site cleft by altering the relative orientations of the two lobes of the kinase domain. Release of the auto-inhibitory interactions between the CAD and kinase domain also allows repositioning of the α-C helix and rearrangement of the P-loop and the activation loop regions into an active conformation as seen in other kinase structures.
Fig. 1.
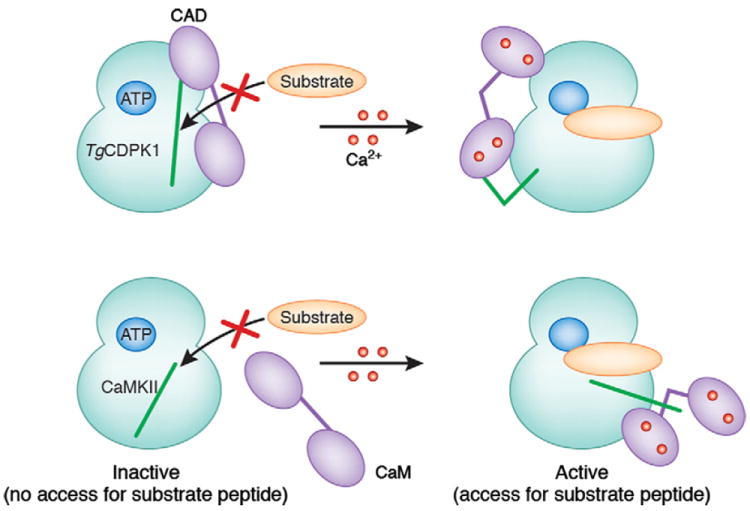
Comparative schematic of activation of Toxoplasma gondii calcium-dependent protein kinases (CDPKs) and calmodulin-dependent kinases (CaMKs). The green lines represent the auto-inhibitory region. CaM, Calmodulin; CAD, CDPK-activating domain. See Section 2 for details. [Reprinted from Doerig and Billker (2010) with permission from Macmillan Publishers Ltd]
Apicomplexan CDPKs have no orthologs in humans and are most closely related to CDPKs in plants. The essential functions of TgCDPK1 and the lack of orthologs in humans make it an ideal target for novel anti-Toxoplasma therapeutic drugs. In particular, TgCDPK1 is unique in having a glycine residue at the gatekeeper position, which is located at the rear of the adenine binding pocket. ATP analogues with bulky substituents on the purine group can be accommodated by analogue-sensitive kinases with small gatekeeper residues (with glycine being the smallest), while kinases with larger gatekeeper residues cannot accommodate these molecules (Bishop et al., 2000; Blethrow et al., 2004). Since few human kinases contain a small gatekeeper residue (no known mammalian protein kinase has a glycine gatekeeper), TgCDPK1 provides a promising analogue-sensitive target for bumped kinase inhibitors (BKIs) that should have relatively little off-target effects on the human kinome (Manning et al., 2002).
A number of BKIs have been developed for chemical genetic studies with engineered analogue-sensitive kinase alleles in human cells. BKI analogues of 4-amino-1-tert-butyl-3-phenylpyrazol-o[3,4-d]pyrimidine, with bulky aromatic substituents at the C3 position, inhibit TgCDPK1 at sub-micromolar concentrations (Ojo et al., 2010; Sugi et al., 2010). Consistent with conditional TgCDPK1 knockout studies, BKIs greatly reduced host cell invasion by T. gondii and modestly affected intracellular growth. Parasites expressing an analogue-resistant TgCDPK1, in which the gatekeeper glycine is mutated to methionine (G128M), were significantly less sensitive to BKIs and thus validated TgCDPK1 as the main BKI target in vivo. Crystal structures of calcium-free TgCDPK1 in complex with NA-PP2 and NM-PP1 confirm the structural basis for the selectivity of these BKIs for a small gatekeeper residue and will facilitate optimisation of these BKIs to further improve their affinity and specificity (Ojo et al., 2010). Of the 114 functional kinases predicted in the T. gondii genome, 12 are expected to be analogue-sensitive based on their gatekeeper residue (Sugi et al., 2010). Thus, BKIs provide a promising approach to targeting key processes in T. gondii with potentially fewer side effects on the host.
That host cell invasion can be inhibited by staurosporine and involves calcium signalling has also been known to be true for malaria parasites for 2–3 decades (Scheibel et al., 1987; Ward et al., 1994). More recently, a role for a CDPK (PfCDPK1) in this process has been revealed (Green et al., 2008; Kato et al., 2008), highlighting a definite level of unity in the invasion strategies across the Apicomplexa. In addition to invasion, malaria parasites exploit their repertoire of CDPKs to mediate several other developmental processes (see Billker et al. (2009) for a comprehensive review). Briefly, these include male gametogenesis, shown in the rodent-infecting parasite P. berghei to be dependent on PbCDPK4 (Billker et al., 2004); gliding of ookinetes, the motile form that escapes the mosquito midgut during infection of the insect vector, a process blocked in P. berghei parasites lacking PbCDPK3 (Ishino et al., 2006); and switching of parasite behaviour from cell traversal to cell invasion at the early stage of liver infection by P. berghei sporozoites, which requires PbCDPK6 (Coppi et al., 2007). There is unity across the Apicomplexa not only with respect to the involvement of CDPKs in invasion, but also in the mechanism of activation of these enzymes by calcium (see above). A recent structural study of a Plasmodium CDPK (PfCDPK3) (Wernimont et al., 2011) revealed that the residues mediating the largest conformational change following calcium binding are the most conserved across the Apicomplexa, leading the authors to propose that the mechanism described in the section above is conserved, presumably throughout the CDPK family. Another interesting observation described in this report is that although the CAD shares similar motifs and behaviour with calmodulin, there are sufficient structural differences to consider the CDPK activation domain as a distinctive member of the large EF-hand family of calcium-binding proteins. Given their key regulatory roles, malaria CDPKs also present attractive drug targets. However, in contrast to the TgCDPKs, the presence of non-glycine residues at the gatekeeper position in malaria CDPKs will preclude the use of BKIs.
3. Host modification by secreted Plasmodium and Toxoplasma protein (pseudo)kinases
Unlike most other ePKs, most members of the Plasmodium FIKK and Toxoplasma ROPK families have signal peptides and are predicted to be secreted into the host cell. A recent analysis of the completed genome sequence of the three most common strains of T. gondii by Roos and colleagues found that the Toxoplasma kinome consists of 108 predicted active kinases and 51 predicted pseudokinases (Peixoto et al., 2010) (Fig. 2). Compared with the human kinome, which only contains ~10% pseudokinases (Manning et al., 2002), there is a significant enrichment of pseudokinases in the Toxoplasma kinome. This is mainly due to an expansion of the Toxoplasma-specific ROPK family, consisting of ~44 genes of which approximately half are pseudokinases. It must be considered in this context that some proteins thought to be pseudokinases on the basis of their sequence characteristics were actually shown to be competent for phosphorylation (Kannan and Taylor, 2008; Taylor and Kornev, 2010). This may be particularly relevant to sequences that are relatively distant from classical ePKs, such as the ROPKs and FIKKs (see below).
Fig. 2.
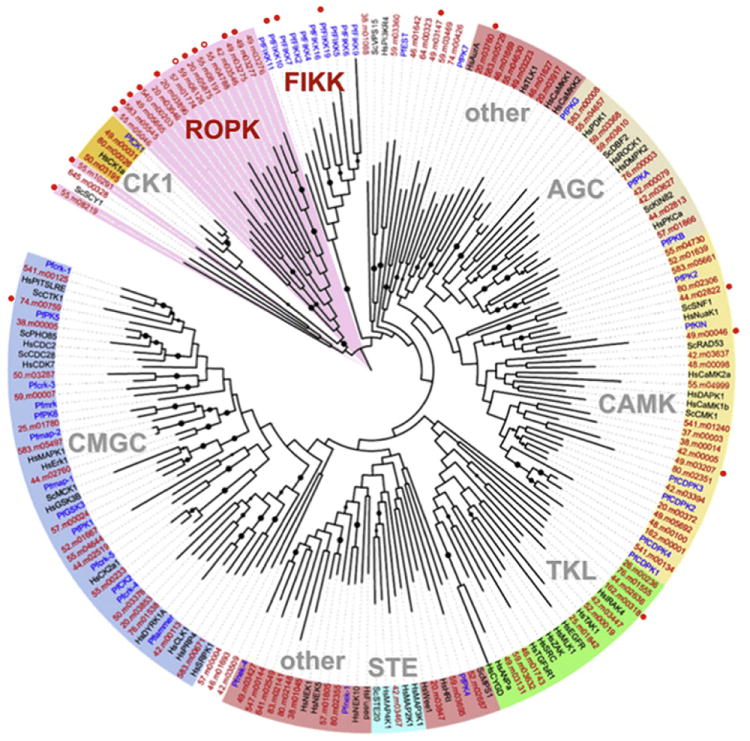
The Toxoplasma kinome. Classification of 108 active kinases predicted from the Toxoplasma gondii genome. Black, human and yeast; blue, Plasmodium falciparum; red, T. gondii. Coloured arcs highlight major kinase groups: AGC, CMGC, CAMK, TKL, CK1 and STE. Red lettering, apicomplexan-specific groups ROPK (pink) and FIKK. Red circles, kinases with predicted secretory signal sequence or signal anchor (open, newly recognised); black dots, bootstrap support >50%. [Reprinted from Peixoto et al. (2010) with permission from Elsevier.]
Members of both the ROPK and FIKK families are characterised by a domain structure consisting of an N-terminal signal peptide, a low complexity region and a conserved C-terminal domain that adopts a canonical protein kinase fold. However, Toxoplasma ROP kinase domains constitute a phylogenetically-distinct group from other ePKs, including other apicomplexan kinases such as the malaria FIKK family. Proteins of the ROPK family have roles in host cell modulation and virulence, although the molecular details of how most of these proteins function are poorly understood. While some members of the ROPK family have been shown to be active kinases (such as ROP16 (Yamamoto et al., 2009; Ong et al., 2010), and ROP18 (El Hajj et al., 2007; Khan et al., 2009b)), others appear to be pseudokinases with substitutions at key residues that abrogate catalytic activity (but see our comment above). The inability of ROP2 to bind ATP has also been reported and may be true for other ROPK family pseudokinases (Labesse et al., 2009).
Twelve sequence motifs or sub-domains are highly conserved in protein kinase domains (Hanks et al., 1988). Most of these sub-domains are clustered within the active site cleft region and are important for ATP binding and catalysis, while others are crucial for structural stability of the protein. The crystal structures of the ROP2 (Protein Data Bank (PDB) accession codes 2W1Z and 3DZO) and ROP8 (3BYV) pseudokinase domains revealed a number of structural features within their active site region that are incompatible with catalysis and ATP binding (Labesse et al., 2009; Qiu et al., 2009). Substitution of the VAIK motif with FEVH in both ROP2 and ROP8 results in loss of the critical lysine side chain that normally coordinates the α- and β-phosphates of ATP, however, WNK is an example of a kinase that lacks this lysine residue but is nevertheless active, through recruiting another residue to substitute for the missing one (Xu et al., 2000). Replacement of the DFG motif with GFE alters the magnesium coordination geometry, while substitution of the HRD motif with HTY results in replacement of the catalytic base (asparagine) with tyrosine.
3.1. Cellular destination of Toxoplasma rhoptry kinases
Although it is now well established that upon invasion Toxoplasma secretes the contents of its rhoptries directly into the host cytosol, it is still unclear how these secreted proteins cross the host membrane. Patch clamp experiments have shown a short spike in conductivity at the time of invasion, which is suggestive of a break in the host membrane followed by resealing (Suss-Toby et al., 1996). The Toxoplasma protein(s) that mediate this break remain unknown. Recently, two Toxoplasma proteins with a Membrane Attack Complex/Perforin (MACPF) domain were identified, one of which (TgPLP1) is necessary for egress, which involves rupture of the PVM (Kafsack et al., 2009). TgPLP2 could be a candidate for mediating the break in the host cell plasma membrane. Once rhoptry proteins are secreted into the host cell cytosol they traffic to distinct cellular destinations based on other signals. For example the rhoptry kinase ROP16 and the rhoptry protein phosphatase 2 C (PP2C-hn) carry a nuclear localisation signal (NLS), which mediates their trafficking to the host nucleus (Gilbert et al., 2007; Saeij et al., 2007). Many ROPKs traffic back to the PVM and it was recently demonstrated that an arginine-rich amphipathic helix (RAH) domain is necessary and sufficient for targeting ROPKs to the PVM (Reese and Boothroyd, 2009). Transgenic expression of the RAH domain in host cells results in punctuate staining with some overlap with the nuclear envelope. However, after Toxoplasma infection of those cells, most of the staining relocated to the PVM (Reese and Boothroyd, 2009). The fact that the RAH domain only interacts weakly with a variety of host membranes but strongly with the PVM, which is derived from the host membrane, is puzzling. It has been suggested that the RAH domain has no special affinity for a particular lipid composition but rather for negative membrane curvature as the host nuclear envelope, which is negatively curved, associated somewhat with RAH domain (Reese and Boothroyd, 2009). Indeed, it is well known that Toxoplasma secretes proteins from its dense granules that are involved in the formation of a tubulovesicular network consisting of elongated negatively curved nanotubules of 60–90 nm in diameter, which are topologically cytosolic and connect with the vacuole-delimiting membrane. Consistent with this, parasites without the dense granule protein GRA2, which have an attenuated PVM tubular network, have attenuated recruitment of the RAH domain to the PVM (Reese and Boothroyd, 2009). Thus, an important function of dense granule proteins might be the formation of the extremely negatively curved tubular network, which subsequently functions to attract rhoptry proteins with a RAH domain from the cytosolic face of the PVM. Rhoptry proteins without a RAH domain or an NLS, such as Toxofilin, stay in the host cell cytosol (Lodoen et al., 2010).
3.2. Toxoplasma rhoptry kinase ROP16 modulates STAT signalling
Although it has been known for some time that Toxoplasma ROPKs can be secreted into the host cytosol, their function has remained elusive. ROP2 was thought to mediate the recruitment of mitochondria to the PVM, but a recent study showed that a ROP2 knockout has no defect in mitochondrial recruitment (Pernas and Boothroyd, 2010). One of the first ROPKs for which a particular function was ascribed was ROP16. Expression profiling of human foreskin fibroblasts (HFFs) infected with types I, II or III parasites demonstrated that these strains differ significantly in the modulation of host gene expression (Saeij et al., 2007). To identify the Toxoplasma genomic loci involved, expression profiles of HFFs infected with F1 progeny derived from crosses between types II and III were determined. Quantitative trait locus (QTL) analysis, using host gene expression as the quantitative trait, determined that the genotype at a locus on Toxoplasma chromosome VIIb correlated with the strain-specific regulation of >1,000 human genes. Analyses for enrichment in functional annotation of these genes implicated the STAT signalling pathway. Indeed HFFs infected with type I or type III parasites have strong and sustained activation of STAT3 and STAT6 while the activation by type II is much weaker and only transient. A candidate gene approach identified ROP16 as the Toxoplasma gene responsible for the strain-specific activation of STAT3 and STAT6. ROP16 is secreted into the host cytosol upon infection and subsequently traffics to the host nucleus. This nuclear translocation is dependent on an NLS in ROP16 but its nuclear localisation was not important for the activation of STAT3 and STAT6. Subsequently, these experiments were confirmed by other groups using a type I strain where the ROP16 gene was removed by double homologous recombination (Yamamoto et al., 2009; Ong et al., 2010). As expected, ROP16 kinase activity is necessary for its effect on STAT3 and STAT6 (Yamamoto et al., 2009; Ong et al., 2010). Initially it was predicted from sequence homology that ROP16 was a serine/threonine kinase and was therefore unlikely to be responsible for the tyrosine phosphorylation of STAT3 and STAT6. However, it has recently been shown that it can in fact directly phosphorylate these targets (Yamamoto et al., 2009; Ong et al., 2010). One group showed that immunoprecipitated ROP16 can phosphorylate Tyr705 of recombinant STAT3 and subsequent co-immunoprecipitation experiments determined that the N-terminal region (amino acid 223–303) of ROP16 is required for this interaction with STAT3. By constructing chimeras between the types I and II ROP16 proteins, the difference in STAT activation by these two alleles could be attributed to a single amino acid substitution; a conversion of serine to leucine at position 503 also made type II ROP16 a potent activator of STAT3.
Similarly, Boothroyd and colleagues showed that ROP16 has intrinsic tyrosine kinase activity as determined by phosphoamino acid analysis of auto-phosphorylated ROP16. In vitro kinase assays with recombinant ROP16 and recombinant STAT6-GST as a substrate also showed efficient phosphorylation of Tyr641 on STAT6 by wild-type but not K404N (kinase-dead) recombinant ROP16. The pan-JAK inhibitor and the tyrosine kinase inhibitor K-252a severely impaired the kinase activity of ROP16, providing further evidence that ROP16 is a tyrosine kinase (Yamamoto et al., 2009; Ong et al., 2010). Thus, ROP16 can directly phosphorylate Tyr705 and Tyr641 from STAT3 and STAT6, respectively. This could explain why the up-regulation of the suppressor of cytokine signalling (SOCS) genes, which normally down-regulate the STAT pathway (Dalpke et al., 2008), do not down-regulate the STAT3/6 activation by ROP16 (Saeij et al., 2007). SOCS proteins bind to phosphorylated tyrosine residues on Janus kinases (JAKs) and/or cytokine receptor subunits through a central SH2 domain and subsequently mediate their degradation (Dalpke et al., 2008). Because the JAKs and cytokine receptors are bypassed by the direct phosphorylation of STAT3 and STAT6 by ROP16, the inhibitory function of SOCS proteins is abrogated. By directly phosphorylating STAT3 Toxoplasma could mimic the effects of IL-10, which also maintains constitutive activation of STAT3 because its receptor does not interact with the SOCS proteins (Yasukawa et al., 2003). IL-10 is a potent anti-inflammatory cytokine which can down-regulate the production of IFN-γ, the main mediator of resistance to Toxoplasma. The prediction would therefore be that ROP16 can enhance parasite virulence by suppressing IFN-γ production.
Indeed, ROP16 was also identified as one of the loci involved in the difference in virulence between types II and III (Saeij et al., 2006). However, addition of ROP16 from type I or type III into a type II strain made it less virulent. Why type II + ROP16I/III parasites have reduced virulence is currently unknown. Although ROP16I/III can lower the level of secretion of IL-12 by macrophages infected with type II strains, which could lead to subsequent lower induction of IFN-γ secretion, one would predict that this would lead to an enhanced and not a reduced virulence phenotype.
A potential explanation for this apparent contradiction is emerging from our recent studies showing that macrophages infected with type I or type III strains are converted into an alternatively activated or M2 macrophage (Jensen et al., 2011). These M2 macrophages are characterised by high level expression of arginase-I and several lectin receptors such as the mannose receptor and macrophage galactose specific lectin. Type II infected macrophages are converted into classically activated or M1 macrophages and secrete large amounts of proinflammatory cytokines such as IL-12p70 and IL-23. Activation of STAT6 by ROP16 is necessary to convert macrophages to the M2 phenotype, while the M1 phenotype is due to the fact that the type II strains, but not the types I and III, activate NFκB, a phenotype due to a new polymorphic dense granule protein GRA15 (Rosowski et al., 2011). Thus, it is possible that one of the roles of ROP16 is to limit the inflammation induced by the strong Th1 induction by Toxoplasma and the reduction of virulence of the II + ROP16I might be due to reduced immune pathology. We have evidence that ROP16-mediated reduction of type II virulence is especially pronounced in Th1-prone mice such as C57/BL6 (J. Saeij, unpublished data). Alternatively, the high induction of the arginase enzyme by ROP16 could deplete l-arginine, for which T. gondii is an auxotroph, and thereby inhibit Toxoplasma growth (Butcher et al., 2011).
3.3. Toxoplasma rhoptry kinase ROP18
ROP18 was identified as a rhoptry kinase involved in virulence by two different groups (Saeij et al., 2006; Taylor et al., 2006). Sibley and colleagues were interested in the genes involved in the difference in virulence between the highly virulent type I strain (LD100 = 1) and the avirulent type III strains (LD50 ~ 100,000) (Taylor et al., 2006). Boothroyd and colleagues used the difference in virulence between types II (LD50 ~ 500) and III as the basis for their studies (Saeij et al., 2006). Both groups used crosses between these strains to generate F1 progeny which were subsequently injected into mice to determine their virulence phenotypes. A genetic map was previously generated for Toxoplasma (Khan et al., 2005) and this was used to determine which Toxoplasma loci correlated with virulence. Interestingly, the only locus identified using the Ix-III cross F1 progeny was ROP18 and indeed a type III strain expressing type I ROP18 (III + ROP18I) was as virulent as a wild-type type I strain (Taylor et al., 2006). ROP18 kinase activity was necessary to confer virulence as the type III expressing the type I kinase dead mutant was avirulent. The difference in virulence between types II and III was more complicated and implicated five different loci, one of which was ROP18. A type III strain expressing type II ROP18 (III + ROP18II) became extremely virulent (LD50 ~ 5) (Saeij et al., 2006). The expression level of ROP18 seems to be a key determinant in the strain-specific differences in virulence. Type III strains have an extremely low level of ROP18 expression, probably due to an extra 2.1 kb sequence 85 bp upstream of the ATG start codon while types I and II express ROP18 at a high level (Saeij et al., 2006).
Although both studies demonstrated that ROP18 is secreted into the host cytosol and subsequently traffics back to the PVM, its substrate(s) remained unidentified. When ROP18 is over-expressed in host cells it also traffics to the PVM of invading parasites, showing that it has a strong affinity for the PVM (El Hajj et al., 2007; Reese and Boothroyd, 2009). Like many rhoptry proteins, ROP18 is proteolytically processed. It is initially present as a 60 kDa protein but is subsequently processed to a 56 kDa protein. It contains the conserved S-Phe-X-E consensus motif (Phe represents bulky hydrophobic residues and X is any amino acid) for recognition by TgSUB2, a subtilisin-like serine proteinase (El Hajj et al., 2007). When comparing the ROP18 genes from multiple strains it was noted that it is one of most polymorphic proteins, with evidence for strong diversifying selection. For example, the sequences of types I and II ROP18 are very different, with almost no synonymous changes between the types I and II alleles. Currently there is no evidence for a different function of the different ROP18 alleles, because quite diverse ROP18 alleles from several strains all confer virulence to type III parasites when expressed in this avirulent strain (Khan et al., 2009b). It is also unknown whether over-expression of the type III ROP18 itself would also confer virulence. Comparison of the ROP18 genomic region between Toxoplasma and Neospora caninum, a close relative of T. gondii, demonstrated that Neospora also has the extra region in the promoter of its ROP18. Thus, the most parsimonious explanation is that the ancestral ROP18 gene contained the extra sequence in its promoter, resulting in low level expression and subsequently this region was deleted to give rise to the more recently derived types I and II ROP18 alleles (Khan et al., 2009b). It was also hypothesized that strains carrying the type I ROP18 allele might be more successful because more strains have a ROP18 allele similar to the type I strain (Khan et al., 2009b); however this could also be due to other strains being more similar to type I strains compared with type II strains.
3.3.1. Proposed regulatory mechanism of ROP18
Protein kinases display a diverse array of regulatory mechanisms that exploit multiple ways of impeding or distorting the binding of ATP, magnesium or protein substrates to prevent catalysis. This often involves a distortion of the inter-lobe orientation, conformational change of the activation loop to block ATP and/or substrate binding, or occlusion of the active site. Activation of the kinase domain often involves phosphorylation (particularly of the activation loop), but may also involve other mechanisms such as binding or dissociation of regulatory domains or subunits (Huse and Kuriyan, 2002; Dar et al., 2005).
The ROP2 and ROP8 kinase domain crystal structures (PDB accession codes 3DZO and 3BYV) revealed a unique N-terminal subdomain that is conserved in other ROPK family kinase domains and contains features that preclude ATP binding in two of the ROPK pseudokinase structures determined. This ~40 residue N-terminal extension consists of an α-helical segment (packed against the C-terminal lobe) connected via a short linker region to an α-helix and β-strand packed against the N-terminal lobe. In both 3DZO and 3BYV, an arginine side chain from the N-terminal subdomain linker region (Arg 219 in 3DZO and Arg 228 in 3BYV) forms a salt bridge interaction with the side chain of Glu 284 in 3DZO and Glu 293 in 3BYV. The glutamate and arginine side chains occupy the expected position of the ATP adenine group. Based on a homology model of the ROP18 kinase domain (with the ROP8 crystal structure as a template), a novel kinase regulatory mechanism was proposed for ROP18, in which the N-terminal subdomain was predicted to impede ATP-binding (as observed in 3DZO and 3BYV). Furthermore, phosphorylation at sites within and near the ROP18 N-terminal subdomain was proposed to regulate this auto-inhibition (Qiu et al., 2009).
Similar to other active protein kinases and in contrast to the ROP2 and ROP8 structures, the modelled active site of ROP18 was free of bulky side chains that would preclude ATP binding, with the exception of Gln 214 and Gln 216. The side chains of both residues were predicted to protrude into the ATP binding pocket from the N-terminal subdomain linker region (analogous to Arg 219 in 3DZO and Arg 228 in 3BYV). Consistent with the homology model, mutation of Gln 214 and Gln 216 to Ala resulted in a three to four fold increase in in vitro auto-phosphorylation and phosphorylation of myelin basic protein by recombinant ROP18 kinase domain. Activation was then suggested to involve a displacement or conformational change of the N-terminal subdomain to move the side chains of Gln 214 and Gln 216 out of the ATP binding pocket. ROP18 kinase domain expressed in Escherichia coli was auto-phosphorylated on Ser 221 and Thr 229 (second helix of the N-terminal subdomain), and on Thr 249 and Thr 251 (in contact with the N-terminal subdomain β-strand). Mutation of these Ser and Thr residues to Ala, reduced in vitro kinase activity by up to 90% (Qiu et al., 2009). It was therefore proposed that phosphorylation of one or more of these Ser/Thr residues activated ROP18 by altering the interaction of the N-terminal subdomain with the N-terminal lobe to shift the linker region containing Gln 214 and Gln 216 away from the ATP binding pocket. However, several aspects of this proposed auto-inhibitory mechanism require further investigation. Comparison of the available crystal structures, suggests that loss of the salt bridge interaction between the glutamate side chain within the adenine binding pocket (Glu 284 in 3DZO and Glu 293 in 3BYV) and the arginine side chain on the N-terminal subdomain linker (Arg 219 in 3DZO and Arg 228 in 3BYV) displaces the N-terminal subdomain linker away from the active site as can be seen in 2W1Z. Since this salt bridge interaction is also not conserved in ROP18, it is unclear if the N-terminal subdomain linker in ROP18 adopts the auto-inhibitory conformation as seen in 3DZO and 3BYV. The auto-phosphorylation sites within the ROP18 N-terminal subdomain were mapped using recombinant protein, and whether these sites are phosphorylated in vivo and even the extent to which these sites were auto-phosphorylated in vitro were not reported. Kinase assays of recombinant ROP18 following phosphatase treatment were not presented, but may help to verify the importance of auto-phosphorylation in activating ROP18. Given the importance of the N-terminal subdomain to the proper folding of ROP18, it remains formally possible that the N-terminal Ser/Thr to Ala mutants decreased kinase activity by destabilising ROP18 structure. Conversely, the effect of phospho-mimicking mutations of these N-terminal Ser/Thr residues to Asp or Glu on kinase activity were also not reported, but may provide a useful alternative means to test the importance of these phosphorylation sites in activating ROP18.
3.3.2. What are the ROP18 substrates?
The effect of ROP18I on type III virulence could be due to its effect on parasite growth; vacuoles with type III + ROP18I have on average twice as many parasites at 40 h p.i. compared with vacuoles with wild-type type III strain parasites (El Hajj et al., 2007). This effect of ROP18 on parasite growth seems to be vacuole autonomous as only parasites in vacuoles of type III + ROP18I have a shorter replication time even if they are next to a vacuole with a wild-type type III strain. Because this result was obtained in HFFs, possible host targets for ROP18 should be present in non-stimulated HFFs and are most likely present on the vacuole itself. Besides parasite proteins such as rhoptry proteins and dense granule proteins, several host proteins have also been described to relocalise to the PVM.
The most interesting potential substrates, at least in murine cells, would be the immunity-related GTPases (IRGs). These small GTPases are related to dynamins but are highly induced upon IFN-γ stimulation of cells and subsequently traffic to the PVM where they mediate dynamin-like vesiculation and subsequent rupture of the PVM and eventually the destruction of the parasite (Martens et al., 2005; Ling et al., 2006; Melzer et al., 2008). The accumulation of IRGs on the parasite PVM is also a strain-specific phenotype; type I vacuoles seem to resist high accumulation of these IRGs and type I vacuoles are resistant to destruction, while types II and III vacuoles accumulate high levels of the IRGs and are effectively destroyed by IFN-γ (Zhao et al., 2009a,b). To investigate whether ROP18 had a role in this strain-specific difference, the accumulation of IRGs on vacuoles of type III infected cells was compared with vacuoles of type III + ROP18I but no difference in accumulation of ROP18 was observed (Zhao et al., 2009a). Similarly, transfection of ROP18I or ROP18II into IFN-γ-induced L929 fibroblasts had no effect on the accumulation of IRGs on type II vacuoles (Khaminets et al., 2010).
In contrast to the above mentioned studies it was recently reported that ROP18I can bind to and directly phosphorylate Irga6, Irgb6 and Irgb10 (Fentress et al., 2011; Steinfeldt et al., 2011), thereby preventing their accumulation on the PVM. For Irga6 it was demonstrated that ROP18 can phosphorylate two threonine residues in the switch I loop (T102 and T108) (Steinfeldt et al., 2011), while Irgb6 was mainly phosphorylated on a single threonine residue in its switch I loop (Fentress et al., 2011). Because these threonine residues are at the catalytic interface that is essential for GTP-dependent active dimer formation, it is likely that their phosphorylation inactivates the protein. Indeed, phospho-mimetic mutants of Irga6 were inefficiently loaded onto the PVM and could inhibit loading of wild-type Irga6 (Steinfeldt et al., 2011). Antibodies specific for pT102 and pT108 demonstrated that type I vacuoles contained significant amounts of pT102 and pT108. Type II vacuoles did not contain any pT102 but did contain pT108, although significantly less compared with type I strains. Whether the phosphorylation of T108 is mediated by type II ROP18 remains to be determined. The phosphorylation of Irgb6 is of particular interest as this is one of the first IRGs recruited to the vacuole and it has been proposed to mediate the subsequent recruitment of other IRGs. Knockdown of Irgb6 resulted in significantly less killing of Toxoplasma by IFN-γ stimulated macrophages.
It was speculated that the difference between the studies reporting an effect of ROP18 and studies reporting no effect (all studies were performed by the same two groups) was the IFN-γ concentration; only at a low (<1 U/ml) IFN-γ concentration was an effect of ROP18I expressed in type III on Irga6 loading on the vacuole detectable. This probably indicates that other polymorphic parasite proteins play a role in resistance to the IRG system, because type I strains can inhibit IRG loading even after stimulation of 200 U/ml IFN-γ. The specific cell type used might also be important. Sibley and colleagues demonstrated that prevention of IRG accumulation on vacuoles of parasites expressing ROP18I prevents the killing of Toxoplasma by GR1+ inflammatory monocytes or IFN-γ stimulated macrophages (Fentress et al., 2011).
That ROP18 is not the only factor responsible for reduced IRG loading might also explain why type III + ROP18II are as virulent as type III + ROP18I, while the PVM of the type II strain itself is efficiently coated by the IRGs. Possibly the type II strain lacks another polymorphic Toxoplasma protein, present in both types I and III, that is necessary for ROP18 activity in vivo or it could contain a protein that somehow blocks ROP18 activity. Furthermore, it is unlikely that the IRGs are the only substrates for ROP18 because these IRGs are not expressed in human cells or non-stimulated cells and the effect of ROP18 on parasite growth was reported in un-stimulated HFFs. Based on the ability of bacterially-expressed and re-folded ROP18 kinase to phosphorylate 70 kDa and 68 kDa proteins in heat-inactivated parasite lysates, it has been suggested that ROP18 can also phosphorylate parasite-derived proteins (El Hajj et al., 2007). It is possible that these proteins are in fact ROP2, ROP8 or ROP18 itself, as bacterially-expressed versions of those proteins were also shown to be phosphorylated by ROP18. However, whether these proteins are bona fide substrates in an infected cell remains unknown.
A number of unbiased strategies may be pursued for the identification of substrates or interactors of ROPK family active kinases and pseudokinases (Sopko and Andrews, 2008). Engineering a kinase of interest (by mutation of its gatekeeper residue to Gly or Ala, see below) to accept ATPγS analogues containing bulky substituents, allows chemical tagging of its direct substrates. Subsequent alkylation of the thiophosphate groups allows isolation of the tagged substrates by a thiophosphate ester-specific antibody for identification by mass spectrometry (Allen et al., 2007). Due to the cell impermeability of ATP analogues, substrates must be thiophosphorylated in lysates, where the relative spatial organisation of kinases and their substrates can be lost, potentially leading to false positives (Blethrow et al., 2004). Additionally, not all kinases are compatible with gatekeeper mutations required for analogue sensitivity and/or with thiophosphorylation. A potential problem for the Toxoplasma kinome discussed earlier in the present review is the presence of a number of endogenous analogue-sensitive kinases, such as TgCDPK1, that inherently have small gatekeeper residues, which may further complicate this approach (Sugi et al., 2010). An alternative method of kinase substrate identification utilises mass spectrometry to detect changes in the abundance of phospho-sites due to the knockout of a kinase or treatment with a specific small molecule inhibitor, if available (Sopko and Andrews, 2008). Due to the effects on possible downstream kinases, substrates identified by this approach may not be direct, although it may be possible to filter out such indirect “false positives” with knowledge of the phosphorylation motif of the kinase of interest. The use of a positional-scanning peptide array (Hutti et al., 2004) to define the substrate preference of ROP18 was recently published (Fentress et al., 2011). Other approaches to identify kinase substrates that are also amenable to identifying interactors of pseudokinases include yeast 2-hybrid and co-purification of ligands/substrates with a tagged kinase or pseudokinase from cell lysates (Sopko and Andrews, 2008). The sensitivity of yeast 2-hybrid can facilitate the detection of transient kinase–substrate interactions, which may be stabilised by the use of a catalytically-inactive kinase bait.
3.4. ROP5, a pseudokinase essential for Toxoplasma virulence
The analysis of virulence of the F1 progeny from II × III crosses identified a locus on Toxoplasma chromosome XII as having the largest contribution to the strain-specific differences in virulence. Interestingly, the avirulent type III strain contributed to the enhanced virulence while the more virulent type II strain contained the avirulent locus (Saeij et al., 2006).
It was recently demonstrated that the pseudokinase ROP5 is responsible for this difference in virulence (Reese et al., 2011). Initially, a completely avirulent F1 progeny (S22, LD50 >1 × 106 para-sites) was complemented with a type I cosmid containing ROP5 and a four log increase in virulence was noted. However, because the cosmid contained two other genes, a refined analysis was needed. Therefore the ROP5 locus was deleted in a type I strain, and surprisingly this strain was completely avirulent (LD50 >1 × 106) (Reese et al., 2011). The ROP5 locus is quite complicated and it seems that type II strains contain 10 copies of ROP5, type I: four copies and type III: six copies. These copies consist of at least three different alleles for each strain, with the types I and III alleles being the most similar. Complementation of the type I knockout with one allele increased virulence, while only complementation with two or more alleles converted the virulence to wild-type virulence. The crystal structure of the ROP5BI kinase domain showed a canonical protein kinase domain fold and is highly similar to the crystal structures of the ROP2 and ROP8 pseudokinase domains (Reese and Boothroyd, 2011). However ROP5 was predicted to be inactive due to the substitution of the Asp residue within the HRD motif (catalytic base) with a basic residue (Lys or His depending on the ROP5 allele). Consistent with this, no detectable kinase activity was reported for recombinant ROP5 protein in vitro. The ROP5 pseudoactive site is able to bind ATP and the crystal structure revealed a distorted ATP binding mode, due to an unusual positioning of magnesium coordination sites. Comparison of the ROP5BI pseudokinase domain crystal structures with and without ATP bound revealed no major conformational changes upon ATP binding. ROP5 is also not predicted to be processed because it lacks the subtilisin cleavage motif. Sibley and colleagues recently made a cross between a strain from types I and II and reported that a single QTL, containing the ROP5 gene cluster, controls virulence difference between these strains (Behnke et al., 2011). Indeed, through similar studies as described above they determined that ROP5 determines the differences in virulence between the types I and II. The fact that ROP18 did not determine virulence differences between types I and II indicates that types I and II ROP18 seem to be functionally equivalent. Although only a single QTL was identified, this does not mean that other genes are not involved in determining virulence differences between the types I and II. For example, some of the F1 progeny from the I×II cross contain both the virulent ROP5 and ROP18 alleles but are not 100% lethal, indicating that other genes must be involved (Behnke et al., 2011).
The mechanism underlying ROP5-mediated virulence is currently unknown. Restoration of the catalytic base within the HRD motif by substitution of the basic residue in the ROP5AIII allele with Asp resulted in significantly reduced virulence in mice. However the equivalent substitution in the ROP5BI allele did not restore in vitro catalytic activity in recombinant protein, and the basis for the reduced virulence conferred by the ROP5AIII R389D mutant is unclear (Reese and Boothroyd, 2011). To compare the contribution of ROP5 to virulence with that of ROP18, a ROP18 knockout strain was also created. Surprisingly, the type I ROP18 knockout strain was as virulent as the wild-type strain and only a slight delay until death was noted. This seems to contradict the important role of ROP18 in virulence that was previously demonstrated by complementation of the avirulent type III strain, and it also raises the question of the importance of evasion of the IRG system. Sibley and colleagues also found that although the RHROP18 KO had a delayed time-till-death phenotype, it was still 100% lethal (Behnke et al., 2011). It should be noted, however, that ROP18 was knocked out in the RH type I strain while the QTL analysis of virulence was performed using the GT1 type I strain. It was recently noted that these strains behave quite differently; RH has a faster duplication time, it survives much better extracellularly and a significant number of parasite genes is differentially expressed compared with GT1 (Khan et al., 2009a). It would therefore be interesting to remove ROP18 in the GT1 background and investigate its virulence phenotype.
Although the ROPK family pseudokinases are not expected to directly phosphorylate target proteins, they may play scaffolding roles in allosteric regulation of active kinases or mediating kinase–substrate interactions (Zeqiraj and van Aalten, 2010). The crystal structure of the LKB1-STRADα-MO25α ternary complex revealed the structural basis of how the STRADα pseudokinase helps to maintain the LKB1 kinase in an active conformation. LKB1 interacts as a pseudosubstrate with regions of STRADα that correspond to the substrate binding site in active protein kinases (Zeqiraj et al., 2009). Consistent with this theme, the substrate binding loops of ROP2 and ROP8 are well ordered and structurally conserved between the 3DZO and 3BYV crystal structures. However, differences in the surface electrostatic potentials in the ROP2 (partly neutral and negative) and ROP8 (highly positively charged) pseudosubstrate binding regions suggest different ligand specificities (Qiu et al., 2009).
3.5. Toxoplasma rhoptry kinase ROP38 modulated MAPK signalling
After analysing the complete Toxoplasma kinome, Roos and colleagues further characterised the ROP38 gene for the following reasons: it was predicted to be an active kinase, it was differentially expressed between strains (up >64 fold in type III; >8 fold in type II compared with type I), it was induced upon differentiation from tachyzoites to bradyzoites, it was triplicated in both Toxoplasma and Neospora, and there was evidence for convergent evolution of Toxoplasma ROP38 and its Neospora homologue (Peixoto et al., 2010). Because its expression is very low in type I strains they decided to study its function by over-expressing HA-tagged type I ROP38 using a tubulin promoter in a type I strain. HA-tag staining of infected cells confirmed co-localisation of ROP38 with ROP2, indicating that it is indeed a rhoptry protein. ROP38 was also observed on the PVM, which contrasts with a previously published report that it does not traffic to the PVM (ROP38 corresponds to ROP2L5 in that paper) (Reese and Boothroyd, 2009). It is possible that the over-expression of ROP38 resulted in mislocalisation and definitive localisation will depend on staining with an antibody recognising endogenous ROP38. To determine if ROP38, like ROP16, modulates host cell gene expression, host microarrays were performed after infection with type I + ROP38I or wild-type type I. Interestingly, expression of the ROP38 transgene was able to suppress a large proportion of genes that are normally regulated by type I infection. Preliminary experiments suggested that ROP38 might modulate the MAPK pathway, as the kinetics of ERK phosphorylation were different in type I-infected versus type I + ROP18I infected cells. However, this difference in the modulation of host gene expression seemed to have no consequences for virulence in mice as type I over-expressing ROP38 was as virulent as wild-type type I.
3.6. The Plasmodium kinome
The P. falciparum kinome has been reported to comprise 85 or 99 enzymes, depending on the stringency applied for inclusion of borderline sequences (Ward et al., 2004; Anamika and Krupa, 2005). Both studies concur to present a picture of the Plasmodium kinome as being characterised by significant divergences from the yeast or metazoan kinomes (reviewed in Doerig and Meijer (2007) and Doerig et al. (2010).
Most established ePK groups have members in Plasmodium, although TyrK (tyrosine protein kinases) and STEs (a group of PKs involved in mitogen-activated protein kinase (MAPK) pathways) are not represented. The Plasmodium kinome comprises two atypical MAPKs, but the absence of typical MAPKKs suggests that activity of these two enzymes is regulated in a way that differs from that found in classical eukaryotic MAPK pathways. These atypical MAPKs are but an example of a number of plasmodial PKs that can be classified as belonging to established ePK families, but cannot be assigned precise orthology with specific mammalian enzymes (for examples, see Dorin et al. (1999, 2001), Reininger et al. (2005, 2009, 2011), Fennell et al. (2009), Abdi et al. (2010), Halbert et al. (2010), Agarwal et al. (2011)). The Plasmodium kinome also includes many enzymes that do not cluster with any of the PK groups and families established from the yeast and mammalian kinomes, such as the FIKK kinases, which are specific to apicomplexan parasites (Schneider and Mercereau-Puijalon, 2005), and the CDPKs (see above). Interestingly, individual PKs displaying sequence features that are characteristic of distinct ePK groups are found in the P. falciparum kinome, illustrating its evolutionary divergence from other kinomes (Bracchi-Ricard et al., 2000; Dorin et al., 2001, 2005). A notable example of such “composite kinases” is Pfnek-1, which has a clear relatedness to the NIMA family despite the presence of a MAPKK-like putative activation site (Dorin et al., 2001).
A systematic kinome-wide knockout approach in P. berghei (Tewari et al., 2010) identified several PKs as essential for development in the mosquito (genetic manipulations are significantly more straightforward in the rodent malaria P. berghei than in P. falciparum, because (i) gene replacement by double cross-over occurs at a much higher rate in the former than in the latter, and (ii) the selection of transformed parasites occurs in the mouse and is much faster than the in vitro cultivation of P. falciparum (Carvalho and Menard, 2005)). A similar study has recently addressed essentiality of 62 of the 65 P. falciparum ePKs (this study excluded the FIKKs, see above). The inability to knockout a given locus, together with the ability to modify the allele in a way that does not cause loss-of-function of the gene product (e.g. tagging the C-terminal end with HA epitopes or with GFP), is interpreted as strongly indicative of an essential role of that locus during schizogony. Thirty-six kinases were thus identified as playing a crucial role in erythrocytic schizogony, while 26 are most likely dispensable for this part of the life cycle (with cloned parasite lines having thus far been obtained for 12 of these) (Solyakov et al., 2011).
3.6.1. Plasmodium exported kinases
Plasmodium expresses a set of unique kinases, the FIKKs (Ward et al., 2004), 16 of which are predicted to be exported into the RBC due to the presence of a PEXEL motif in their N-terminal region (Schneider and Mercereau-Puijalon, 2005). The roles played by FIKK in the RBC are much less well understood than those of some of the ROPKs in Toxoplasma-infected cells discussed above. We know that a significant subset of FIKKs are indeed exported into the RBC shortly following parasite invasion (Nunes et al., 2007, 2010) and that at least one member of the family remains within the parasite, despite possessing a recognisable PEXEL (Nunes et al., 2007). Most fikk genes are located in the sub-telomeric regions of 10 of the 14 chromosomes in P. falciparum, in the vicinity of the var genes that mediate cytoadherence and antigenic variation. The var genes are under allelic exclusion control, with a single PfEMP1 (var-encoded) protein expressed at a time; this appears not to be the case for the fikk genes. Interestingly, however, expression of a subset of fikk genes was modulated in parasites undergoing a switch in their cytoadherence phenotype (Nunes et al., 2007). Knockout studies implicated two FIKKs in the remodelling of the RBC membrane skeleton, as distinct phosphorylation profiles in ghost preparations of parasite-infected RBCs were detected for each of the knockout parasite clones (Nunes et al., 2010). In addition to FIKKs, a number of Plasmodium kinases have been reported to be exported into the RBC (Kun et al., 1997; Droucheau et al., 2004; Vaid et al., 2010) or even secreted to the extracellular milieu (Singh et al., 2009).
Recent data demonstrate that infection with malaria parasites causes a dramatic activation of a signalling pathway involving host RBC PAK and MEK kinases, and that pharmacological interference with these enzymes using highly selective allosteric inhibitors is lethal for the parasite (Sicard et al., 2011). The parasite is also known to interfere with host cell signalling in the liver stages (reviewed in Luder et al. (2009)), resulting in down-regulation of the NFκB pathway (Singh et al., 2007) and in prevention of apoptosis of host hepatocytes (van de Sand et al., 2005).
4. Conclusion
Thus, it appears that in both Toxoplasma and Plasmodium, two apicomplexan parasites that are phylogenetically widely divergent from each other (Kuo et al., 2008), calcium-regulated protein phosphorylation mediated by CDPKs plays central roles in regulating their entry into and egress from the host and other developmental processes, and that both parasites export proteins into their host cell to tailor it to their own needs. The secretion of virulence factors into the host is a strategy widely used not only by the Apicomplexa (a striking further apicomplexan-based example of this concept is the reversible transformation of leucocytes by Theileria (reviewed in Shiels et al. (2006)), but also by many, if not all, intracellular parasites (reviewed in Munter et al. (2006)). This not only provides a fascinating fundamental glimpse into unique signalling mechanisms operating within parasites and between parasites and their hosts, but also suggests opportunities for novel therapeutic intervention.
Acknowledgments
J. Saeij is supported by the American Heart Association (0835099N), by a Massachusetts Life Sciences Center New Investigator Award, by the Singapore-MIT Alliance for Research and Technology (SMART), by the PEW charitable trust, by a NERCE Developmental Grant and by NIH RO1-AI080621. B.M.C. is supported by a Senior Research Fellowship and Project Grants from The National Health and Medical Research Council of Australia (NHMRC). D. Lim was supported in part by a postdoctoral fellowship from Merck Research Laboratories and the MIT Department of Biology. Work in the CD laboratory was supported by Inserm, EPFL, the Wellcome Trust and the FP6 (SIGMAL and ANTIMAL projects) and FP7 (MALSIG project and EviMalaR Network of Excellence) Framework Programmes of the European Commission.
References
- Abdi A, Eschenlauer S, Reininger L, Doerig C. SAM domain-dependent activity of PfTKL3, an essential tyrosine kinase-like kinase of the human malaria parasite Plasmodium falciparum. Cell Mol Life Sci. 2010;67:3355–3369. doi: 10.1007/s00018-010-0434-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal S, Kern S, Halbert J, Przyborski JM, Baumeister S, Dandekar T, Doerig C, Pradel G. Two nucleus-localized CDK-like kinases with crucial roles for malaria parasite erythrocytic replication are involved in phosphorylation of splicing factor. J Cell Biochem. 2011;112:1295–1310. doi: 10.1002/jcb.23034. [DOI] [PubMed] [Google Scholar]
- Allen JJ, Li M, Brinkworth CS, Paulson JL, Wang D, Hubner A, Chou WH, Davis RJ, Burlingame AL, Messing RO, Katayama CD, Hedrick SM, Shokat KM. A semisynthetic epitope for kinase substrates. Nat Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anamika SN, Krupa A. A genomic perspective of protein kinases in Plasmodium falciparum. Proteins. 2005;58:180–189. doi: 10.1002/prot.20278. [DOI] [PubMed] [Google Scholar]
- Behnke MS, Khan A, Wootton JC, Dubey JP, Tang K, Sibley LD. Virulence differences in Toxoplasma mediated by amplification of a family of polymorphic pseudokinases. Proc Natl Acad Sci U S A. 2011;108:9631–9636. doi: 10.1073/pnas.1015338108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell. 2004;117:503–514. doi: 10.1016/s0092-8674(04)00449-0. [DOI] [PubMed] [Google Scholar]
- Billker O, Lourido S, Sibley LD. Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe. 2009;5:612–622. doi: 10.1016/j.chom.2009.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, Wood JL, Morgan DO, Shokat KM. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Blethrow J, Zhang C, Shokat KM, Weiss EL. Design and use of analog-sensitive protein kinases. Curr Protoc Mol Biol. 2004;Chapter 18(Unit 18.11) doi: 10.1002/0471142727.mb1811s66. [DOI] [PubMed] [Google Scholar]
- Bracchi-Ricard V, Barik S, Delvecchio C, Doerig C, Chakrabarti R, Chakrabarti D. PfPK6, a novel cyclin-dependent kinase/mitogen-activated protein kinase-related protein kinase from Plasmodium falciparum. Biochem J. 2000;347:255–263. [PMC free article] [PubMed] [Google Scholar]
- Bradley PJ, Ward C, Cheng SJ, Alexander DL, Coller S, Coombs GH, Dunn JD, Ferguson DJ, Sanderson SJ, Wastling JM, Boothroyd JC. Proteomic analysis of rhoptry organelles reveals many novel constituents for host–parasite interactions in Toxoplasma gondii. J Biol Chem. 2005;280:34245–34258. doi: 10.1074/jbc.M504158200. [DOI] [PubMed] [Google Scholar]
- Butcher BA, Fox BA, Rommereim LM, Kim SG, Maurer KJ, Yarovinsky F, De’Broski RH, Bzik DJ, Denkers EY. Toxoplasma gondii rhoptry kinase ROP16 activates STAT3 and STAT6 resulting in cytokine inhibition and arginase-1-dependent growth control. PLoS Pathog. 2011;7:e1002236. doi: 10.1371/journal.ppat.1002236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carruthers VB, Giddings OK, Sibley LD. Secretion of micronemal proteins is associated with Toxoplasma invasion of host cells. Cell Microbiol. 1999;1:225–235. doi: 10.1046/j.1462-5822.1999.00023.x. [DOI] [PubMed] [Google Scholar]
- Carvalho TG, Menard R. Manipulating the Plasmodium genome. Curr Issues Mol Biol. 2005;1:39–55. [PubMed] [Google Scholar]
- Cooke BM, Mohandas N, Coppel RL. The malaria-infected red blood cell: structural and functional changes. Adv Parasitol. 2001;50:1–86. doi: 10.1016/S0065-308X(01)50029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooke BM, Lingelbach K, Bannister LH, Tilley L. Protein trafficking in Plasmodium falciparum-infected red blood cells. Trends Parasitol. 2004;20:581–589. doi: 10.1016/j.pt.2004.09.008. [DOI] [PubMed] [Google Scholar]
- Coppi A, Tewari R, Bishop JR, Bennett BL, Lawrence R, Esko JD, Billker O, Sinnis P. Heparan sulfate proteoglycans provide a signal to Plasmodium sporozoites to stop migrating and productively invade host cells. Cell Host Microbe. 2007;2:316–327. doi: 10.1016/j.chom.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpke A, Heeg K, Bartz H, Baetz A. Regulation of innate immunity by suppressor of cytokine signaling (SOCS) proteins. Immunobiology. 2008;213:225–235. doi: 10.1016/j.imbio.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Dar AC, Wybenga-Groot LE, Sicheri F. The Eukaryotic Protein Kinase Domain. Wiley-VCH; Weinheim: 2005. [Google Scholar]
- Doerig C, Meijer L. Antimalarial drug discovery: targeting protein kinases. Expert Opin Ther Targets. 2007;11:279–290. doi: 10.1517/14728222.11.3.279. [DOI] [PubMed] [Google Scholar]
- Doerig C, Abdi A, Bland N, Eschenlauer S, Dorin-Semblat D, Fennell C, Halbert J, Holland Z. Malaria: targeting parasite and host cell kinomes. Biochim Biophys Acta. 2010;1804:604–612. doi: 10.1016/j.bbapap.2009.10.009. [DOI] [PubMed] [Google Scholar]
- Doerig C, Billker O. A parasite calcium switch and Achilles’ heel revealed. Nat Struct Mol Biol. 2010;17:541–543. doi: 10.1038/nsmb0510-541. [DOI] [PubMed] [Google Scholar]
- Dorin D, Alano P, Boccaccio I, Cicéron L, Doerig C, Sulpice R, Parzy D, Doerig C. An atypical mitogen-activated protein kinase (MAPK) homologue expressed in gametocytes of the human malaria parasite Plasmodium falciparum. J Biol Chem. 1999;274:29912. doi: 10.1074/jbc.274.42.29912. [DOI] [PubMed] [Google Scholar]
- Dorin D, Le Roch K, Sallicandro P, Alano P, Parzy D, Poullet P, Meijer L, Doerig C. Pfnek-1, a NIMA-related kinase from the human malaria parasite Plasmodium falciparum biochemical properties and possible involvement in MAPK regulation. Eur J Biochem. 2001;268:2600–2608. doi: 10.1046/j.1432-1327.2001.02151.x. [DOI] [PubMed] [Google Scholar]
- Dorin D, Semblat JP, Poullet P, Alano P, Goldring JP, Whittle C, Patterson S, Chakrabarti D, Doerig C. PfPK7, an atypical MEK-related protein kinase, reflects the absence of classical three-component MAPK pathways in the human malaria parasite Plasmodium falciparum. Mol Microbiol. 2005;55:184. doi: 10.1111/j.1365-2958.2004.04393.x. [DOI] [PubMed] [Google Scholar]
- Droucheau E, Primot A, Thomas V, Mattei D, Knockaert M, Richardson C, Sallicandro P, Alano P, Jafarshad A, Baratte B, Kunick C, Parzy D, Pearl L, Doerig C, Meijer L. Plasmodium falciparum glycogen synthase kinase-3: molecular model, expression, intracellular localisation and selective inhibitors. Biochim Biophys Acta. 2004;1697:181–196. doi: 10.1016/j.bbapap.2003.11.023. [DOI] [PubMed] [Google Scholar]
- El Hajj H, Lebrun M, Arold ST, Vial H, Labesse G, Dubremetz JF. ROP18 is a rhoptry kinase controlling the intracellular proliferation of Toxoplasma gondii. PLoS Pathog. 2007;3:e14. doi: 10.1371/journal.ppat.0030014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fennell C, Babbitt S, Russo I, Wilkes J, Ranford-Cartwright L, Goldberg DE, Doerig C. PfeIK1, a eukaryotic initiation factor 2alpha kinase of the human malaria parasite Plasmodium falciparum, regulates stress-response to amino-acid starvation. Malar J. 2009;8:99. doi: 10.1186/1475-2875-8-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fentress SJ, Behnke MS, Dunay IR, Mashayekhi M, Rommereim LM, Fox BA, Bzik DJ, Taylor GA, Turk BE, Lichti CF, Townsend RR, Qiu W, Hui R, Beatty WL, Sibley LD. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe. 2011;8:484–495. doi: 10.1016/j.chom.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Ravindran S, Turetzky JM, Boothroyd JC, Bradley PJ. Toxoplasma gondii targets a protein phosphatase 2C to the nuclei of infected host cells. Eukaryot Cell. 2007;6:73–83. doi: 10.1128/EC.00309-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green JL, Rees-Channer RR, Howell SA, Martin SR, Knuepfer E, Taylor HM, Grainger M, Holder AA. The motor complex of Plasmodium falciparum. J Biol Chem. 2008;283:30980. doi: 10.1074/jbc.M803129200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueirard P, Tavares J, Thiberge S, Bernex F, Ishino T, Milon G, Franke-Fayard B, Janse CJ, Ménard R, Amino R. Development of the malaria parasite in the skin of the mammalian host. Proc Natl Acad Sci U S A. 2010;107:18640–18645. doi: 10.1073/pnas.1009346107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halbert J, Ayong L, Equinet L, Le Roch K, Hardy M, Goldring D, Reininger L, Waters N, Chakrabarti D, Doerig C. A Plasmodium falciparum transcriptional cyclin-dependent kinase-related kinase with a crucial role in parasite proliferation associates with histone deacetylase activity. Eukaryot Cell. 2010;9:952–959. doi: 10.1128/EC.00005-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Quinn AM, Hunter T. The protein kinase family: conserved features and deduced phylogeny of the catalytic domains. Science. 1988;241:42–52. doi: 10.1126/science.3291115. [DOI] [PubMed] [Google Scholar]
- Huse M, Kuriyan J. The conformational plasticity of protein kinases. Cell. 2002;109:275–282. doi: 10.1016/s0092-8674(02)00741-9. [DOI] [PubMed] [Google Scholar]
- Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- Ishino T, Orito Y, Chinzei Y, Yuda M. A calcium dependent protein kinase regulates Plasmodium ookinete access to the midgut epithelial cell. Mol Microbiol. 2006;59:1175–1184. doi: 10.1111/j.1365-2958.2005.05014.x. [DOI] [PubMed] [Google Scholar]
- Jensen KD, Wang Y, Wojno ED, Shastri AJ, Hu K, Cornel L, Boedec E, Ong YC, Chien YH, Hunter CA, Boothroyd JC, Saeij JP. Toxoplasma polymorphic effectors determine macrophage polarization and intestinal inflammation. Cell Host Microbe. 2011;9:472–483. doi: 10.1016/j.chom.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafsack BFC, Pena J, Coppens I, Ravindran S, Boothroyd JC, Carruthers VB. Rapid membrane disruption by a perforin-like protein facilitates parasite exit from host cells. Science. 2009;323:530–533. doi: 10.1126/science.1165740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan N, Taylor SS. Rethinking pseudokinases. Cell. 2008;133:204–205. doi: 10.1016/j.cell.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato N, Sakata T, Breton G, Le Roch KG, Nagle A, Andersen C, Bursulaya B, Henson K, Johnson J, Kumar KA. Gene expression signatures and small-molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat Chem Biol. 2008;4:347–356. doi: 10.1038/nchembio.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Hunn JP, Könen Waisman S, Zhao YO, Preukschat D, Coers J, Boyle JP, Ong YC, Boothroyd JC, Reichmann G. Coordinated loading of IRG resistance GTPases on to the Toxoplasma gondii parasitophorous vacuole. Cell Microbiol. 2010;12:939–961. doi: 10.1111/j.1462-5822.2010.01443.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Taylor S, Su C, Mackey AJ, Boyle J, Cole R, Glover D, Tang K, Paulsen IT, Berriman M, Boothroyd JC, Pfefferkorn ER, Dubey JP, Ajioka JW, Roos DS, Wootton JC, Sibley LD. Composite genome map and recombination parameters derived from three archetypal lineages of Toxoplasma gondii. Nucleic Acids Res. 2005;33:2980–2992. doi: 10.1093/nar/gki604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Behnke MS, Dunay IR, White MW, Sibley LD. Phenotypic and gene expression changes among clonal type I strains of Toxoplasma gondii. Eukaryot Cell. 2009a;8:1828–1836. doi: 10.1128/EC.00150-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan A, Taylor S, Ajioka JW, Rosenthal BM, Sibley LD. Selection at a single locus leads to widespread expansion of Toxoplasma gondii lineages that are virulent in mice. PLoS Genet. 2009b;5:e1000404. doi: 10.1371/journal.pgen.1000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieschnick H, Wakefield T, Narducci CA, Beckers C. Toxoplasma gondii attachment to host cells is regulated by a calmodulin-like domain protein kinase. J Biol Chem. 2001;276:12369–12377. doi: 10.1074/jbc.M011045200. [DOI] [PubMed] [Google Scholar]
- Kun JF, Hibbs AR, Saul A, McColl DJ, Coppel RL, Anders RF. A putative Plasmodium falciparum exported serine/threonine protein kinase. Mol Biochem Parasitol. 1997;85:41–51. doi: 10.1016/s0166-6851(96)02805-8. [DOI] [PubMed] [Google Scholar]
- Kuo CH, Wares JP, Kissinger JC. The apicomplexan whole-genome phylogeny: an analysis of incongruence among gene trees. Mol Biol Evol. 2008;25:2689–2698. doi: 10.1093/molbev/msn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labesse G, Gelin M, Bessin Y, Lebrun M, Papoin J, Cerdan R, Arold ST, Dubremetz JF. ROP2 from Toxoplasma gondii: a virulence factor with a protein-kinase fold and no enzymatic activity. Structure. 2009;17:139–146. doi: 10.1016/j.str.2008.11.005. [DOI] [PubMed] [Google Scholar]
- Ling YM, Shaw MH, Ayala C, Coppens I, Taylor GA, Ferguson DJ, Yap GS. Vacuolar and plasma membrane stripping and autophagic elimination of Toxoplasma gondii in primed effector macrophages. J Exp Med. 2006;203:2063–2071. doi: 10.1084/jem.20061318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodoen MB, Gerke C, Boothroyd JC. A highly sensitive FRET based approach reveals secretion of the actin binding protein toxofilin during Toxoplasma gondii infection. Cell Microbiol. 2010;12:55–66. doi: 10.1111/j.1462-5822.2009.01378.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature. 2010;465:359–362. doi: 10.1038/nature09022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luder CG, Stanway RR, Chaussepied M, Langsley G, Heussler VT. Intracellular survival of apicomplexan parasites and host cell modification. Int J Parasitol. 2009;39:163–173. doi: 10.1016/j.ijpara.2008.09.013. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Martens S, Parvanova I, Zerrahn J, Griffiths G, Schell G, Reichmann G, Howard JC. Disruption of Toxoplasma gondii parasitophorous vacuoles by the mouse p47-resistance GTPases. PLoS Pathog. 2005;1:e24. doi: 10.1371/journal.ppat.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melzer T, Duffy A, Weiss LM, Halonen SK. The gamma interferon (IFN-gamma)-inducible GTP-binding protein IGTP is necessary for Toxoplasma vacuolar disruption and induces parasite egression in IFN-gamma-stimulated astrocytes. Infect Immun. 2008;76:4883–4894. doi: 10.1128/IAI.01288-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munter S, Way M, Frischknecht F. Signaling during pathogen infection. Sci STKE. 2006:re5. doi: 10.1126/stke.3352006re5. [DOI] [PubMed] [Google Scholar]
- Nagamune K, Moreno SN, Chini EN, Sibley LD. Calcium regulation and signaling in apicomplexan parasites. Subcell Biochem. 2008:70–81. doi: 10.1007/978-0-387-78267-6_5. [DOI] [PubMed] [Google Scholar]
- Nunes MC, Goldring JP, Doerig C, Scherf A. A novel protein kinase family in Plasmodium falciparum is differentially transcribed and secreted to various cellular compartments of the host cell. Mol Microbiol. 2007;63:391–403. doi: 10.1111/j.1365-2958.2006.05521.x. [DOI] [PubMed] [Google Scholar]
- Nunes MC, Okada M, Scheidig-Benatar C, Cooke BM, Scherf A, Nielsen K. Plasmodium falciparum FIKK kinase members target distinct components of the erythrocyte membrane. PLoS ONE. 2010;5:e11747. doi: 10.1371/journal.pone.0011747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojo KK, Larson ET, Keyloun KR, Castaneda LJ, Derocher AE, Inampudi KK, Kim JE, Arakaki TL, Murphy RC, Zhang L, Napuli AJ, Maly DJ, Verlinde CL, Buckner FS, Parsons M, Hol WG, Merritt EA, Van Voorhis WC. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol. 2010;17:602–607. doi: 10.1038/nsmb.1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong YC, Reese ML, Boothroyd JC. Toxoplasma rhoptry protein 16 (ROP16) subverts host function by direct tyrosine phosphorylation of STAT6. J Biol Chem. 2010;285:28731–28740. doi: 10.1074/jbc.M110.112359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons M, Worthey E, Ward P, Mottram J. Comparative analysis of the kinomes of three pathogenic trypanosomatids: Leishmania major, Trypanosoma brucei and Trypanosoma cruzi. BMC Genomics. 2005;6:127. doi: 10.1186/1471-2164-6-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peixoto L, Chen F, Harb OS, Davis PH, Beiting DP, Brownback CS, Ouloguem D, Roos DS. Integrative genomic approaches highlight a family of parasite-specific kinases that regulate host responses. Cell Host Microbe. 2010;8:208–218. doi: 10.1016/j.chom.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L, Boothroyd JC. Association of host mitochondria with the parasitophorous vacuole during Toxoplasma infection is not dependent on rhoptry proteins ROP2/8. Int J Parasitol. 2010;40:1367–1371. doi: 10.1016/j.ijpara.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu W, Wernimont A, Tang K, Taylor S, Lunin V, Schapira M, Fentress S, Hui R, Sibley LD. Novel structural and regulatory features of rhoptry secretory kinases in Toxoplasma gondii. EMBO J. 2009;28:969–979. doi: 10.1038/emboj.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese ML, Boothroyd JC. A helical membrane-binding domain targets the Toxoplasma ROP2 family to the parasitophorous vacuole. Traffic. 2009;10:1458–1470. doi: 10.1111/j.1600-0854.2009.00958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese ML, Boothroyd JC. A conserved noncanonical motif in the pseudoactive site of the ROP5 pseudokinase domain mediates its effect on Toxoplasma virulence. J Biol Chem. 2011;33:29366–29375. doi: 10.1074/jbc.M111.253435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese ML, Zeiner GM, Saeij JPJ, Boothroyd JC, Boyle JP. Polymorphic family of injected pseudokinases is paramount in Toxoplasma virulence. Proc Natl Acad Sci U S A. 2011;108:9625–9630. doi: 10.1073/pnas.1015980108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reininger L, Billker O, Tewari R, Mukhopadhyay A, Fennell C, Dorin-Semblat D, Doerig C, Goldring D, Harmse L, Ranford-Cartwright L. A NIMA-related protein kinase is essential for completion of the sexual cycle of malaria parasites. J Biol Chem. 2005;280:31957–31964. doi: 10.1074/jbc.M504523200. [DOI] [PubMed] [Google Scholar]
- Reininger L, Tewari R, Fennell C, Holland Z, Goldring D, Ranford-Cartwright L, Billker O, Doerig C. An essential role for the Plasmodium Nek-2 Nima-related protein kinase in the sexual development of malaria parasites. J Biol Chem. 2009;284:20858–20868. doi: 10.1074/jbc.M109.017988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reininger L, Wilkes JM, Bourgade H, Miranda-Saavedra D, Doerig C. An essential Aurora-related kinase transiently associates with spindle pole bodies during Plasmodium falciparum erythrocytic schizogony. Mol Microbiol. 2011;79:205–221. doi: 10.1111/j.1365-2958.2010.07442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg OS, Deindl S, Sung RJ, Nairn AC, Kuriyan J. Structure of the autoinhibited kinase domain of CaMKII and SAXS analysis of the holoenzyme. Cell. 2005;123:849–860. doi: 10.1016/j.cell.2005.10.029. [DOI] [PubMed] [Google Scholar]
- Rosowski EE, Lu D, Julien L, Rodda L, Gaiser RA, Jensen KD, Saeij JP. Strain-specific activation of the NF-kappaB pathway by GRA15, a novel Toxoplasma gondii dense granule protein. J Exp Med. 2011;208:195–212. doi: 10.1084/jem.20100717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij JP, Boyle JP, Coller S, Taylor S, Sibley LD, Brooke-Powell ET, Ajioka JW, Boothroyd JC. Polymorphic secreted kinases are key virulence factors in toxoplasmosis. Science. 2006;314:1780–1783. doi: 10.1126/science.1133690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeij JP, Coller S, Boyle JP, Jerome ME, White MW, Boothroyd JC. Toxoplasma co-opts host gene expression by injection of a polymorphic kinase homologue. Nature. 2007;445:324–327. doi: 10.1038/nature05395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibel LW, Colombani PM, Hess AD, Aikawa M, Atkinson CT, Milhous WK. Calcium and calmodulin antagonists inhibit human malaria parasites (Plasmodium falciparum): implications for drug design. Proc Natl Acad Sci U S A. 1987;84:7310. doi: 10.1073/pnas.84.20.7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider AG, Mercereau-Puijalon O. A new Apicomplexa-specific protein kinase family: multiple members in Plasmodium falciparum, all with an export signature. BMC Genomics. 2005;6:30. doi: 10.1186/1471-2164-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiels B, Langsley G, Weir W, Pain A, McKellar S, Dobbelaere D. Alteration of host cell phenotype by Theileria annulata and Theileria parva: mining for manipulators in the parasite genomes. Int J Parasitol. 2006;36:9–21. doi: 10.1016/j.ijpara.2005.09.002. [DOI] [PubMed] [Google Scholar]
- Sicard A, Semblat JP, Doerig CM, Spicer JA, Srivastava A, Retzlaff S, Heussler V, Waters AP, Doerig C. Activation of a host erythrocyte protein kinase signaling pathway is critical for malaria parasite survival. Cell Microbiol. 2011;13:836–845. doi: 10.1111/j.1462-5822.2011.01582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AP, Buscaglia CA, Wang Q, Levay A, Nussenzweig DR, Walker JR, Winzeler EA, Fujii H, Fontoura BM, Nussenzweig V. Plasmodium circumsporozoite protein promotes the development of the liver stages of the parasite. Cell. 2007;131:492–504. doi: 10.1016/j.cell.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Singh M, Mukherjee P, Narayanasamy K, Arora R, Sen SD, Gupta S, Natarajan K, Malhotra P. Proteome analysis of Plasmodium falciparum extracellular secretory antigens at asexual blood stages reveals a cohort of proteins with possible roles in immune modulation and signaling. Mol Cell Proteomics. 2009;8:2102–2118. doi: 10.1074/mcp.M900029-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solyakov L, Halbert J, Alam MM, Semblat JP, Dorin-Semblat D, Reininger L, Bottrill AR, Mistry S, Abdi A, Fennell C, Holland Z, Demarta C, Bouza Y, Sicard A, Nivez MP, Eschenlauer S, Lama T, Thomas DC, Sharma P, Agarwal S, Kern S, Pradel G, Graciotti M, Tobin AB, Doerig C. Global kinomic and phospho-proteomic analyses of the human malaria parasite Plasmodium falciparum. Nat Commun. 2011;29:565. doi: 10.1038/ncomms1558. [DOI] [PubMed] [Google Scholar]
- Sopko R, Andrews BJ. Linking the kinome and phosphorylome – a comprehensive review of approaches to find kinase targets. Mol Biosyst. 2008;4:920–933. doi: 10.1039/b801724g. [DOI] [PubMed] [Google Scholar]
- Steinfeldt T, Könen-Waisman S, Tong L, Pawlowski N, Lamkemeyer T, Sibley LD, Hunn JP, Howard JC. Phosphorylation of mouse immunity-related GTPase (IRG) resistance proteins is an evasion strategy for virulent Toxoplasma gondii. PLoS Biol. 2011;8:3–16. doi: 10.1371/journal.pbio.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugi T, Kato K, Kobayashi K, Watanabe S, Kurokawa H, Gong H, Pandey K, Takemae H, Akashi H. Use of the kinase inhibitor analog 1NM-PP1 reveals a role for Toxoplasma gondii CDPK1 in the invasion step. Eukaryot Cell. 2010;9:667–670. doi: 10.1128/EC.00351-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suss-Toby E, Zimmerberg J, Ward GE. Toxoplasma invasion: the parasitophorous vacuole is formed from host cell plasma membrane and pinches off via a fission pore. Proc Natl Acad Sci U S A. 1996;93:8413–8418. doi: 10.1073/pnas.93.16.8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Hajj HE, Jerome M, Behnke MS, White M, Wootton JC, Sibley LD. A secreted serine–threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science. 2006;314:1776–1780. doi: 10.1126/science.1133643. [DOI] [PubMed] [Google Scholar]
- Taylor SS, Kornev AP. Yet another “active” pseudokinase, Erb3. Proc Natl Acad Sci U S A. 2010;107:8047–8048. doi: 10.1073/pnas.1003436107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tewari R, Straschil U, Bateman A, Bohme U, Cherevach I, Gong P, Pain A, Billker O. The systematic functional analysis of Plasmodium protein kinases identifies essential regulators of mosquito transmission. Cell Host Microbe. 2010;8:377–387. doi: 10.1016/j.chom.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaid A, Ranjan R, Smythe WA, Hoppe HC, Sharma P. PfPI3K, a phosphatidylinositol-3 kinase from Plasmodium falciparum, is exported to the host erythrocyte and is involved in hemoglobin trafficking. Blood. 2010;115:2500–2507. doi: 10.1182/blood-2009-08-238972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sand C, Horstmann S, Schmidt A, Sturm A, Bolte S, Krueger A, Lutgehetmann M, Pollok JM, Libert C, Heussler VT. The liver stage of Plasmodium berghei inhibits host cell apoptosis. Mol Microbiol. 2005;58:731–742. doi: 10.1111/j.1365-2958.2005.04888.x. [DOI] [PubMed] [Google Scholar]
- Ward GE, Fujioka H, Aikawa M, Miller LH. Staurosporine inhibits invasion of erythrocytes by malarial merozoites. Exp Parasitol. 1994;79:480–487. doi: 10.1006/expr.1994.1109. [DOI] [PubMed] [Google Scholar]
- Ward P, Equinet L, Packer J, Doerig C. Protein kinases of the human malaria parasite Plasmodium falciparum: the kinome of a divergent eukaryote. BMC Genomics. 2004;5:79. doi: 10.1186/1471-2164-5-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernimont AK, Artz JD, Finerty P, Jr, Lin YH, Amani M, Allali-Hassani A, Senisterra G, Vedadi M, Tempel W, Mackenzie F, Chau I, Lourido S, Sibley LD, Hui R. Structures of apicomplexan calcium-dependent protein kinases reveal mechanism of activation by calcium. Nat Struct Mol Biol. 2010;17:596–601. doi: 10.1038/nsmb.1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernimont AK, Amani M, Qiu W, Pizarro JC, Artz JD, Lin YH, Lew J, Hutchinson A, Hui R. Structures of parasitic CDPK domains point to a common mechanism of activation. Proteins. 2011;79:803–820. doi: 10.1002/prot.22919. [DOI] [PubMed] [Google Scholar]
- Xu B, English JM, Wilsbacher JL, Stippec S, Goldsmith EJ, Cobb MH. WNK1, a novel mammalian serine/threonine protein kinase lacking the catalytic lysine in subdomain II. J Biol Chem. 2000;275:16795–16801. doi: 10.1074/jbc.275.22.16795. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Standley DM, Takashima S, Saiga H, Okuyama M, Kayama H, Kubo E, Ito H, Takaura M, Matsuda T, Soldati-Favre D, Takeda K. A single polymorphic amino acid on Toxoplasma gondii kinase ROP16 determines the direct and strain-specific activation of Stat3. J Exp Med. 2009;206:2747–2760. doi: 10.1084/jem.20091703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa H, Ohishi M, Mori H, Murakami M, Chinen T, Aki D, Hanada T, Takeda K, Akira S, Hoshijima M. IL-6 induces an anti-inflammatory response in the absence of SOCS3 in macrophages. Nat Immunol. 2003;4:551–556. doi: 10.1038/ni938. [DOI] [PubMed] [Google Scholar]
- Zeqiraj E, Filippi BM, Deak M, Alessi DR, van Aalten DM. Structure of the LKB1-STRAD-MO25 complex reveals an allosteric mechanism of kinase activation. Science. 2009;326:1707–1711. doi: 10.1126/science.1178377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeqiraj E, van Aalten DM. Pseudokinases-remnants of evolution or key allosteric regulators? Curr Opin Struct Biol. 2010;20:772–781. doi: 10.1016/j.sbi.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ferguson DJ, Wilson DC, Howard JC, Sibley LD, Yap GS. Virulent Toxoplasma gondii evade immunity-related GTPase-mediated parasite vacuole disruption within primed macrophages. J Immunol. 2009a;182:3775–3781. doi: 10.4049/jimmunol.0804190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao YO, Khaminets A, Hunn JP, Howard JC. Disruption of the Toxoplasma gondii parasitophorous vacuole by IFNgamma-inducible immunity-related GTPases (IRG proteins) triggers necrotic cell death. PLoS Pathog. 2009b;5:e1000288. doi: 10.1371/journal.ppat.1000288. [DOI] [PMC free article] [PubMed] [Google Scholar]