

Abstract
Metabolic transformations have been reported as involved in neoplasms survival. This suggests a role of metabolic pathways as potential cancer pharmacological targets. Modulating tumor's energy production pathways may become a substantial research area for cancer treatment. The significant role of metabolic deregulation as inducing transcriptional instabilities and consequently whole-system failure, is thus of foremost importance. By using a data integration approach that combines experimental evidence for high-throughput genome wide gene expression, a non-equilibrium thermodynamics analysis, nonlinear correlation networks as well as database mining, we were able to outline the role that transcription factors MEF2C and MNDA may have as main master regulators in primary breast cancer phenomenology, as well as the possible interrelationship between malignancy and metabolic dysfunction. The present findings are supported by the analysis of 1191 whole genome gene expression experiments, as well as probabilistic inference of gene regulatory networks, and non-equilibrium thermodynamics of such data. Other evidence sources include pathway enrichment and gene set enrichment analyses, as well as motif comparison with a comprehensive gene regulatory network (of homologue genes) in Arabidopsis thaliana. Our key finding is that the non-equilibrium free energies provide a realistic description of transcription factor activation that when supplemented with gene regulatory networks made us able to find deregulated pathways. These analyses also suggest a novel potential role of transcription factor energetics at the onset of primary tumor development. Results are important in the molecular systems biology of cancer field, since deregulation and coupling mechanisms between metabolic activity and transcriptional regulation can be better understood by taking into account the way that master regulators respond to physicochemical constraints imposed by different phenotypic conditions.
Introduction
It is known that tumors could depend on energy production pathways that are different from those of normal cells. These unique pathways require in some cases the expression and function of so-called tumor-specific enzymes. Some of these glycolytic enzymes, as well as other modulators of tumor behavior, have recently been analyzed in search for a clue that inhibition of such enzymes or appropriate tuning of such modulators should deprive tumors of energy, while leaving non-transformed cells unaffected. Recent findings seem to point out to several so-called metabolic transformations that permit neoplasms survival, thus suggesting a role of metabolic pathways as potential pharmacological targets [1]. In fact, preliminary experiments on animals with hepatocellular carcinoma have indeed shown very encouraging results. It appears that modulating the energy production pathways of tumors is poised to become a substantial research area for cancer treatment [2]. The role of perturbed local cell energetics in association with cancer is not new. In the past, under several instances, relationships seem to appear between metabolic variation and tumorigenesis, spread and dissemination of malignancy. In recent times a growing interest (or best a revival of it) has taken place and evidence seem to suggest closer connections than those suspected. For instance, the importance of glycolysis in cancer development [3]. It has been discussed how a combination of agents that inhibit both energy production and cell signaling may provide a novel and effective approach to target pancreatic cancer effectively.
Thermodynamic studies at the transcriptional [4], epigenetic [5], [6], and metabolic [7] levels have pointed out to energetics as playing a non-trivial role in the onset and development of malignancy. In the particular case of this paper, we will focus on the relationship between transcriptional de-regulation of a set of genes that present transcription factor (TF) and metabolic activity (some of them) while at the same time have been associated with the presence of breast cancer. We will then study its regulatory and thermodynamical behavior by means of gene expression data obtained from genome-wide analysis experiments in RNA from biopsy-captured tissue of both primary breast cancer and normal breast.
The role of gene interaction networks have also been extensively mentioned in relation to cancer phenomenology, it has been claimed that these network effects are, in fact much more important than individual gene contributions [8]. Some of these networks are indeed related to energetic and metabolic processes [9], tyrosine-related deregulation [10], and immunity weakening [11]. One usually think of tumor cells as having successful mechanisms to evade normal control and cell regulation of proliferation and apoptosis. Alterations in gene expression have become a better (but far from completely) understood component of normal development and disease progression. In particular, TFs have become a promising target for therapy. In brief, gross alterations in TF regulation would result in cascade triggering affecting both the whole cell cycle and the metabolic activity thus resulting in possible development of cancer. Many people have come to conclude that cancer is a transcriptional disorder disease [12]–[15], while, as we have mentioned other authors have recently turned their attention to the metabolic and energetic component [7], [9], hence a possible connection between these two approaches could be found in the energetic deregulation
 transcriptional disorder leading both to cascade triggering and metabolic disorders related to neoplasm formation and development. For these reasons this paper will attempt to model the role of TFs at both the energetics (thermodynamic) level and the network approach.
transcriptional disorder leading both to cascade triggering and metabolic disorders related to neoplasm formation and development. For these reasons this paper will attempt to model the role of TFs at both the energetics (thermodynamic) level and the network approach.
Analysis
One of the cornerstones of contemporary genomic studies, in particular of the systems biology approach, is data integration (DI). DI is useful to make sense out of the extremely large corpus of experimental evidence given, for instance by genome-wide expression analysis. With the continuous advent of novel techniques in high throughput molecular biology and the ‘omics maybe just one thing has been established: Complex biological systems need to be studied from several standpoints to unveil the actual mechanisms behind them. In the present case, our aim is to sketch some hints for a proposal of functional mechanisms behind gene expression in cancer and cell energetics. The analysis work-flow for the present study was as follows (see also Figure 1):
Figure 1. Flow chart design for this work.

It is noticeable that this is an hybrid model which incorporates both data-driven discovery (gene expression analysis, gene-set enrichment and probabilistic network inference) and hypothesis driven inquiries (data mining and non-equilibrium thermodynamics modeling).
Statistical pre-processing of the microarray gene expression data.
Determination of differentially expressed genes and statistical significance assessment.
Data mining for functional features within the statistically significant differential expression gene set.
Non-equilibrium thermodynamics calculations (Figures 2, 3, 4, 5).
Probabilistic inference of gene regulatory networks.
Pathway statistical enrichment analysis.
Search for common non-linear correlations found for human MEF2C in this work (Figure 6) that are present also in a highly curated A. Thaliana transcription factor database, indicating modular conservation among species.
Gene Set Enrichment Analysis applied to the 1191 samples expression matrix to look up for dysregulated functions and pathways as a complement for the gene analysis in cancer and metabolic pathways (Figure 7 and Figure 8).
Figure 2. Gene Expression Intensity profile.

We can notice strong stochasticity in the signals. However, a definite background tendency may be identified, as it may be clearer when examining the concentration profile.
Figure 3. mRNA concentration profile.

In spite of strong stochasticity, we can notice some trends. For instance, there are some genes with a low concentration and low variability while others present larger concentrations and variabilities.
Figure 4. Transcriptional affinities profile.

, a strong stochastic behavior can be noted, however when considering the associated chemical potentials of transcription (see Fig. 5) definite trends arise.
Figure 5. Chemical potentials of transcription profile.

As in the case of the concentration profiles we can notice some trends in spite of stochasticity. There are some genes with a low concentration and low variability while others present larger concentrations and variabilities.
Figure 6. Inferred gene regulatory network [10], [16].
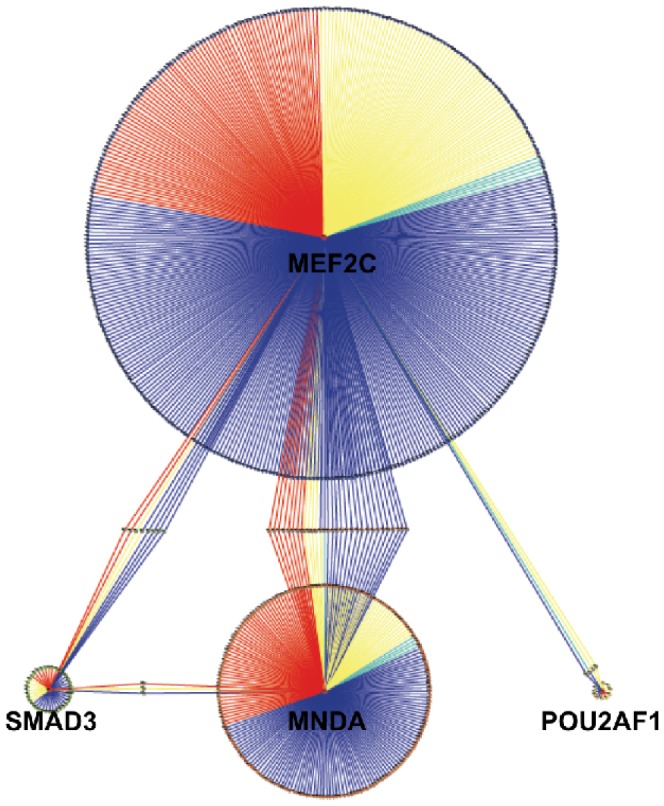
Links colored in red represent interactions associated (in the literature, from data mining) with breast cancer, yellow links are interactions associated with other types of cancer, turquoise links are interactions associated with metabolic disorders and navy blue links are otherwise. We could notice that some genes are regulated by more than one of these transcription factors as is the case with RAD52, ADH1C, OIP5, ELK4, PEX10, GAA, FTHP1 and ADAP1 which are co-regulated by MEF2C and SMAD3; of STMN1, STAU1 and the c10ORF10 transcript which are co-regulated by SMAD3 and MNDA; of CSH1, FANCI, FHIT, CDKN2A, PRC1, CENPF, MAP3K1, BNIP3, HCLS1, TAF15, PCBP2, SPAI7, SLC38A1, PASK, HIST1H2BD, POLR2I, UPK3B, EHD1, PRICKLE3, LOC91316, ANAPC2, LST1, RRP12, c9ORF6, BRP44, CLEC4A, TMEM194A, TPO3B, HIST1H2AM and ZFP37 which are co-regulated by MEF2C and MNDA; and by c14ORF1, DOK5, AFF1, and CADM4 which are co-regulated by MEF2C and POU2AF1. In the case of these double-regulatory interactions there are some cases in which both regulatory interactions are associated with a certain phenotype and other cases in which different (or no) phenotype is associated for each link.
Figure 7. Cancer related deregulated pathways.

Statistical enrichment of deregulated pathways within our differential gene-sets include canonical cancer pathways such as DNA damage repair, AMPK and RAS. Molecules marked with a red star correspond to differentially expressed genes in the cancer/control contrast.
Figure 8. Metabolism related deregulated pathways.

Statistical enrichment of deregulated pathways within our differential gene-sets include metabolic pathways. For instance, both branches of the cholesterol biosynthesis pathway are affected. Molecules marked with a red star correspond to differentially expressed genes in the cancer/control contrast.
Differentially expressed genes
After pre-processing (background correction, normalization and summarization) of the samples [16] according to the RMA algorithm [17], we proceeded to implement a statistical analysis by using linear modeling (limma) to look up for significant differentially expressed genes (full data matrix available upon request). Empirical Bayes and other shrinkage methods are used to borrow information across genes making the analyses stable even for experiments with small number of arrays. This method allows very general experiments to be analyzed as easily as single replica experiments. The approach requires two matrices, the first one is called the design matrix which gives a representation of the different RNA targets which have been hybridized to the arrays. The second one, or contrast matrix which allows the coefficients defined by the design matrix to be combined into contrasts of interest. Each contrast corresponds to a comparison of interest between the RNA targets [18].
Data mining for metabolic and transcription factor activity
Once we had a set of differentially expressed genes, we proceeded to implement a data mining search over it. Search parameters include the following constraints:
Genes that are well known transcription factors, reported not only by sequence homology but also by actual experimental evidence.
Genes that have been associated in the literature with the presence of breast cancer (higher scores) or any other tumors -liquid neoplasms were excluded- (lower scores).
Genes whose protein products are related to cellular level metabolic pathways.
Genes whose transcripts possess a complete physicochemical characterization, e.g. Affymetrix® calibration probes have reported free energies of formation.
From the set of genes included in the GeneChip® under study (namely Affymetrix HGU133-A) which were statistically significant in their differential expression between tumors and controls, we built sets that satisfy the aforementioned constraints. Then we made the intersection set of all these. This set, that we will call hereon a Core set consisted in four genes, namely MNDA, POU2AF1, MEF2C and SMAD3. In what follows we will analyze in detail the non-equilibrium thermodynamics of transcription as well as the regulatory network structure of such genes within a sample set of 1191 microarrays.
Non-equilibrium thermodynamic model
On general grounds, the finding of relevant genes associated with a cancer phenotype is based on determining features such as differential expression patterns. However, two important issues that should be also taken into account besides the gene expression levels, are the energetics within the cell and the physicochemical properties of the biomolecules involved in transcriptional regulation (transcription factors in particular). Both features could be responsible for TF activity since they affect the mechanism behind the activation of target genes according to previous specific cellular energetic conditions.
An interesting trend in the transcriptional energetics in some well-studied genes [19] is that, the values of the activation energies are in general lower for genes that act as transcription factors and higher for genes with no-known TF-activity. The physicochemical meaning of this finding seem to point-out to transcription factors as genes whose expression is regulated by lower activation-energy barriers. Since TF's are involved in the transcriptional activation of other genes, it is expected that they are synthesized first when energy is started to being released by metabolic processes in the cell. Transcriptional targets should, in general be synthesized later and with higher activation energies. These higher saturation limits for the chemical potentials of TFs suggest both stability and spontaneity in the expression of these as compared to target genes.
In brief, since master TFs are needed in early stages of whole-genome transcription (i.e. upstream in the regulatory cascade) in order to kick-start such processes and, by taking into account that transcriptional regulation has been characterized as an activated process [20]; a hierarchy in the synthesis of mRNA templates (and, further on, in their product proteins) is established in terms of the corresponding activation energies -as given by their chemical potentials- and of the availability of free energy in the cellular environment. Then, by calculating thermodynamic properties for TFs, it is possible to unveil the order or priority in their activation which is dependent on energy accessibility.
A non-equilibrium thermodynamical theory of gene regulation has already been proposed [20]. As it was shown the thermodynamic analysis of transcriptional regulation presents several challenges, in particular associated with the fact that the cell is a small system, in the sense that the role of fluctuations and noise plays a rather fundamental role for its characterization. Systems outside the domain of the thermodynamic limit are characterized by large fluctuations and hence stochastic effects need to be taken into account. An extremely important conundrum in contemporary thermal physics lies in the connections between probability and thermodynamics. A developed theory exists however, called mesoscopic nonequilibrium thermodynamics (MNET) [21] which specifically addresses the issue by considering the stochastic nature of the time evolution of small non-equilibrium systems, in a context which is extremely close to our work. To account for stochasticity one needs to recognize that scaling down the description of a physical system brings up energy contributions that are commonly neglected in thermodynamical descriptions. The time-evolution of these systems could be described as a generalized diffusion process over a potential landscape in the space of mesoscopic variables. This process is driven by a generalized mesoscopic-thermodynamic force whose stochastic origin could be tracked back by means of, for example, a Fokker-Planck-like analysis [21]. These classes of formalisms are appropriated in the case of activated processes, for instance, a system crossing a potential barrier. The complex biochemical reactions involved in transcriptional regulation belong also to this category.
The present theoretical framework [20] shares similar ideas with MNET (although treated in a less formal way due to present unavailability of information regarding the non-local probability distributions) and deals with intensity levels of gene activity as measured in whole genome gene expression profiling on GeneChips. It is based on the thermodynamics of hybridization [22] that considers a basic two-state model that quantifies mRNA concentrations by competitive hybridization [23], [24]. This non-equilibrium thermodynamical theory has been used to study the role of transcription factors in the phenomena of anomalous transcriptional bursts [19], [25].
As is usual in non-equilibrium thermodynamics we will assume that a generalized entropy-like function 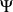 exists, which may be written in the form [26], [27]:
exists, which may be written in the form [26], [27]:
| (1) |
Eq. 1 is a formal extension of the Gibbs relation of equilibrium thermodynamics. The quantities appearing are as usual: 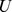 is the internal energy,
is the internal energy, 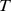 is the local temperature,
is the local temperature, 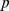 and
and  the pressure and volume,
the pressure and volume, 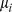 is the chemical potential for the
is the chemical potential for the  -th mRNA species.
-th mRNA species. 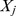 and
and 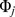 are extended thermodynamical fluxes and forces [26]. For a multicomponent mRNA mixture (under fixed volume and pressure), the set of relevant variables consists in the temperature
are extended thermodynamical fluxes and forces [26]. For a multicomponent mRNA mixture (under fixed volume and pressure), the set of relevant variables consists in the temperature 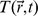 and concentration of each gene species
and concentration of each gene species 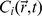 as the slow varying (classical) parameters set and the mass flux of these species
as the slow varying (classical) parameters set and the mass flux of these species 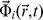 and their corresponding forces
and their corresponding forces 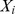 as fast variables. These latter variables will take into account the presence of inhomogeneous regions (concentration domains formed because of the gene regulatory interactions) to correct the predictions based on the local equilibrium hypothesis.
as fast variables. These latter variables will take into account the presence of inhomogeneous regions (concentration domains formed because of the gene regulatory interactions) to correct the predictions based on the local equilibrium hypothesis.
The non-equilibrium Gibbs free energy for a mixture of 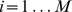 , mRNA transcripts reads:
, mRNA transcripts reads:
| (2) |
Quantities are local fields defined as usual, (e.g. 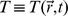 ), within the mentioned formalism one can consider that a generalized entropy-like function
), within the mentioned formalism one can consider that a generalized entropy-like function 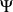 exists [26], [27], also T is the temperature, p the pressure,
exists [26], [27], also T is the temperature, p the pressure, 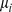 the chemical potential, etc.;
the chemical potential, etc.; 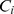 is the concentration for species
is the concentration for species  ,
, 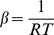 , with
, with 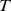 the absolute temperature and
the absolute temperature and 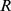 the gas constant,
the gas constant, 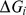 is the free energy of hybridization of
is the free energy of hybridization of  ,
, 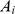 is a parameter that sets the scale of intensity [22] corresponding to the saturation limit
is a parameter that sets the scale of intensity [22] corresponding to the saturation limit 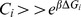 ;
; 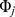 and
and 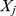 are extended thermodynamical fluxes and forces that take into account non-local effects.
are extended thermodynamical fluxes and forces that take into account non-local effects.
If we recall from reference [20] given the relation between gene expression intensity 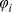 and concentration
and concentration 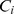 , we have that:
, we have that:
| (3) |
After this, a proposal on the form for the extended fluxes and forces should be given. Hence, we are proposing a system of linear (in the forces) coupled fluxes with memory [20].
The constitutive equations are,
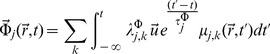 |
(4) |
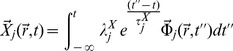 |
(5) |
The 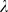 's are time-independent amplitudes,
's are time-independent amplitudes, 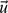 is a unit vector in the direction of mass flow (the nature of
is a unit vector in the direction of mass flow (the nature of 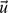 will not affect the rest of our description, since we will be dealing with the magnitude of the mass flux
will not affect the rest of our description, since we will be dealing with the magnitude of the mass flux 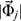 ) and
) and  's are relaxation times considered path-independent scalars. Since we have a linear relation between thermodynamic fluxes and forces some features of the Onsager-Casimir formalism will still hold.
's are relaxation times considered path-independent scalars. Since we have a linear relation between thermodynamic fluxes and forces some features of the Onsager-Casimir formalism will still hold.
Irreversible coupling is given by Eq. 4 and 5, nevertheless due to the fact that actual transcription measurement experiments are made either on homeostasis (steady state) settings or within time series designs with intervals several orders of magnitude larger than the associated relaxation times (which are of the order of a few molecular collision times) it is possible to take the limits 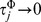 and
and 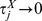 , then the integrals become evaluated delta functions to give:
, then the integrals become evaluated delta functions to give:
| (6) |
| (7) |
Also due to the spatial nature of the experimental measurements (either RNA blots or DNA/RNA chips measure space-averaged mRNA concentrations) it is possible to work with the related scalar quantities instead, to give:
| (8) |
| (9) |
Substituting Eq. 8 and 9 into Eq. 2 one gets:
| (10) |
If we assume that the generalized transport coefficient 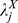 is independent of the flux
is independent of the flux 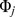 we can write:
we can write:
| (11) |
Or in terms of the transcription regulation chemical potentials
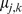
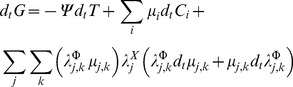 |
(12) |
In the constant transport coefficient approximation, Eq. 12 reads:
| (13) |
Defining generalized transport coefficients 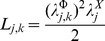 .
.
If we change variables in equation 2 from concentration to gene expression intensity (by using equation 3) and introduce the fluxes, forces and generalized transport coefficients, we could rephrase it as follows:
| (14) |
The resulting affinity of transcription i.e. the thermodynamic conjugate variable to the probe intensity 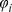 is given by:
is given by:
| (15) |
The quantity 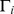 plays the role of a chemical affinity for the gene expression process. As gene expression is a process that follows thermal activation kinetics,
plays the role of a chemical affinity for the gene expression process. As gene expression is a process that follows thermal activation kinetics, 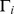 is a temperature dependent variable. This dependency shows up both explicitly (by the value
is a temperature dependent variable. This dependency shows up both explicitly (by the value 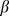 ) and also due to indirect temperature dependency given by the saturation constants
) and also due to indirect temperature dependency given by the saturation constants 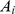 .
. 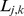 are coefficients related to gene cross-regulation and
are coefficients related to gene cross-regulation and 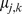 are the chemical potentials associated with transcriptional regulation [19], [20].
are the chemical potentials associated with transcriptional regulation [19], [20].
The related chemical potential of transcription [20] is given by:
| (16) |
Since we have reliable experimental data for the expression levels 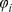 from 1191 whole-genome gene expression experiments, it is possible to calculate the gene transcriptional affinities and chemical potentials for the set of genes of interest from equations 15 and 16 (we also have good values for the constants
from 1191 whole-genome gene expression experiments, it is possible to calculate the gene transcriptional affinities and chemical potentials for the set of genes of interest from equations 15 and 16 (we also have good values for the constants 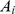 taken from spike-in experimental data provided by the gene-chip manufacturer). Since these experiments have been made without the use of any knock-out or knock-down techniques (i.e. all genes are subject to their corresponding regulatory interactions), the gene expression levels
taken from spike-in experimental data provided by the gene-chip manufacturer). Since these experiments have been made without the use of any knock-out or knock-down techniques (i.e. all genes are subject to their corresponding regulatory interactions), the gene expression levels 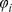 used to calculate the chemical potentials and transcriptional affinities have already incorporated (although in an implicit way) the effect of transcriptional regulation as given by the gene regulatory mechanisms depicted in the third term at the r.h.s. of equation 14.
used to calculate the chemical potentials and transcriptional affinities have already incorporated (although in an implicit way) the effect of transcriptional regulation as given by the gene regulatory mechanisms depicted in the third term at the r.h.s. of equation 14.
Non-linear correlation inference of regulatory networks
To deconvolute a Gene Regulatory Network (GRN) related to primary breast cancer we applied a methodology based on a local pattern-sharing measure as surrogate to actual gene-gene interactions to our dataset(see Materials and Methods -  Experimental datasets). The goal of deconvolution methods is inferring GRNs based on statistical dependencies within the joint probability distribution of gene expression for all genes within a given gene set. Typical means to reach this goal consist in the quantification of the new information content that arise when we look at the full joint probability distribution when compared to a series of successive independence approximations. According with the method given in reference [10], we calculated measures of non-linear correlation between the normalized expression values (22238 probesets) and the core set of 4 genes. Threshold-analysis was made on such measures to look up for statistical significance in the inference and based on their IBS index value we selected a set of 712 genes. The optimal network (within the given approximations) was found by a Maximum Entropy Method as is shown elsewhere [10] and was validated by several (mostly in silico and database-mining) methods [16].
Experimental datasets). The goal of deconvolution methods is inferring GRNs based on statistical dependencies within the joint probability distribution of gene expression for all genes within a given gene set. Typical means to reach this goal consist in the quantification of the new information content that arise when we look at the full joint probability distribution when compared to a series of successive independence approximations. According with the method given in reference [10], we calculated measures of non-linear correlation between the normalized expression values (22238 probesets) and the core set of 4 genes. Threshold-analysis was made on such measures to look up for statistical significance in the inference and based on their IBS index value we selected a set of 712 genes. The optimal network (within the given approximations) was found by a Maximum Entropy Method as is shown elsewhere [10] and was validated by several (mostly in silico and database-mining) methods [16].
Biochemical pathway statistical enrichment analysis
Reactome [28] biological/biochemical pathway over-representation analysis was performed to determine the Reactome pathways in which gene IDs in our list were strongly enriched. Reactome is a an open-source, open access, manually curated and peer-reviewed pathway database that may help to understand the biological context of genomic data. Significance assessment was also made by means of ‘urn model’ hypergeometric distribution tests.
Materials and Methods
Experimental datasets
A curated set of 1191 whole genome gene expression profiles was generated [16] from datasets for several publicly available experiments deposited in the GEO database [29]. These experiments were performed in total mRNA extracted under the GPL96 protocol [30] which is based on the Affymetrix HGU133A microarray GeneChip platform. In the case of experiments including some kind of treatment or cell modification we only took the unaltered samples to include them in our analyses. Details are given in Table 1. Further information is available in the corresponding GEO entries and/or may be available upon request. In the case of human mRNA samples taken directly from organ tissue (by a biopsy) and not from cultured cell-lines, it is extremely difficult to design time-course experiments. Thus, in order to study a surrogate model of transcriptional de-regulation, we proposed the following alternative to look for correlations: After quality control pre-processing, background correction and normalization of the microarrays, the samples were prioritized (ordered) according to their BNIP3 (Affymetrix-probe ID 201848_s_at) expression level. BNIP3 is a well known marker of progression and malignancy in primary breast cancer that correlates both with lab tests and clinical trials [31]. By ordering the independent, steady-state samples in this manner it is now possible to look up for correlation patterns of gene expression.
Table 1. GEO [29] identifier and references for the Microarray experiments used here, first column is GEO key ID, second and third columns are the corresponding number of samples cases/controls resp.
| GEO ID Series | Tumors | Controls | Reference |
| GSE1456 | 159 | [68] | |
| GSE4922 | 249 | [69] | |
| GSE7390 | 198 | [70] | |
| GSE2603 | 99 | [71] | |
| GSE2990 | 125 | [72] | |
| GSE3494 | 251 | [73] | |
| GSE1561 | 49 | [74] | |
| GSE15852 | 43 | [75] | |
| GSE9574 | 15 | [76] | |
| GSE6883 | 3 | [77] |
Fourth column is the reference entry.
Statistical and Computational tools
Microarray pre-processing of the data was performed by using the affy library in BioConductor running under [R] on a 128 Gb RAM 8-Power5+ dual core-processor, symmetric multiprocessing (SMP) unit by IBM. Whereas all statistical tests were performed on a Dell Precision Series 8 Gb RAM QuadCore Workstation by using limma package in [R]/BioConductor. Information theoretical measures (Information Based Similarity) were calculated with the ibs program. Such information theoretical measures were used to infer regulatory interactions between TFs and target genes, i.e. to deconvolute the associated gene regulatory network [10], [16]. As is thoroughly discussed in reference [10], genes highly correlated in their expression patterns are likely to be transcriptional partners. Since dynamic correlations between genes and their TFs are of a nonlinear character, we have used mutual-information related measures instead of linear measures such as Pearson's correlations and covariances. Graphical depiction and network analyses were performed with Cytoscape. Non-equilibrium thermodynamics calculations and other analyses were performed with custom [R] and shell scripts. Pathway enrichment analysis was made by means of hypergeometric testing of databases by Reactome [28]. Gene Set Enrichment Analyses were performed with the GSEA Java library [32].
Results
Core regulation genes
From the set of differentially expressed genes, data mining techniques were implemented to determine a set of genes that at the same time were involved in metabolic activity at the cell level and/or in cancer; and possess experimental data to accurately determine the parameters involved in our non-equilibrium thermodynamic model. We have then come to investigate cell energetics in relation with mRNA expression of the following TFs: MNDA, POU2AF1, MEF2C and SMAD3.
MNDA acts as a transcriptional activator/repressor in the myeloid lineage [33]. Also plays a role in the granulocyte/monocyte cell-specific response to interferon and stimulates the DNA binding of the transcriptional repressor protein YY1
[34]. It belongs to a family of P200 proteins that inhibit cell cycle progression and modulates cell survival. POU2AF1 is a transcriptional coactivator [35]–[37] that specifically associates with either OCT1 or OCT2. It is located in a so-call Tumor suppressor region in 11q22-23 and it is suspected to modify non-coding effects on gene expression [35]. POU2AF1 amplification has been detected in multiple myeloma cells and this copy number variation also reflected in over-expression at both the mRNA and protein levels [37]. MEF2C is a transcription activator which binds specifically to the MEF2 element present in the regulatory regions of many muscle-specific genes. In fact, p38, ERK5, and MEF2C itself have been recently described as novel downstream Brk (PTK6) effector pathways [38] supposedly playing a role in primary breast cancer. The actual mechanism seems to be related with ERK5 being an input to cyclin D1 transcriptional up-regulation, maybe following MEF2C-dependent up-regulation and recruitment of c-JUN to the cyclin D1 promoter [38]. MEF2C is a transcriptional enhancer whose biological function in human breast cancer is still unknown. However, it has been shown that its chromosomal localization is assigned to the so-called mammary cancer susceptibility 1 locus (Mcs1) on chromosome 2q1 segregating with the sensitivity to mammary cancer development in a murine model [39]. SMAD3 is a transcriptional modulator activated by TGF-
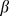 (transforming growth factor) and activin type 1 receptor kinase. TGF-
(transforming growth factor) and activin type 1 receptor kinase. TGF-
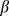 induces a cytostatic response in most normal cell types, but in cancer cells promotes metastasis, and its high expression is correlated with poor prognosis [40]. Knocking-off experiments have showed that the TGF-
induces a cytostatic response in most normal cell types, but in cancer cells promotes metastasis, and its high expression is correlated with poor prognosis [40]. Knocking-off experiments have showed that the TGF-
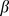 -induced SMAD3-mediated transcriptional response, was mitigated and enhanced by SMAD3 and SMAD2 knockdown, respectively, and this could be directly correlated with divergence in the regulation of tumor angiogenesis in vivo [41].
-induced SMAD3-mediated transcriptional response, was mitigated and enhanced by SMAD3 and SMAD2 knockdown, respectively, and this could be directly correlated with divergence in the regulation of tumor angiogenesis in vivo [41].
In view of the importance of these genes in the onset and development of breast cancer, we have decided to investigate both the energetics and connectivity of their functions in both normal and neoplastic cells. We will discuss the role of TF activity, activation energies and chemical potentials of transcription as outlined and we will suggest some routes to follow to further understand the role of metabolic changes (both at energetics and pathways level) in breast malignancy.
Thermodynamic analysis of MNDA, POU2AF1, MEF2C and SMAD3 transcription factors
The role of integrative analysis in modern (high throughput) genomics is to present a basis for hypotheses generation that may be tested in more specific and detailed studies. In this sense our non-equilibrium thermodynamics calculations (plus some assumptions regarding energy release within the cell) supply a means to try to unveil causal structure of the regulatory interactions from correlation analysis (such as the network study presented here), thus providing a more appropriate frame for study.
The parameters needed to calculate the intensity-dependent concentration, the expression affinity and the chemical potential of transcription for MNDA, POU2AF1, MEF2C and SMAD3 are shown in Table 2 and the explicit calculations are in Table 3. If we look at the gene expression profiles in the surrogate model (Figure 2), we notice the presence of stochastic components with a high variance. In the mRNA concentration representation (Figure 3) although we retain stochastic evolution, it is easier to notice differences between the concentrations for the considered transcripts. Lowest concentration values of MEF2C are present in almost all stages of tumor progression. This gene also showed the lowest variance between sampling points as it could be seen in figure 3. Even if POU2AF1 expression levels were almost as low as those of MEF2C, its variance was much greater and in some instances mRNA concentrations doubling its baseline level are present. SMAD3 was in general found to present medium expression levels and medium variance. MNDA showed the highest concentrations (about 3.5 times that of MEF2C in some instances) and also showed the greater variability. With regards to transcriptional affinities (Figure 4) similar comments can be made as with expression levels (these are after all thermodynamic conjugate variables). We can however notice higher variability around the mean behavior which points out to the possible presence of activation processes taking place. If we consider that transcriptional affinity and transcription level are conjugate (i.e. its product is an energy term), the fact that affinities (for example in the case of MNDA) present a pattern of variance different from that of the expression levels, imply the existence of energy fluctuations that may be due to activation processes. This assertion becomes clearer when we consider the time evolution of the chemical potential (Figure 5). The chemical potential associated with MNDA transcription presents the lowest values (thus it is easier to synthesize because of its lower activation energy barrier) as well as the higher variance. SMAD3 also presented relatively low chemical potentials and medium values of variance. In accordance with mRNA concentration profiles, POU2AF1 showed medium chemical potentials but large variance and MEF2C showed the largest mean values of chemical potentials but a lower variability.
Table 2. Thermodynamic parameters needed for the calculations of 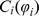 ,
, 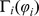 and
and 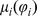 at physiological temperature (
at physiological temperature (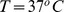 or, 310.15 K).
or, 310.15 K).
| Gene |
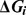 (kcal/mol) (kcal/mol) |
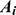
|
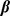 (mol/kcal) (mol/kcal) |
| MNDA | 433.97 | 3105 |
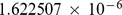
|
| POU2AF1 | 473.5 | 4684 |
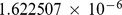
|
| MEF2C | 472.81 | 5110 |
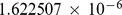
|
| SMAD3 | 465.08 | 4497 |
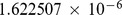
|
Table 3. Thermodynamic calculations of 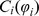 ,
, 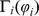 and
and 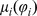 at physiological temperature (
at physiological temperature (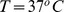 or, 310.15 K).
or, 310.15 K).
| Gene |
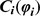
|
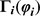
|
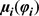
|
| MNDA |
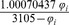
|
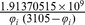
|
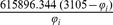
|
| POU2AF1 |
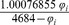
|
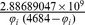
|
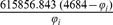
|
| MEF2C |
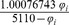
|
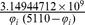
|
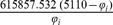
|
| SMAD3 |
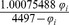
|
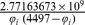
|
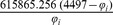
|
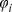 is the corresponding gene expression intensity value, concentration [ = ] picomolar, chemical potentials [ = ]
is the corresponding gene expression intensity value, concentration [ = ] picomolar, chemical potentials [ = ] 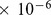 kcal/mol.
kcal/mol.
Non-linear correlation networks
The results of the inferred non-linear correlation network centered in the transcriptional regulation partners for the core regulation genes could be seen in Figure 6. Indirect correlations between these 4 genes were not pruned (e.g. by using the Data Processing Inequality [42]) since it is precisely through these links (and their corresponding regulatory and signalling pathways) that the interconnection between metabolism and transcriptional regulation is more clear, as it may be evident later. Referring to Figure 6, it may be noticed that genes participating in the interactions represented by links colored in red have been related in the literature with breast cancer, whereas yellow links are associated with other types of cancer-related genes; turquoise links are interactions associated with genes in metabolic disorders and navy blue links are otherwise (complete list of references available upon request). Some genes are transcriptionally correlated with more than one of these transcription factors, as is it can be seen in the network and noticed in the caption of Figure 6. Hence, close to a half (about 45% indeed) of the regulatory interactions found in this network analysis have been reported to play a role either in cancer, metabolic deregulation or both.
The structure of the GRN (Figure 6) resembles a dual-control loop centered in core genes MEF2C and MNDA (and its targets-interactors) that may be fine-tuned by the action of POU2AF1, SMAD3 and its associated genes. The pathway analysis performed (see the following subsection) presents also indirect evidence pointing in this direction. However, conclusive assertions could only be made after more detailed and specific studies in regulatory dynamics are performed by means of actual time-course experiments.
The somewhat special role of MEF2C related interactions is worth-mentioning, in particular with regards to the regulatory effects of its MADS-box structure. It was previously shown that MADS-box gene transcription factor is a common regulator extremely conserved across plant and animal kingdoms. This is particularly the case in either H. Sapiens and A. Thaliana. In particular, from our core genes, human MEF2C belongs to the MADS-box genes family [43]. A representative short aminoacid sequence (60 aa) from MEF2C human protein (MEF2C_Hs) was taken from reference [43] and used for searching conserved domain sequences within A. Thaliana's MADs family members, by using NCBI Blast protein tool [44]. Protein conserved domains from Putative MADS-box family transcription and Mef2 myocyte enhancer factor 2 families sharing domain-architecture were found, 50 and 101 respectively (E-value 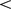 1E-07).
1E-07).
From the inferred gene regulatory network, MEF2C target genes: TAF12 and POLR2I correspond to be Arabidopsis homolog genes with TAF12 and NRPB9A/B. The one-to-one gene interaction in human between MEF2C and POLR2I founded in the present work corresponds to 25 MEF2C-MADS-box conserved domain genes and NRPB9A interactions as results from a search in the Arabidopsis thaliana root transcriptional interactions database [45].
Pathway analysis
In order to sketch the role of specific biochemical sets of reactions related to energetic and transcriptional deregulation processes in the regulatory networks just referred, we performed pathway-related analyses in the list of 712 genes obtained from the IBS calculations. In particular, we made a pathway enrichment analysis for the statistical over-representation of pathways in this gene set.
Significantly enriched pathways include cancer related pathways such as Activation of ATR in response to replication stress, Association of RAD51 with RAD52:DNA double-strand break ends, as well as metabolic activity related pathways like Cholesterol biosynthesis and Addition of galactose by beta 4-galactosyltransferases. With regards to the activation of ATR in response to replication stress [46]; ATR belongs the PI3/PI4-kinase family, and is most closely related to ATM sharing similarity with rad3, a cell cycle checkpoint gene required for cell cycle arrest and DNA damage repair in response to DNA damage [47]. This kinase has been shown to phosphorylate checkpoint kinase CHK1, checkpoint proteins RAD17, and RAD9, as well as tumor suppressor protein BRCA1 [48]. Deregulation of the ATR signaling pathway has been related with various instances of breast cancer development [49]–[51]. Figure 7 shows some prototypical cancer pathways that are deregulated (abnormally expressed genes are marked with a red star) in the 1191 whole-genome gene expression experiments analyzed. Those genes are transcriptionally correlated with one or more of the genes in our core set as it could be verified in a closer analysis of the network depicted in Figure 6 (Cytoscape .cys file for Figure 6 available upon request). We can see that mRNA levels for CDC25A, CDC25C, BRCA1, PRKAB2, S6K, ROCK1 and RASGRP2 are highly correlated with MEF2C expression; whereas levels of RAD51, PRKAA2, HRASLS2 correlate with MNDA. ROCK2 is correlated with SMAD3. Finally expression of RAD52 is associated both with those of MEF2C and SMAD3.
Transcriptional deregulation also occurred in genes participating in metabolic pathways, for instance, Figure 8 depicts the cholesterol biosynthesis pathway, again genes marked with a red star are abnormally expressed. Cholesterol biosynthesis and in particular the so-called Mevalonate pathway is a central process in the metabolic functioning: the starting point is Acetyl CoA which is a product of the metabolism of namely any source of energy -being carbohydrates, fats or proteins-. In the mevalonate branch of the cholesterol biosynthesis pathway (depicted in Figure 8), HMGCS1 and HMGCR proteins are enzymatically involved in the synthesis of mevalonate. Our analysis has shown that both genes HMGCS1 and HMGCR are abnormally regulated at the transcriptional level. Also in the reaction from  -isopentyl 5 pyrophosphate to trans-trans farnesyl pyrophosphate an auxiliary enzyme GGPS1 is abnormally expressed. In the Lathosterol synthesis both TM7SF2 and NSDHL present affected mRNA expression levels. Expression levels of HMGCS1, HMGCR, GGPS1, NSDHL, TM7SF2 are all correlated with MEF2C expression in our analysis. It is noticeable that deregulation of the mevalonate pathway -and in particular of HMGCR - has been recently correlated with primary breast carcinoma [52]. Hypotheses relating metabolic dysfunction of the mevalonate pathway with primary breast cancer is also supported by the effect that cholesterol-controlling drugs have in patients with breast cancer [53], [54].
-isopentyl 5 pyrophosphate to trans-trans farnesyl pyrophosphate an auxiliary enzyme GGPS1 is abnormally expressed. In the Lathosterol synthesis both TM7SF2 and NSDHL present affected mRNA expression levels. Expression levels of HMGCS1, HMGCR, GGPS1, NSDHL, TM7SF2 are all correlated with MEF2C expression in our analysis. It is noticeable that deregulation of the mevalonate pathway -and in particular of HMGCR - has been recently correlated with primary breast carcinoma [52]. Hypotheses relating metabolic dysfunction of the mevalonate pathway with primary breast cancer is also supported by the effect that cholesterol-controlling drugs have in patients with breast cancer [53], [54].
Further insight in the mechanisms that connect transcriptional deregulation in cancer and metabolic abnormalities could be found if we bring to attention a set of genes, consisting in PRKAA2, PRKAB2, CREB1, MAP3K1, DUSP4, TLR3, JUN, UBE2V1, TLR2 and MEF2C. All these genes turned out to be involved in the enrichment analyses both in the case of cancer-related pathways as well as metabolism-related pathways. PRKAA2 and PRKAB2 are AMPK subunits, CREB1 is a MAP kinase target and the rest are related with Toll-like receptor activity. Hence, kinase activity and signalling dysfunction seem likely to play a central role in cancer development, well beyond the usual RAS-ERK pathway. For more information, please refer to Table 4.
Table 4. Main REACTOME [28] pathways enriched in differentially expressed genes.
| Un-adjusted probability | REACTOME Pathways | Gene ID |
| 1.13E-03 | Activation of ATR in response to replication stress | RFC2, RAD17, CDC25A, |
| ORC5L, CDC6, CDC25C, | ||
| MCM4 | ||
| 2.53E-03 | Cholesterol biosynthesis | HMGCR, HMGCS1, TM7SF2, |
| NSDHL, GGPS1 | ||
| 2.81E-03 | G2/M Checkpoints | RFC2, RAD17, CDC25A, |
| ORC5L, CDC6, CDC25C, | ||
| MCM4 | ||
| 5.92E-03 | Phospholipase C-gamma1 binds to the activated EGF receptor | PLCG1, EGFR |
| 5.92E-03 | EGFR activates PLC-gamma1 by phosphorylation | PLCG1, EGFR |
| 5.92E-03 | Active PLC-gamma1 dissociates from EGFR | PLCG1, EGFR |
| 5.92E-03 | EGFR interacts with phospholipase C-gamma | PLCG1, EGFR |
| 1.48E-02 | Myosin regulatory light chain phosphorylation by ROCK | ROCK1, ROCK2, MYH10 |
| 1.86E-02 | PLC-gamma binds to the active receptor | PDGFRB, PLCG1 |
| 1.86E-02 | PLC-gamma hydrolyses PIP2 | PDGFRB, PLCG1 |
| 1.86E-02 | ROCK activation by Rho | ROCK1, ROCK2 |
| 1.86E-02 | CREB phosphorylation through the activation of Adenylate Cyclase | ADCY8, CREB1 |
| 2.07E-02 | DNA Repair | POLB, APEX1, RFC2, |
| POLR2F, POLR2I, BRCA1, | ||
| FANCI, RAD51,RAD52, | ||
| POLR2C | ||
| 2.43E-02 | MAPK targets/Nuclear events mediated by MAP kinases | DUSP4, JUN, CREB1, |
| MEF2C | ||
| 2.70E-02 | Crk binds to the active PDGF receptor | PDGFRB, CRK |
| 2.70E-02 | LIM kinase phosphorylation by ROCK | ROCK1, ROCK2 |
| 2.77E-02 | CREB phosphorylation through the activation of CaMKII | NEFL, GRIN1, CREB1 |
| 2.78E-02 | Cell Cycle, Mitotic | BUB1B, RFC2, CEP250, |
| AURKA, CDKN2A, ANAPC2, | ||
| ORC5L, CENPF, TYMS, | ||
| MCM4, PSMD14,CKS1B, | ||
| DNA2, GMNN, CCNE2, | ||
| CDC25C, CDC6, ODF2, | ||
| CDC25A, ERCC6L, GORASP1 | ||
| 3.15E-02 | E2F mediated regulation of DNA replication | CDC25A, ORC5L, CDC6, |
| TYMS | ||
| 3.67E-02 | Interaction of MyoD-E protein with MEF2 | MYOD1, MEF2C |
Gene Set Enrichment Analysis
Previous analysis in this work were mostly derived from the list of differentially expressed genes i.e. single-gene analysis. In order to include the collective behavior of genes within pathways and functional modules, we performed Gene Set Enrichment Analysis (GSEA) to determine whether members of gene sets 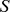 tend to occur at the top or bottom within a ranked list
tend to occur at the top or bottom within a ranked list 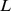 (genes showing largest difference between phenotypes) [55], [56]. GSEA was then applied to our expression data set (1191 samples) considering two sub-collections: Canonical Pathways (CP) and Cancer Modules (CM). Relevant parameters used for both sub-collections were the following: permutations - 1000, scoring scheme - weighted and metric - Signal2Noise.
(genes showing largest difference between phenotypes) [55], [56]. GSEA was then applied to our expression data set (1191 samples) considering two sub-collections: Canonical Pathways (CP) and Cancer Modules (CM). Relevant parameters used for both sub-collections were the following: permutations - 1000, scoring scheme - weighted and metric - Signal2Noise.
Among the resulting statistically significant enriched gene sets (Nominal p-value 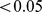 and FDR q-value
and FDR q-value 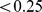 ), we obtain 77 pathways and 38 modules from CP and CM respectively. Table 5 shows some selected modules and pathways up-regulated in cancer that also involve genes with metabolic activity. These selected over-represented gene sets support our results from previous section in which we found deregulation of other genes beyond oncogenes and tumor suppressor genes (i.e. metabolic genes) promoting changes at the transcriptional level.
), we obtain 77 pathways and 38 modules from CP and CM respectively. Table 5 shows some selected modules and pathways up-regulated in cancer that also involve genes with metabolic activity. These selected over-represented gene sets support our results from previous section in which we found deregulation of other genes beyond oncogenes and tumor suppressor genes (i.e. metabolic genes) promoting changes at the transcriptional level.
Table 5. Gene sets from the Molecular Signatures Database (MSigDB) corresponding to 2 of the 5 major collections [56].
| Collection | No. gene | Enriched sets | Nominal | FDR | NES |
| sets | p-value | q-value | |||
| C2 Curated | 3272 | ||||
| Canonical Pathways | 880 | mTOR pathway | 0.0 | 0.191 | 1.72 |
| Rho pathway | 0.004 | 0.197 | 1.74 | ||
| Integration of energy metabolism | 0.008 | 0.219 | 1.783 | ||
| Metabolism of proteins | 0.0 | 0.188 | 1.809 | ||
| C4 Computational | 881 | ||||
| Cancer Modules | 454 | module 159 | 0.002 | 0.148 | 1.61 |
| module 273 | 0.002 | 0.158 | 1.62 | ||
| module 346 | 0.013 | 0.177 | 1.67 |
Summary of the the GSEA results: some of the enriched gene sets involved in cancer and metabolism are shown (Nom. pval 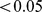 and FDR
and FDR 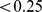 ).
).
Enrichment plots for selected cancer modules, in particular modules 159 (Nom. p-value = 0.002 and FDR = 0.148) and 273 (Nom. p-value = 0.002 and FDR = 0.158) are shown in Figure 9-A, B. Additionally, Figure 9-C, D show enrichment plots for cancer-metabolism related pathways i.e. mTOR (Nom. p-value 0.0 and FDR = 0.191) and integration of energy metabolism (Nom. p-value = 0.008 and FDR = 0.219) pathways. Within module 159, isoforms members from the RAS oncogene family (RAB10, RAB11A, RAB13, RAN, etc.) were found, additionally, genes CDC42 (not shown) and RHOA including in DNA damage (ATP dependent), Rho pathway and cell cycle progression (see Figure 7). Module 273 includes genes from the ATP synthase family, related to metabolic pathways, oxidative phosphorylation, CBFB and FOXO4 genes that participate in the regulation of nuclear SMAD2/3 signaling, MEF2C and POLR2 isoforms in the DNA repair pathway dependent on ATM. In module 346, ARSA, ASAH1, B4GALNT1 genes involved in metabolism of lipids and lipoproteins, B4GALNT1, phospholipase members of families A2, C and D (participates in mTOR pathway) were found.
0.0 and FDR = 0.191) and integration of energy metabolism (Nom. p-value = 0.008 and FDR = 0.219) pathways. Within module 159, isoforms members from the RAS oncogene family (RAB10, RAB11A, RAB13, RAN, etc.) were found, additionally, genes CDC42 (not shown) and RHOA including in DNA damage (ATP dependent), Rho pathway and cell cycle progression (see Figure 7). Module 273 includes genes from the ATP synthase family, related to metabolic pathways, oxidative phosphorylation, CBFB and FOXO4 genes that participate in the regulation of nuclear SMAD2/3 signaling, MEF2C and POLR2 isoforms in the DNA repair pathway dependent on ATM. In module 346, ARSA, ASAH1, B4GALNT1 genes involved in metabolism of lipids and lipoproteins, B4GALNT1, phospholipase members of families A2, C and D (participates in mTOR pathway) were found.
Figure 9. Enrichment score behavior.

Show the distribution of four gene sets from the: A,B) Cancer Modules and C,D) Canonical Pathways; all of them were found up-regulated in cancer samples. Statistical significance of the shown plots can be found in Table 5.
Core Gene Validation
Since the present work is a theoretical-computational analysis based on several sources for experimental data, no wet-lab validation on biological samples could be done, due to lack of sample-availability. However, an indirect validation procedure is performed by means of data mining for experimental results in the literature supporting our findings. For instance, MEF2C was identified as a candidate gene/molecular marker for the transition from ductal carcinoma in situ (DCIS) to invasive ductal carcinoma (IDC) and evaluated by RT-PCR. In reference [57], patient-matched DCIS/IDC samples were used to test expression profiling in Affymetrix oligonucleotide microarrays (GeneChip HG U133A and HG U133 plus 2.0). MEF2C was shown to be up-regulated in IDC compared with DCIS (p-value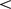 0.01 and FDR
0.01 and FDR 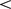 0.01). The average fold change for MEF2C in IDC vs DCIS was 2.03
0.01). The average fold change for MEF2C in IDC vs DCIS was 2.03 1.41 (see Table 6). In order to validate the previously mentioned result, real time PCR was performed in tumor specimens T796, T661, T787 and T808 showing also up-regulation of MEF2C and in situ hybridization was done to confirm its cellular specificity. in situ hybridization showed that MEF2C is present in the cytoplasm of DCIS and IDC, indicating its expression in epithelial tumor cells [57].
1.41 (see Table 6). In order to validate the previously mentioned result, real time PCR was performed in tumor specimens T796, T661, T787 and T808 showing also up-regulation of MEF2C and in situ hybridization was done to confirm its cellular specificity. in situ hybridization showed that MEF2C is present in the cytoplasm of DCIS and IDC, indicating its expression in epithelial tumor cells [57].
Table 6. Gene expression validation for MEF2C and MNDA in breast cancer samples.
| Gene symbol | Fold change from microarray data | Fold change from real-time PCR | Experiment(reference) |
| MEF2C | 2.03 | T796 (1.69); T661 (1.79) | [57] |
| T787 (2.89); T808 (1.38) | |||
| MNDA | S1 (1.74); S2 (1.93) | [59] | |
| S3 (2.67); S4 (3.12) | *NB |
S1–S4 refer to different samples in microarray analysis, T796, T661, T787 and T808 refer to different tumor samples in RT-PCR.
NB refers to experimental determination by Northern blot instead of RT-PCR.
Additionally, in ref. [58], protein expression of MEF2C in breast cancer cell lines was examined. Cellular lysates from exponentially growing MCF-10A, HMEC, MCF-7, T47D, ZR-75-1, and SKBR3 cells were analyzed by Western blot with antibodies specific to MEF2C. MEF2C expression in normal mammary epithelial cells and all breast cancer cell lines examined was observed. Results from this work suggest that Brk inputs to p38 MAPK-dependent activation of MEF2 transcription factors in breast cancer cells.
In a study of differential expression in human breast epithelial cell lines irradiated with low doses of high linear energy transfer radiation, and treated with estrogen assessed with cDNA expression arrays, activity of MNDA was analyzed. MNDA showed a high level of altered expression (see table 6) and it was confirmed by gene-specific semiquantitative reverse transcription polymerase chain reaction, followed by Northern blot analysis. The results showed that the mRNA expression patterns for MNDA was consistent with the expression pattern seen on the array. MNDA showed upregulation in the transformed and tumorigenic cell lines compared a control (MCF-10F cell line) [59].
Expression of MEF2C, SMAD3 and POU2AF1 can be found as deregulated within two different platforms (HGU133A & HGU133plus2) in the BarCode database [60] for breast epithelium, stroma and lobular tissues. As can be seen in Table 7, MEF2C was differentially expressed in more than 60% of breast epithelium tumors samples (HGU133A) and more than 95% of breast stroma tumors samples (HGU133plus2). MNDA expression in breast carcinomas was examined on the EMBL-EBI database. Results from two experiments are shown in Table 8; MNDA was reported up-regulated in both studies.
Table 7. Barcode validation results for differential over-expression of MEF2C, SMAD3 and POU2AF1 within two platforms for several gene-probes in different breast cancer tissues [60], [78].
| Gene symbol/Affy ID | Array | Tissue | % samples |
| MEF2C/209200_at | HGU133A | breast epithelium tumor | 60% |
| MEF2C/209199_s_at | HGU133plus2 | breast stroma tumor | 95% |
| MEF2C/209199_s_at | HGU133plus2 | breast tumor | 60% |
| MEF2C/209200_at | HGU133plus2 | breast tumor | 60% |
| SMAD3/218284_at | HGU133plus2 | breast lobular tumor | 60% |
| POU2AF1/205267_at | HGU133A | breast epithelium tumor | 60% |
Table 8. MNDA expression status in two different microarray experiments also validate our finding [81].
Discussion
We have found an interesting trend in the transcriptional activity coefficients for and other well characterized gene probes. It could be seen [19] that the values of the chemical potentials of transcription 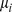 are in general lower for genes that act as transcription factors (such as MNDA, POU2AF1, MEF2C and SMAD3) and high for genes with no-known TF-activity (such as, for example, IL2RB, CD69, TNFRSF1B and TNFRSF14). One possible exception in the group of genes studied is GLDC which codes for a glycine-dehydrogenase enzyme that is anchored to the mitochondrion and has no reported evidence of transcription factor activity yet is grouped with the TF genes in the set of low chemical potential. This fact supports the TF
are in general lower for genes that act as transcription factors (such as MNDA, POU2AF1, MEF2C and SMAD3) and high for genes with no-known TF-activity (such as, for example, IL2RB, CD69, TNFRSF1B and TNFRSF14). One possible exception in the group of genes studied is GLDC which codes for a glycine-dehydrogenase enzyme that is anchored to the mitochondrion and has no reported evidence of transcription factor activity yet is grouped with the TF genes in the set of low chemical potential. This fact supports the TF low
low 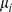 hypothesis. The physicochemical meaning of this finding seem to point-out to transcription factors as genes whose expression is regulated by lower activation-energy barriers. Since TF's are involved in the transcriptional activation of other genes, it is expected that they are synthesized first when energy is started to being released by metabolic processes in the cell. Transcriptional targets should, in general be synthesized later and with higher activation energies. Even between transcription factors, we hypothesize that genes upstream in the transcriptional cascade must in general present lower chemical potentials of transcription. This mechanism would function thus as a lock-in-the-trigger for transcriptional cascading. It is important to stress that the chemical potentials
hypothesis. The physicochemical meaning of this finding seem to point-out to transcription factors as genes whose expression is regulated by lower activation-energy barriers. Since TF's are involved in the transcriptional activation of other genes, it is expected that they are synthesized first when energy is started to being released by metabolic processes in the cell. Transcriptional targets should, in general be synthesized later and with higher activation energies. Even between transcription factors, we hypothesize that genes upstream in the transcriptional cascade must in general present lower chemical potentials of transcription. This mechanism would function thus as a lock-in-the-trigger for transcriptional cascading. It is important to stress that the chemical potentials 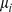 for spontaneous transcription do not show a big difference between TFs and Target Genes (TGs) at low gene expression intensity levels (
for spontaneous transcription do not show a big difference between TFs and Target Genes (TGs) at low gene expression intensity levels (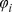 ) but show a significant difference at high values of
) but show a significant difference at high values of 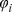 (recall that the values of
(recall that the values of 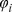 at which the chemical potential becomes negative are the saturation constants
at which the chemical potential becomes negative are the saturation constants 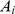 and these show statistical significant differences between TFs and TGs). Thus, higher saturation limits for the chemical potentials of TFs suggest both stability and spontaneity in the expression of these as compared to TGs. Pathway analysis also make evident the fact that transcriptional deregulation occurs not only at the single-gene level, since in some cases several genes in the same pathway are affected.
and these show statistical significant differences between TFs and TGs). Thus, higher saturation limits for the chemical potentials of TFs suggest both stability and spontaneity in the expression of these as compared to TGs. Pathway analysis also make evident the fact that transcriptional deregulation occurs not only at the single-gene level, since in some cases several genes in the same pathway are affected.
With regards to more particular findings, we have already discussed that deregulation seem to be present at the pathway level. Specifically we have found two related sets of pathways affected by deregulation in several of their genes. These two sets correspond grossly with cancer-related pathways (cell cycle, DNA repair, apoptosis) and metabolic control pathways (cholesterol synthesis, AMPK release, etc.). Some affected genes are actually part of both kinds of pathways. These genes are correlated with MEF2C expression (Figure 6). As we mentioned, the network structure in Figure 6 suggest some kind of control between MEF2C and MNDA, however since our inference method is based in (nonlinear) correlations, causality (i.e. directionality of the transcriptional regulation interactions) could not be ascribed to either of the genes. Bayesian and other inference approaches could be useful for that means, but they would require actual time-course experiments in many, many samples to attain statistical significance. In the case of human samples, for logistic and financial reasons, this does not seem at hand in the near future. A thermodynamic analysis -additional to the correlation network study- such as the one we presented, supplies us with an alternative, that although it involves some assumptions, could give us some hints on the causal structure. It should be stressed however, that these clues should be considered hypotheses to be experimentally verified or disregarded, either on the light of more detailed experiments or more specific thermodynamic assumptions and not conclusive results nor proven facts.
Let us recall the results of the thermodynamic analysis and the correlation network analyses. In Figure 3 we observed that whereas MEF2C presents comparatively low mRNA concentrations and low concentration variances, along with a very high connectivity (degree), MNDA presents both a high concentration and high variability in the sample-set, as well as the second highest degree. In the other hand, SMAD3 and POU2AF1 present medium-to-low concentrations, and much smaller connectivities. However, POU2AF1 presents a high variability, while SMAD3 has a medium-to-small variance. If we assume that, in effect, there is a kind of control loop between MNDA and MEF2C somehow fine-tuned by the action of POU2AF1 and SMAD3 (and their interactors); then we may sketch a possible causal scenario if we consider that energy is been released gradually within the cells. MNDA is present at high concentrations (Figure 3) almost always because its associated chemical potential (i.e. free energy of formation) is low (Figure 5) and hence it is easier to synthesize, in the other hand MEF2C is present in small amounts (Figure 3) because its chemical potential is high (Figure 5) so it is more difficult to synthesize. This seem to suggest that we have a balance between the concentrations of MNDA and MEF2C. If energy release within the tumor cells occurs as a consequence of gradual activated processes, then MNDA should be transcribed first and MEF2C expression may be regulated by MNDA and its interactors. Once MEF2C is being produced, then activation of its many transcription targets should cause abnormal regulation in the already mentioned cancer and metabolic related pathways, thus causing diseased cellular states and, ultimately, carcinogenesis.
With regards to the design of possible experimental protocols to verify some of these findings, we must notice that experimental techniques in genomics are rapidly evolving, in such a way that probing the cell in real time under almost in vivo conditions is now becoming possible. In particular with regards to experimental verification of our findings, there have been several instances in which related work has been done. One approach to provide real-time semi-quantitative analysis of transcription is the imaging of reporter gene expression, for example, using firefly luciferase [61], [62]. There is also the need for protein production/degradation rates to experimentally assess gene expression dynamics [63]. Nevertheless, in the most successful cases it has been even possible to account for fluctuations and stochastic components in the dynamics of gene expression [64]. Other modern techniques to monitor the dynamics of gene expression are based in quantitative measurements of polymerase chain reaction (q-PCR) often used in conjunction with immunoprecipitation [65], [66]. In the near future it is also very likely that techniques such as microcalorimetry at a single cell level, and especially Isothermal Titration Calorimetry (ITC) could be applied on a real-time basis to monitor changes in local thermodynamics within the cell [67]. These experiments will also shed light in the thermal component of messenger RNA dynamics, and thus will serve to fine-tune the predictions of the model presented here.
Conclusions
In this paper, we have analyzed the role that thermodynamic fluctuations in energy at the cell-level play in the synthesis of transcription factors MNDA, POU2AF1, MEF2C and SMAD3 and how can this energetic constrains be related with the presence of primary breast carcinomas. In doing so we studied systematically mRNA levels for high throughput, whole-genome gene expression profiling. A set of 1191 publicly available microarrays was first studied by inferring gene regulatory networks based in a non-linear correlation measure between gene expression vectors arranged in a surrogate dynamic model of tumor progression. In the other hand, a non-equilibrium thermodynamic formalism was used to calculate the concentration-dependent gene expression intensity, the chemical potentials of transcription and their associated affinities in order to establish energetic constraints that help us to evaluate the biological hypotheses. Hence, a connection was established between mRNA concentration patterns -as given by experimental gene expression profiles- and local cell energetics - by means of these irreversible thermodynamical quantities. By analyzing the different patterns of gene expression for the selected genes, the corresponding non-equilibrium energetics, as well as their correlation structure as given by network analysis, an integrative model for the action of a core set of master regulator genes was developed. We analyzed in the surrogate dynamic model already mentioned to look up for transcriptional regulation paths. All these analyses suggest a novel potential role of transcription factor energetics in tumor development.
In particular, by using a data integration approach that combines experimental evidence for high throughput genome wide gene expression, a non-equilibrium thermodynamics analysis, nonlinear correlation networks as well as database mining, we were able to hypothesize about the role that transcription factors MEF2C and MNDA may have as master regulators in primary breast cancer phenomenology, as well as the possible interrelationship between malignancy and metabolic dysfunction. Nevertheless, we can never emphasize enough that these findings should be regarded as hypothesis generators rather than as conclusive results. However, we believe that systematic studies relying in data integration guided by well-founded physical principles rather than intuition may become mandatory in time. This work intends to point-out in such direction. However, further, deeper investigations are needed in this direction in order that our understanding of these extremely complex phenomena will be substantially increased.
Acknowledgments
We thank Professor Elena R. Álvarez-Buylla for her useful comments.
Funding Statement
The authors gratefully acknowledge support by grant: PIUTE10-92 El Instituto de Ciencia y Tecnología del Distrito Federal (ICyT-DF) [Contract 281-2010], as well as federal funding from the National Institute of Genomic Medicine (Mexico). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Tennant DA, Durn RV, Gottlieb E (2010) Targeting metabolic transformation for cancer therapy,. Nature Reviews Cancer 10, 4: 267–77. [DOI] [PubMed] [Google Scholar]
- 2. Geschwind JF, Georgiades CS, Ko YH, Pedersen PL (2004) Recently elucidated energy catabolism pathways provide opportunities for novel treatments in hepatocellular carcinoma,. Expert Reviews of Anticancer Therapy 4, 3: 449–57. [DOI] [PubMed] [Google Scholar]
- 3. Bhardwaj V, Rizvi N, Lai MB, Lai JC, Bhushan A (2010) Glycolytic enzyme inhibitors affect pancreatic cancer survival by modulating its signaling and energetics,. Anticancer Research 30, 3: 743–9. [PubMed] [Google Scholar]
- 4. Dennison JB, Balakrishnan K, Gandhi V (2009) Preclinical activity of 8-chloroadenosine with mantle cell lymphoma: roles of energy depletion and inhibition of DNA and RNA synthesis,. British Journal of Haematology 147, 3: 297–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wallace DC, Fan W, Procaccio V (2010) Mitochondrial energetics and therapeutics,. Annual Re-views of Pathology 5: 297–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wallace DC, Fan W (2010) Energetics, epigenetics, mitochondrial genetics,. Mitochondrion 10, 1: 12–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Galant NJ, Wang H, Lee DR, Mucsi Z, Setiadi DH, et al. (2009) Thermodynamic role of glutathione oxidation by peroxide and peroxybicarbonate in the prevention of Alzheimer's disease and cancer,. Journal of Physical Chemistry A 113, 32: 9138–49. [DOI] [PubMed] [Google Scholar]
- 8. Tonon G (2008) From oncogene to network addiction: the new frontier of cancer genomics and therapeutics,. Future Oncology 4, 4: 569–77. [DOI] [PubMed] [Google Scholar]
- 9. Sethi JK, Vidal-Puig A (2010) Wnt signalling and the control of cellular metabolism,. Biochemical Journal 427,1: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hernández-Lemus E, Velázquez-Fernández D, Estrada-Gil JK, Silva-Zolezzi I, Herrera-Hernández MF, et al. (2009) Information Theoretical Methods to Deconvolute Genetic Regulatory Networks applied to Thyroid Neoplasms,. Physica A 388: 5057–5069. [Google Scholar]
- 11. Kong YC, Wei WZ, Tomer Y (2010) Opportunistic autoimmune disorders: from immunotherapy to immune dysregulation,. Annals of the New York Academy of Sciences 1183: 222–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piulats J, Tarrason G (2001) E2F transcription factors and cancer,. Clinical and Translational Oncology 3,5: 241–9. [Google Scholar]
- 13. Thompson MR, Xu D, Williams BR (2009) ATF3 transcription factor and its emerging roles in immunity and cancer,. Journal of Molecular Medicine 87, 11: 1053–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Beger M (2004) Expression pattern of AP-2 transcription factors in cervical cancer cells and analysis of their influence on human papillomavirus oncogene transcription,. Journal of Molecular Medicine 79, 5–6: 314–320. [DOI] [PubMed] [Google Scholar]
- 15. Shen Q, Brown PH (2003) Novel Agents for the Prevention of Breast Cancer: Targeting Tran-scription Factors and Signal Transduction Pathways,. Journal of Mammary Gland Biology and Neoplasia 8,1: 45–73. [DOI] [PubMed] [Google Scholar]
- 16. Baca-López K, Hernández-Lemus E, Mayorga M (2009) Information-theoretical analysis of gene expression to infer transcriptional interactions,. Revista Mexicana de Física 55, 6: 456–466. [Google Scholar]
- 17. Bolstad BM, Irizarry RA, Astrand M, Speed TP (2003) A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Bias and Variance,. Bioinformatics 19, 2: 185–193. [DOI] [PubMed] [Google Scholar]
- 18. Smyth G (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments,. Statistical Applications in Genetics and Molecular Biology 3: 3. [DOI] [PubMed] [Google Scholar]
- 19.Hernández-Lemus E (2010) Non-equilibrium of transcriptional bursts, New Trends in Statistical Physics: Festschrift in honor of Leopoldo García-Colín's 80 th Birthday, Macáas, A., Dagdug, L. (eds.), World Scientifi.
- 20. Hernández-Lemus E (2009) Non-Equilibrium Thermodynamics of Gene Expression and Transcrip-tional Regulation,. Journal of Non-equilibrium Thermodynamics 34, 4: 371–394. [Google Scholar]
- 21. Reguera D, Rubí JM, Vilar JMG (2005) The mesoscopic dynamics of thermodynamic systems,. J Phys Chem B 109: 21502–21515. [DOI] [PubMed] [Google Scholar]
- 22. Carlon E, Hein T (2006) Thermodynamics of RNA/DNA hybridization in high density oligonu-cleotide arrays,. Physica A 362: 433–449. [Google Scholar]
- 23. Lu ZJ, Matthews DH (2008) Effint siRNA selection using hybridization thermodynamics,. Nu-cleic Acids Research 36, 2: 640–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hekstra D, Taussig AR, Magnasco M, Naef F (2003) Absolute mRNA concentrations from sequence-specific calibration of oligonucleotide arrays,. Nucleic Acids Research 31, 7: 1962–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hernández-Lemus E, Correa-Rodríguez MD (2011) Non-equilibrium hyperbolic transport in tran-scriptional regulation,. PLoS ONE 6, 7: e21558 http://dx.plos.org/10.1371/journal.pone.0021558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. García-Colín LS, Rodríguez RF, López de Haro M, Jou D, Pérez García C (1985) Generalized hydrodynamics and extended irreversible thermodynamics,. Physical Review A 31: 2502–2508. [DOI] [PubMed] [Google Scholar]
- 27. Chen M, Eu BC (1993) On the integrability of differential forms related to nonequilibrium entropy and irreversible thermodynamics,. Journal of Mathematical Physics 34, 7: 3012–29. [Google Scholar]
- 28.Reactome website: an open-source, open access, manually curated and peer-reviewed pathway database. Available: http://www.reactome.org. Accessed 2012 July 17.
- 29.Gene Expression Omnibus website. Available: http://www.ncbi.nlm.nih.gov/geo/. Accessed 2012 July 17.
- 30.GPL96 protocol NCBI entry. Available: http://www.ncbi.nlm.nih.gov/projects/geo/query/acc.cgi?acc=GPL96. Accessed 2012 July 17.
- 31. Tan EY, Campo L, Han C, Turley H, Pezella F, et al. (2007) BNIP3 as a progression marker in primary human breast cancer; opposing functions in situ versus invasive cancer,. Clin Cancer Res 13, 2: 467–474. [DOI] [PubMed] [Google Scholar]
- 32.Gene Set Enrichment Analysis website. Available: http://www.broadinstitute.org/gsea/downloads.jsp. Accessed 2012 July 17.
- 33. Burrus GR, Briggs JA, Briggs RC (1992) Characterization of the human myeloid cell nuclear differentiation antigen: relationship to interferon-inducible proteins,. J Cell Biochem 48, 2: 190–202. [DOI] [PubMed] [Google Scholar]
- 34. Xie J, Briggs JA, Briggs RC (1998) Human hematopoietic cell specifinuclear protein MNDA interacts with the multifunctional transcription factor YY1 and stimulates YY1 DNA binding,. J Cell Biochem 70, 4: 489506. [PubMed] [Google Scholar]
- 35. Pittman AM, Webb E, Carvajal-Carmona L, Howarth K, Di Bernardo MC, et al. (2008) Re-finement of the basis and impact of common 11q23.1 variation to the risk of developing colorectal cancer,. Human Molecular Genetics 17, 23: 3720–3727. [DOI] [PubMed] [Google Scholar]
- 36. Duan H, Xiang H, Ma L, Boxer LM (2008) Functional long-range interactions of the IgH 3′ enhancers with the bcl-2 promoter region in t(14;18) lymphoma cells,. Oncogene 27, 53: 6720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhao C, Inoue J, Imoto I, Otsuki T, Iida S, et al. (2008) POU2AF1, an amplifition target at 11q23, promotes growth of multiple myeloma cells by directly regulating expression of a B-cell maturation factor, TNFRSF17,. Oncogene 27, 1: 63–75. [DOI] [PubMed] [Google Scholar]
- 38. Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA (2007) Breast tumor kinase (protein tyrosine kinase 6) regulates heregulin-induced activation of ERK5 and p38 MAP kinases in breast cancer cells,. Cancer Research 67, 9: 199–20. [DOI] [PubMed] [Google Scholar]
- 39. Schuetz SC, Bonin M, Clare SE, Nieselt K, Sotlar K, et al. (2006) Progression-Specific Genes Identified by Expression Profiling of Matched Ductal Carcinomas In situ and Invasive Breast Tu-mors, Combining Laser Capture Microdissection and Oligonucleotide Microarray Analysis,. Cancer Research 66, 10: 5278–5286. [DOI] [PubMed] [Google Scholar]
- 40. Matsuura I, Lai CY, Chiang KN (2010) Functional interaction between Smad3 and S100A4 (metastatin-1) for TGF-beta-mediated cancer cell invasiveness,. Biochemical Journal 426, 3: 327–35. [DOI] [PubMed] [Google Scholar]
- 41. Petersen M, Pardali E, van der Horst G, Cheung H, van den Hoogen C, et al. (2009) Smad2 and Smad3 have opposing roles in breast cancer bone metastasis by differentially affecting tumor angiogenesis,. Oncogene 29: 1351–1361. [DOI] [PubMed] [Google Scholar]
- 42. Margolin AA, Nemenman I, Basso K, Wiggins C, Stolovitzky G, et al. (2006) ARACNe: An Algorithm for the Reconstruction of Gene Regulatory Networks in a Mammalian Cellular Context,. BMC Bioinformatics 7 (Suppl I) S7 doi:10.1186/1471–2105-7-S1–S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Álvarez-Buylla ER, Pelaz S, Liljegren SJ, Gold SE, Burgeff C, et al. (2000) An ancestral MADS-box gene duplication occurred before the divergence of plants and animals,. Proc Natl Acad Sci U S A 97, 10: 5328–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.BLAST website. Available: http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi. Accessed 2012 July 17.
- 45.Chávez Montes RA, Huerta-Verde AJ, Coello G, Hernández-Lemus E, Álvarez-Buylla ER Ara- bidopsis thaliana root transcriptional interactions database. Available: http://132.248.224.66.
- 46. Yilmaz S, Sancar A, Kemp MG (2011) Multiple ATR-Chk1 Pathway Proteins Preferentially As-sociate with Checkpoint-Inducing DNA Substrates,. PLoS ONE 6 (7) e22986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cotta-Ramusino C, McDonald ER, Hurov K, Sowa ME, Harper JW, et al. (2011) A DNA damage response screen identifies RHINO, a 9-1-1 and TopBP1 interacting protein required for ATR signaling,. Science 332 (6035) 1313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Merry C, Fu K, Wang J, Yeh IJ, Zhang Y (2010) Targeting the checkpoint kinase Chk1 in cancer therapy,. Cell Cycle 9 (2) 279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, et al. (2011) Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines,. Br J Cancer 105 (3) 372–81 doi: 10.1038/bjc.2011.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wang K, Ye Y, Xu Z, Zhang X, Hou Z, et al. (2010) Interaction between BRCA1/BRCA2 and ATM/ATR associate with breast cancer susceptibility in a Chinese Han population,. Cancer Genet Cytogenet 200 (1) 40–6. [DOI] [PubMed] [Google Scholar]
- 51. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER (2009) Estrogen inhibits ATR signaling to cell cycle checkpoints and DNA repair,. Mol Biol Cell 20 (14) 3374–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Clendening JW, Pandyra A, Boutros PC, El Ghamrasni S, Khosravi F, et al. (2010) Dysregulation of the mevalonate pathway promotes transformation,. Proc Natl Acad Sci U S A 107 (34) 15051–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Knight LA, Kurbacher CM, Glaysher S, Fernando A, Reichelt R, et al. (2009) Activity of meval-onate pathway inhibitors against breast and ovarian cancers in the ATP-based tumour chemosen-sitivity assay,. BMC Cancer 9: 38 doi:10.1186/1471–2407-9–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ressler S, Mlineritsch B, Greil R (2011) Zoledronic acid for adjuvant use in patients with breast cancer,. Expert Rev Anticancer Ther 11 (3) 333–49. [DOI] [PubMed] [Google Scholar]
- 55. Subramanian A, Tamayo P, Mootha KV, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles,. PNAS 102, 43: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molecular Signature Database. Available: http://www.broadinstitute.org/gsea/msigdb/collections.jsp. Accessed 2012 July 17.
- 57. Schuetz CS, Bonin M, Clare SE, Nieselt K, Sotlar K, et al. (2006) Progression-Specific Genes Iden-tified by Expression Profiling of Matched Ductal Carcinomas In situ and Invasive Breast Tumors, Combining Laser Capture Microdissection and Oligonucleotide Microarray Analysis,. Cancer Res 66: 10. [DOI] [PubMed] [Google Scholar]
- 58. Ostrander JH, Daniel AR, Lofgren K, Kleer CG, Lange CA (2007) Breast Tumor Kinase (Protein Tyrosine Kinase 6) Regulates Heregulin-Induced Activation of ERK5 and p38 MAP Kinases in Breast Cancer Cells,. Cancer Res 67: 9. [DOI] [PubMed] [Google Scholar]
- 59. Roy D, Calaf G, Hei TK (2001) Profiling of Differentially Expressed Genes Induced by High Linear Energy Transfer Radiation in Breast Epithelial Cells,. Mol. Carcinog 31: 4. [DOI] [PubMed] [Google Scholar]
- 60.Barcode project website. Available: http://rafalab.jhsph.edu/barcode/. Accessed 2012 July 17.
- 61. White MRH, Masuko M, Amet L, Elliott G, Braddock M, et al. (1995) Real-time analysis of the transcriptional regulation of Hiv and Hcmv promoters in single mammalian-cells,. J Cell Sci 108: 441455. [DOI] [PubMed] [Google Scholar]
- 62. Norris AJ, Stirland JA, McFerran DW, Seymour ZC, Spiller DG, et al. (2003) Dynamic patterns of growth hormone gene transcription in individual living pituitary cells,. Mol Endoc 17: 193–202. [DOI] [PubMed] [Google Scholar]
- 63. Yu J, Xiao J, Ren X, Lao K, Xie XS (2006) Probing gene expression in live cells, one protein molecule at a time,. Science 311: 16001603. [DOI] [PubMed] [Google Scholar]
- 64. Komorowski M, Finkenstadt B, Rand D (2010) Using a single flrescent reporter gene to infer half-life of extrinsic noise and other parameters of gene expression,. Biophys J 98: 2759–2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. John S, Johnson TA, Sung MH, Biddie SC, Trump S, et al. (2009) Kinetic complexity of the global response to glucocorticoid receptor action,. Endocrinology 50, 4: 1766–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Lee AW, Wang N, Hornell TM, Harding JJ, Deshpande C, et al. (2011) Human cytomegalovirus decreases constitutive transcription of MHC class II genes in mature Langerhans cells by reducing CIITA transcript levels,. Mol Immunol 48: 9–10,1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Salim NS, Feig AL (2009) Isothermal Titration Calorimetry of RNA,. Methods 47,3: 198205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pawitan Y, Bjöhle J, Amler L, Borg AL, Egyhazi S, et al. (2005) Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts,. Breast Cancer Res 7,6: R953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Ivshina AV, George J, Senko O, Mow B, Putti TC, et al. (2006) Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer,. Cancer Res 66,21: 10292–301. [DOI] [PubMed] [Google Scholar]
- 70. Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, et al. (2007) Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series,. Clin Cancer Res 13,11: 3207–14. [DOI] [PubMed] [Google Scholar]
- 71. Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, et al. (2005) Genes that mediate breast cancer metastasis to lung,. Nature 436,7050: 518–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, et al. (2006) Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis,. J Natl Cancer Inst 98,4: 262–72. [DOI] [PubMed] [Google Scholar]
- 73. Miller LD, Smeds J, George J, Vega VB, Vergara L, et al. (2005) An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival,. Proc Natl Acad Sci 102,38: 13550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Farmer P, Bonnefoi H, Becette V, Tubiana-Hulin M, Fumoleau P, et al. (2005) Identification of molecular apocrine breast tumours by microarray analysis,. Oncogene 24,29: 4660–71. [DOI] [PubMed] [Google Scholar]
- 75. Pau Ni IB, Zakaria Z, Muhammad R, Abdullah N, Ibrahim N, et al. (2010) Gene expression patterns distinguish breast carcinomas from normal breast tissues: the Malaysian context,. Pathol Res Pract 206,4: 223–8. [DOI] [PubMed] [Google Scholar]
- 76. Tripathi A, King C, de la Morenas A, Perry VK, Burke B, et al. (2008) Gene expression abnormal-ities in histologically normal breast epithelium of breast cancer patients,. Int J Cancer 122,7: 1557–66. [DOI] [PubMed] [Google Scholar]
- 77. Liu R, Wang X, Chen GY, Dalerba P, Gurney A, et al. (2007) The prognostic role of a gene signature from tumorigenic breast-cancer cells,. N Engl J Med 356,3: 217–26. [DOI] [PubMed] [Google Scholar]
- 78. McCall MN, Uppal K, Jaffee HA, Zilliox MJ, Irizarry RA (2011) The Gene Expression Barcode: leveraging public data repositories to begin cataloging the human and murine transcriptomes,. Nu-cleic Acids Research 39: D1011–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.EMBL Breast tumors transcriptional profiling website. Available: http://www.ebi.ac.uk/gxa/experiment/E-TABM-276/ENSG00000163563. Accessed 2012 July 17.
- 80.EMBL Breast carcinomas transcriptional profiling website. Available: http://www.ebi.ac.uk/gxa/experiment/E-GEOD-10780ENSG00000163563. Accessed 2012 July 17.
- 81.EMBL MNDA transcriptional profiling website. Available: http://www.ebi.ac.uk/gxa/gene?gid=P41218#ef=diseasestate&efv=. Accessed 2012 July 17.