

Abstract
Human babesiosis, a blood-borne infection caused by several species of Babesia, including B. microti, is an emerging disease that is endemic in the Northeast, upper Midwest, and Pacific Northwest regions of the United States. Risk factors for babesiosis include exposure to the infected tick vector and blood transfusions from infected donors. In this work, we cloned and expressed two of the immunodominant antigens from B. microti and used them in a multiplex bead format assay (MBA) to detect parasite-specific IgG responses in human sera. The MBA using recombinant B. microti secreted antigen 1 (BmSA1) protein was more specific (100%) and slightly more sensitive (98.7%) than the assay using a truncated recombinant BMN1-17 construct (97.6% and 97.4%, respectively). Although some antibody reactivity was observed among sera from confirmed-malaria patients, only one Plasmodium falciparum sample was simultaneously positive for IgG antibodies to both antigens. Neither antigen reacted with sera from babesiosis patients who were infected with Babesia species other than B. microti. Both positive and negative MBA results were reproducible between assays and between instruments. Additional studies of these recombinant antigens and of the multiplex bead assay using blood samples from clinically defined babesiosis patients and from blood donors are needed to more clearly define their usefulness as a blood screening assay.
INTRODUCTION
Babesia spp. (phylum Apicomplexa, order Piroplasmorida) are intraerythrocytic protozoan parasites that infect a wide range of mammalian hosts, including rodents, dogs, livestock, and humans (21). Babesiosis in humans is a zoonotic disease that is transmitted from animal reservoirs to humans by ticks of the Ixodes genus (21, 25, 52). The first human infections were identified among asplenic patients in Europe (1957) and among normosplenic residents of Nantucket Island, MA, in the United States (1970) (11, 16, 51, 55). More recently, blood transfusions from otherwise healthy donors who harbor occult infections have been recognized as an important potential risk factor in disease transmission (1, 12, 19, 23, 32, 53).
Depending upon the immunologic status of the host and the species or strain of the parasite, babesiosis can vary from a mild and self-limiting flu-like illness to one with severe, life-threatening complications (21). Severe symptoms resulting from high parasitemia and the ensuing circulatory complications are more commonly encountered among the splenectomized, the immunocompromised, and the elderly (15, 48, 56). Asymptomatic infections have been well documented, and seroprevalence estimates in regions of endemicity have ranged from 0 to 10%, depending on the year, location, and season of blood collection (10, 20, 24, 26–28, 33, 49). In the United States, endemic Babesia microti infections have been identified in the Northeast (in Connecticut, Massachusetts, Rhode Island, New York, and New Jersey) and in the upper Midwest (in Minnesota and Wisconsin), while Babesia duncani infections have been found in Washington and California and infections with a Babesia divergens-like parasite have been found in Washington, Kentucky, and Missouri (2, 6, 14, 17, 18, 41, 54). Some evidence suggests that the geographic range and prevalence of babesiosis infections may be increasing in the United States due to an expansion of white-tailed deer populations and the concomitant increases in tick vector densities (25).
The U.S. Food and Drug Administration has recognized the increasing threat posed to the blood supply by the lack of a sensitive and specific blood product screening assay for babesiosis (13, 14). The current “gold standard” diagnostic assay for babesiosis, visual detection of Babesia organisms in blood films, lacks sensitivity, especially in early and subclinical infections (14). Infected individuals can also be identified by using a PCR-based nucleic acid detection assay or by antibody detection using Babesia-infected hamster blood as the antigen in an immunofluorescence assay (IFA), a Western blot assay, or an enzyme-linked immunosorbent assay (ELISA) (14). Although the reported sensitivities and specificities have ranged from 88% to 100% and from 90% to 100%, respectively, none of these assays are ideally suited for blood product screening campaigns (3, 14, 29, 30, 34, 50). Antibody assays can identify asymptomatically infected individuals and individuals in the chronic late phase of infection (14, 28). However, antibody responses among the three dominant human pathogens of humans, B. microti, B. duncani, and B. divergens, appear to be species specific (7, 14, 17, 18, 41), and three separate assays would be required to conclusively rule out a diagnosis of infection with Babesia sp. PCR, while more sensitive than slide microscopy (29), has a reported limit of detection of 10 piroplasms per milliliter of whole blood (50% confidence interval) (57), and cases of transfusion-transmitted babesiosis have been documented from PCR-negative, antibody-positive blood donors (23, 33, 53).
The development of sensitive, specific, and scalable screening assays for babesiosis would be facilitated by the identification of Babesia-specific peptide and protein antigens. In 2000, Lodes et al. (35) used a randomly sheared B. microti DNA expression library to identify potential antigens of interest. Three families of proteins and two additional, unrelated proteins showed promising results by ELISA and Western blotting (35), and epitopes from the two most reactive antigens were later mapped (22). Cross-reactivity with sera from malaria patients was noted with some of the B. microti peptides (22). More recently, Luo et al. (36) screened a cDNA expression library with sera from infected hamsters and identified one clone, B. microti secreted antigen 1 (BmSA1), that showed diagnostic potential in a limited human study. BmSA1 shared 75% of its coding sequence with a clone previously identified as BMN1-9 by Lodes et al. (35). BmSA1 antigen appeared to be secreted into the serum of the host and was not recognized by sera from animals infected with B. bovis, B. bigemina, B. equi, or B. gibsoni (37).
In the current work, we use classical antigen identification techniques to demonstrate that the BMN1-9/BmSA1 antigen is membrane associated and is commonly recognized by sera from humans infected with B. microti. We then compare BmSA1 to members of one of the immunodominant antigen families identified by Lodes et al., BMN1-17 (22, 35), in a multiplex bead format assay. The identification of sensitive and specific antibody assay antigens may help in the development of suitable screening tools for at-risk blood donations.
MATERIALS AND METHODS
Growth of B. microti, protein extraction, and analysis of proteins by Western blotting.
Animal protocols were approved by the CDC Animal Use Committee. Adult female golden hamsters were inoculated with B. microti Gray strain cells. When the parasitemia reached 10% to 40% (3 to 6 weeks after inoculation), the animals were euthanized and infected blood was collected in heparinized tubes by heart puncture. The blood was stored on ice until it could be processed (usually <1 h). After a 10-min centrifugation at 650 × g, the serum and buffy coat were removed, and the packed red blood cells (RBCs) were resuspended and washed twice with ice-cold buffer containing 0.85% NaCl and 10 mM Na2HPO4 at pH 7.2 (phosphate-buffered saline [PBS; buffer containing 0.85% NaCl and 10 mM Na2HPO4 at pH 7.2]).
To make a total cell protein fraction, the washed RBC pellet was resuspended in approximately 10 vol of 0.01% saponin in water and incubated for 30 min at 37°C. The hypotonic saponin lysate was centrifuged at 3,000 × g for 10 min at 4°C. The pellet was washed repeatedly with ice-cold PBS until the majority of the hemoglobin was removed. The resulting white pellet was resuspended in 1.5 ml of 10 mM Tris (pH 8.0) with 1 mM phenylmethylsulfonyl fluoride (PMSF), 5 mM N-ethylmaleimide (NEM), 10 μM E-64, 10 μM leupeptin, and 1 μM pepstatin A and then sonicated on ice for 15 s (Heat Systems model W-225 sonicator with a microtip at setting number 6). The protein concentration was determined using the bicinchoninic acid (BCA) microassay (Pierce, Rockford, IL).
Alternatively, the washed RBC pellet was resuspended in approximately 10 vol of ice-cold PBS with protease inhibitors and sonicated for 1 min on ice as described above. Membranes and other large debris were collected by centrifugation at 25,000 × g for 20 min at 4°C. The pellet was resuspended in 1 ml of ice-cold PBS with protease inhibitors and sonicated on ice (1 min) in the presence of 2% Triton X-114. Insoluble material was removed by centrifugation for 10 min at 25,000 × g. After overnight storage at −20°C, a membrane-associated protein fraction was isolated by repeated Triton X-114 phase partition extraction using 20 mM HEPES buffer (pH 7.4), 150 mM NaCl, and 2% Triton X-114, as previously described (44). When the aqueous phase was no longer tinted with hemoglobin, the proteins in the detergent phase were collected by acetone precipitation, resuspended in buffer containing 0.5% SDS and 20 mM HEPES at pH 7.4, and heated at 95°C for 5 min (44). After a 5-min centrifugation at 17,000 × g to remove insoluble material, the protein concentration was determined using the BCA microassay (Pierce). As a negative control, a Triton X-114 extract was made from uninfected, washed, hamster RBCs using the protocol given above.
Prior to electrophoresis, each protein preparation was diluted with 1 volume of 2× sodium dodecyl sulfate (SDS) sample loading buffer with 10% β-mercaptoethanol (31), heated at 95°C for 5 min, and centrifuged at 17,000 × g for 5 min to remove insoluble material. Proteins were resolved on 15% SDS-polyacrylamide gels (SDS-PAGE) using the discontinuous electrophoresis buffer system of Laemmli (31). Resolved proteins were then electrotransferred onto a polyvinylidene difluoride (PVDF) membrane (Immobilon P; Millipore Corp., Bedford, MA) and exposed to serum dilutions as previously described (1:100 in 0.3% Tween 20-PBS) (44). Western blots to detect human IgG antibodies (mouse monoclonal anti-human IgG clone HP6017; Zymed, South San Francisco, CA) were conducted using the biotin-streptavidin-alkaline phosphatase system previously described (44). Mouse, hamster, and rabbit IgG antibodies were detected using a biotinylated monoclonal rat anti-mouse IgG (Zymed), a biotinylated rabbit anti-hamster IgG (Southern Biotech, Birmingham, AL), and a biotinylated goat anti-rabbit IgG (Zymed), respectively.
To determine whether epitopes were protein or carbohydrate in nature, sodium periodate and proteinase K treatments were performed on blotted B. microti antigens as previously described (45). Treated blots were then reacted with a strong positive human serum as described above to detect antigens that were insensitive to digestion.
B. microti peptide sequencing.
Approximately 140 μg of Triton X-114-extracted proteins from B. microti-infected RBC membranes was resolved under reducing conditions on a 13.5% SDS-polyacrylamide gel using the buffer system of Laemmli (31). After brief staining (20 min) with Coomassie brilliant blue R-250 (Bio-Rad), the protein band of interest at approximately 45 kDa was excised, destained, and submitted to the Harvard Microchemistry Facility for trypsin digestion and tandem mass spectrometry (MS/MS) peptide sequence analysis. Digestion products were analyzed by microcapillary reverse-phase high-performance liquid chromatography (HPLC) nano-electrospray tandem mass spectrometry (μLC/MS/MS) on a Finnigan LCQ DECA XP Plus quadrupole ion trap mass spectrometer. MS/MS spectral data were correlated with sequences in databases using published algorithms (5, 9).
An amount of Triton X-114-extracted protein similar to that described above was resolved under reducing conditions on a 12% SDS-polyacrylamide gel and electrotransferred onto a PVDF membrane. The blot was stained with Coomassie blue, and the protein of interest was excised and submitted to the Biotechnology Core Facility at the Centers for Disease Control and Prevention for amino-terminal peptide sequencing using the Procise protein sequencing system (Applied Biosystems, Foster City, CA).
Isolation of B. microti nucleic acids and antigen cloning.
Genomic DNA was isolated from a hypotonic saponin preparation of B. microti by lysis in buffer containing 10 mM Tris (pH 8.0), 1 mM EDTA, 0.5% SDS, and 1 mg/ml proteinase K for digestion (Fisher, Fair Lawn, NJ), followed by phenol-chloroform extraction, RNase A digestion, proteinase K digestion, and ethanol precipitation (38).
Total RNA was isolated from 2 ml of washed, infected red blood cells using 10 vol of TRIzol reagent as directed by the manufacturer (Invitrogen, Carlsbad, CA). The RNA was digested with RQ1 RNase-free DNase for 1 h at 37°C (Promega Corp., Madison, WI), digested with proteinase K for 1 h at 37°C, extracted with phenol-chloroform, and precipitated with ethanol. Poly(A)+ mRNA was selected using oligo(dT) magnetic particles (Absolutely mRNA purification kit; Stratagene, La Jolla, CA). First- and second-strand cDNAs were made using the Universal Riboclone cDNA synthesis kit (Promega) and an oligo(dT)20 primer with an EcoRI restriction endonuclease site (underlined): 5′-GCG GAA TTC TTT TTT TTT TTT TTT TTT TT-3′.
The BMN1-9 sequence of Lodes et al. (35) (GenBank accession number AF206252) was PCR amplified from double-stranded cDNA using AmpliTaq gold DNA polymerase (Perkin-Elmer Cetus, Foster City, CA) and the amplification protocol previously described by Priest et al. (44). The 5′ primer was based on the published BMN1-9 sequence (5′-GCT TTT GGG AAT ATC TCA CCT G-3′), while the 3′ primer was the EcoRI oligo(dT)20 described above. An ∼1-kb product, purified from a 1.5% agarose gel in TBE buffer (38) was then used in a seminested PCR with the EcoRI oligo(dT)20 primer and a primer based upon the mature amino terminus of the BMN1-9 protein sequence (BamHI restriction endonuclease site underlined): 5′-CGC GGA TCC GCT GGT GGT AGT GGT GGT AAT G-3′. The annealing temperature for the nested PCR was increased from 55°C to 60°C. The final PCR product was purified, doubly digested with EcoRI and BamHI restriction endonucleases (New England BioLabs, Ipswich, MA), ligated into the EcoRI/BamHI-digested pGEX4T-2 vector (GE Healthcare, Piscataway, NJ), which had been dephosphorylated with shrimp alkaline phosphatase (Roche Chemicals, Indianapolis, IN), and cloned into HB101 Escherichia coli cells (Promega) as previously described (46). The plasmid from the HB101 E. coli cell clone was purified, sequenced in both directions, and subcloned into E. coli BL21 Gold cells (Stratagene) for glutathione S-transferase (GST) fusion protein expression. The cDNA clone of BMN1-9 was designated BmSA1 because its sequence matched that reported by Luo et al. (36).
A fragment of the overlapping BMN1-17 and BMN1-20 coding sequences (GenBank accession numbers AF206526 and AF206527) (35) that excluded the putative amino-terminal signal peptide and the hydrophobic sequence at the carboxy terminus was amplified from genomic B. microti DNA using the PCR protocol described above (55°C annealing temperature) with the following forward and reverse primers: 5′-CGC GGA TCC GGG GAT GTA TAT GAG ATA TCT TC-3′ and 5′-GCG GAA TTC TTA ATG AGT GGA TGT TTC AGT CTT GAG-3′. The primers included restriction endonuclease sites for directional cloning (underlined) as well as a translation stop codon (italics). The resulting PCR product was cloned into E. coli and sequenced as described above.
Purification of recombinant parasite antigens.
BL21 E. coli cultures containing the BmSA1 and Bm17N plasmids were grown, induced, and lysed as previously described (39, 44). Recombinant GST-linked BmSA1 (rBmSA1/GST) and Bm17N (rBm17N/GST) fusion proteins were initially purified on a glutathione Sepharose 4B affinity column as directed by the manufacturer (GE Healthcare) using a lysis buffer containing PBS, 1 mM PMSF, 10 μM leupeptin, 1 μM pepstatin A, 5 mM EDTA, 0.3% Tween 20, and 10 mg/ml lysozyme.
Glutathione-eluted rBmSA1/GST protein was desalted into 25 mM Tris buffer at pH 8.0 using a 50-ml Sephadex G-25 desalting column (GE Healthcare) at a flow rate of 2 ml/min. Protein was bound on a Mono Q HR 5/5 strong anion exchange column (GE Healthcare) and eluted with a 20-ml linear gradient from 0 to 0.4 M NaCl in 25 mM Tris at pH 7.4. Pooled protein-containing fractions were concentrated using Centricon-50 centrifugal filter devices (Millipore Corporation, Bedford, MA) and then dialyzed against 2,000 volumes of PBS overnight at 4°C (Spectra/Por3, 3,500-Da cutoff; Spectrum Laboratories, Rancho Dominguez, CA). Protein concentrations were determined using the BCA microassay (Pierce).
Glutathione-eluted rBm17N/GST protein samples were concentrated using Centricon-30 centrifugal filter devices (Millipore) and then desalted into 25 mM Tris-8 M urea buffer at pH 8.0 using a 5-ml HiTrap desalting column as directed by the manufacturer (GE Healthcare). Protein was bound on a Mono Q HR 5/5 column (GE Healthcare) and eluted with a 20-ml linear gradient from 0 to 0.4 M NaCl in 25 mM Tris-8 M urea buffer at pH 8.0. Pooled protein-containing fractions were dialyzed against 1,000 volumes of PBS with 4 M urea overnight at 4°C (Spectra/Por3). Protein concentrations were determined using the BCA microassay (Pierce).
Schistosoma japonicum GST protein was expressed and purified as previously described (39, 44).
Monoclonal antibody.
Recombinant BmSA1 protein lacking the GST fusion partner was generated by cleavage with thrombin (GE Healthcare), while the protein was bound to a glutathione Sepharose 4B affinity column. Cleavage conditions were as directed by the manufacturer. A mouse monoclonal IgG1 isotype antibody to the purified recombinant BmSA1 protein (designated 3A6D) was generated by Southern Biotech (Birmingham, AL).
Antigen coupling to beads.
Antigen coupling to SeroMap beads (Luminex Corporation, Austin, TX) using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (Calbiochem/EMD Biosciences, La Jolla, CA) and N-hydroxysulfosuccinimide (Pierce) has been previously described (39, 40). For both recombinant B. microti proteins and the GST control, 120 μg of protein was used for coupling to 12.5 × 106 beads. To assess the efficiency of the coupling reactions, beads were assayed (procedure described below) using a goat anti-GST antibody (GE Healthcare) and a biotinylated rabbit anti-goat IgG secondary antibody (Invitrogen). GST control, rBmSA1/GST-coupled, and rBm17N/GST (two reactions)-coupled beads had anti-GST median fluorescence intensity minus background (MFI−BG) values of 29,374, 28,451, and 23,153/28,580, respectively. Coupled beads from both rBm17N/GST reaction mixtures behaved similarly with Babesia positive-control sera (average MFI−BG values of 26,178 versus 26,039 and 13,587 versus 12,662), and both bead sets were used in the study. After being coupled, the beads were quantified with a hemocytometer and stored at 4°C in PBS containing 1.0% bovine serum albumin (BSA), 0.05% Tween 20, 0.02% sodium azide, and protease inhibitors as follows: 200 μg/ml Pefabloc (Roche Diagnostics, Indianapolis, IL), 200 μg/ml EDTA, and 1 μg/ml each of leupeptin and pepstatin A.
Multiplex bead assay (MBA).
A 1:400 dilution of serum in PBS containing 0.5% BSA, 0.05% Tween 20, 0.02% sodium azide, 0.5% poly(vinyl alcohol), and 0.8% polyvinylpyrrolidone was incubated for 1 h at 37°C and stored overnight at 4°C. In order to reduce nonspecific binding, extract from an isopropyl-β-d-thiogalactopyranoside (IPTG)-induced culture of pGEX4T-2 in BL21 E. coli cells was included in each serum diluent at a final concentration of 3 μg/ml (40). After centrifugation at 17,000 × g for 5 min, 50 μl of the clarified serum dilution was added to each well of a 96-well filter bottom plate (Millipore, Bedford, MA). All sera were assayed in duplicate using approximately 2,500 beads from each antigen-coupling reaction mixture/well. The beads were suspended in the serum dilutions and allowed to gently shake for 1.5 h at room temperature. The development protocol using biotinylated, monoclonal anti-IgG (clone H4; Southern Biotech) and anti-IgG4 (clone HP6025; Zymed) antibodies and R-phycoerythrin-labeled streptavidin (Invitrogen) has been previously described (40). After the final wash step, the beads in each well were suspended in 125 μl PBS, and the plate was analyzed using a Bio-Plex 200 or 100 instrument (Bio-Rad, Hercules, CA) equipped with Bio-Plex Manager 6.0 software (Bio-Rad). Gated data were acquired on at least 100 monodispersed beads of each spectral classification, and the median fluorescence intensity for each analyte was calculated. After subtraction of the background blank, the mean of results from the duplicate wells was reported as the median fluorescence intensity minus background (MFI−BG). Two positive-control sera, one with a high fluorescence intensity and one with a midrange fluorescence intensity, were included on each plate, along with a negative control. Testing of a sample with a positive response was repeated if the standard deviation values for the duplicate wells of both the rBmSA1/GST and rBm17N/GST assays were >15% of the respective mean values. After the completion of the initial Bio-Plex analysis of the positive-control plates, assay wells were evacuated, the beads in each well were resuspended in 125 μl PBS, and the plates were reanalyzed using a different instrument (intra-assay repeats; same plate read twice). Samples used in the specificity study were assayed on two separate occasions (approximately 2 weeks apart) for rBmSA1/GST IgG reactivity (interassay repeats).
Human serum specimens.
Endemic babesiosis caused by B. microti has been reported in the Northeast and upper Midwest regions of the United States (54). As likely negative controls, we used sera from two U.S. cryptosporidiosis outbreaks: 44 from adult residents of Texas collected in 1998 and 8 from adult residents of Kansas collected in 2003. Blood samples were collected from donors by venipuncture, and the resulting sera were stored at −20°C. Written, informed consent was obtained from these U.S. study participants, and the protocol was approved by the Centers for Disease Control and Prevention Institutional Review Board. Also included in the negative panel were 30 anonymous samples from healthy adult Haitian blood donors collected in 1998 from a region with a low prevalence of Plasmodium falciparum malaria.
Residual serum specimens that had been submitted to the Centers for Disease Control and Prevention for babesiosis and malaria diagnostic testing between 1985 and 2011 were aliquoted and stripped of personal identifiers prior to testing. Serum samples from 78 confirmed B. microti-infected individuals who were positive by both slide microscopy and B. microti IFA antibody testing (3) (4, 14, 37, and 23 individuals at IFA titers of 1:64, 1:256, 1:1,024, and ≥1:4,096, respectively) were used in the sensitivity analysis. IFA-negative/microscopy-positive babesiosis samples were not available for testing. To address issues of species specificity, 10 sera from B. duncani- and B. duncani-like organism-infected individuals (isolates CA and WA) (6, 47) and 15 sera from B. divergens- and B. divergens-like organism-infected individuals (isolates MO1 and EU) were tested in the recombinant-protein MBA (7, 17). All of the B. duncani group sera and 53% of the B. divergens group sera were previously shown to have Babesia species-specific antibody responses by IFA with titers of ≥1:64. To determine whether cross-reactivity with malaria antigens was present (4), we assayed 81 sera from microscopy-positive patients infected with Plasmodium malariae (n = 6), Plasmodium ovale (n = 7), Plasmodium falciparum (n = 33), or Plasmodium vivax (n = 35).
Data analysis.
Statistical analyses were conducted using the Wilcoxon signed-rank test, the Kruskal-Wallis test, or the Spearman rank correlation test (SigmaStat for Windows version 2.03.0; SPSS, Inc., Chicago, IL). Statistical significance was set at an alpha level of 0.05.
Reagents.
Unless otherwise stated, reagents were purchased from Sigma Chemicals (St. Louis, MO).
RESULTS
Identification of an immunodominant B. microti antigen.
In a preliminary experiment, a small panel of 10 sera presumed to be negative and 10 sera from patients confirmed to be infected with B. microti was used to screen Western blots of total B. microti proteins for immunodominant antigen candidates. As shown by arrows in Fig. 1, unique B. microti antigen bands were detected in the 90-kDa, 50-kDa, 46-kDa, and 24-kDa regions of the IgG Western blots, with the 46-kDa antigen band being the most consistent. To determine whether any of these antigens were membrane associated, a Triton X-114 detergent-phase extraction was performed on an infected hamster red blood cell particulate fraction. As shown in Fig. 2, both an infected hamster serum (Fig. 2A) and a human patient serum (Fig. 2B) specimen reacted strongly with a detergent-extracted antigen at approximately 40 kDa. The slight downward shift in the apparent molecular mass of the native antigen (from approximately 46 kDa on the 12% polyacrylamide gel to approximately 40 kDa on the 10 to 22.5% gradient polyacrylamide gel) was reproducible on laboratory-prepared gels (data not shown).
Fig 1.
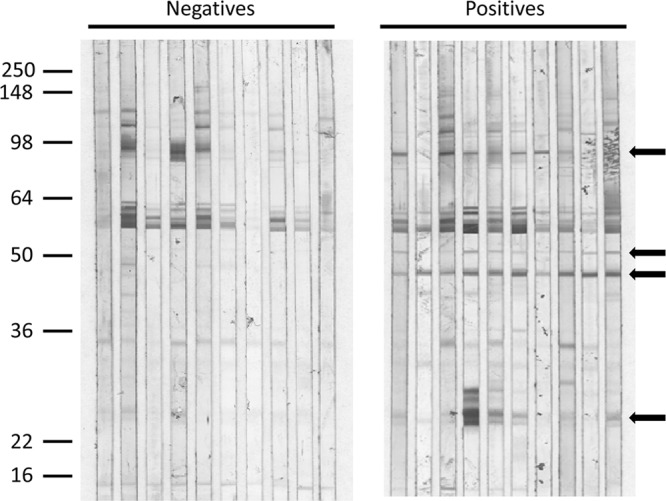
Identification of reactive B. microti antigens. Proteins from a hypotonic saponin lysate of B. microti-infected hamster red blood cells were resolved on a 12% polyacrylamide gel and electrotransferred to a PVDF membrane as described in Materials and Methods. Strips of membrane were incubated with sera from presumed-negative patients or with sera from confirmed-babesiosis patients at a dilution of 1:100. Bound IgG antibodies were visualized using a biotinylated monoclonal mouse anti-human IgG and the streptavidin-alkaline phosphatase system described in Materials and Methods. Reactive antigen bands unique to positive sera are identified with arrows. The positions of molecular mass markers (in thousands) are indicated on the left.
Fig 2.
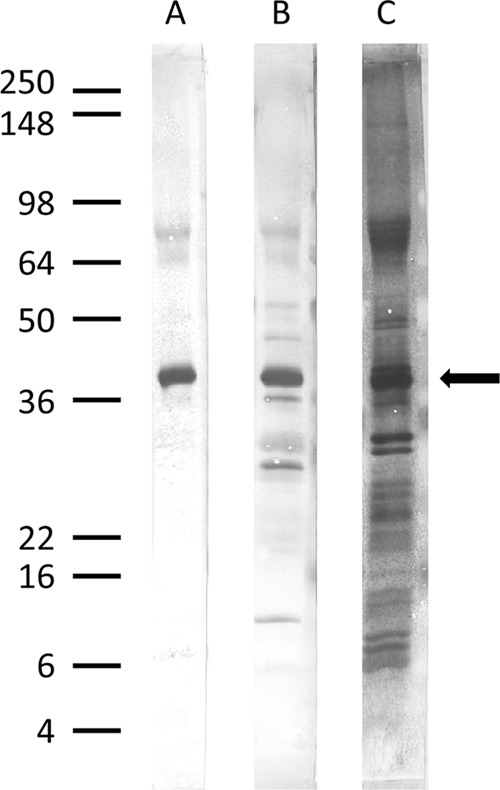
Membrane-associated antigen identified by detergent extraction. A postsonication particulate fraction from B. microti-infected hamster red blood cells was extracted using Triton X-114 detergent as described in Materials and Methods. Extracted proteins were resolved on a 10 to 22.5% polyacrylamide gel and transferred to PVDF. Strips of membrane were incubated with sera from an infected hamster (A), sera from a babesiosis-positive patient (B), or AuroDye Forte colloidal gold stain (C). Bound IgG antibodies were visualized as described for Fig. 1. The positions of molecular mass markers (in thousands) are indicated on the left. A major immunodominant band is indicated by an arrow just above the 36-kDa marker.
We determined that the epitopes on the detergent-extracted antigen were likely protein rather than carbohydrate in nature; proteinase K (24 h at 37°C) completely degraded the epitopes recognized by human serum, while sodium periodate treatment (25 mM for 24 h at room temperature) had no effect on IgG antibody recognition (data not shown). AuroDye staining of a blot strip (Fig. 2C) suggested that the detergent-extracted antigen band was sufficiently isolated to allow for peptide sequencing. The region containing the band of interest was excised from a PVDF blot for amino-terminal Edman degradation sequencing and from a Coomassie blue-stained polyacrylamide gel for trypsin digestion and mass spectral analysis. The 46/40-kDa protein was identified as BMN1-9/BmSA1 (35, 36), with an amino-terminal sequence of Ala Gly Gly Ser. This mature amino terminus was correctly predicted by the SignalP cleavage analysis described by Luo et al. (36).
Cloning and expression of immunodominant B. microti antigens.
Because the BMN1-9-containing clone reported by Lodes et al. (35) appeared to contain two overlapping open reading frames (GenBank accession number AF206252), we decided to PCR amplify the gene from oligo(dT)-primed cDNA using two sequence-specific 5′ primers and a 3′ primer to the poly(A) tail. Except at the 3′ end (where we identified an additional 5′-TG(A)20-3′ sequence), the 1,014-bp product that we obtained from the seminested cDNA amplification (GenBank accession number JX112361) was identical in sequence to the BmSA1 clone recently reported by Luo et al. (36). The predicted 302-amino-acid protein (from the mature amino terminus to the cDNA-encoded stop codon) had a calculated molecular mass of 32.7 kDa and included a carboxy-terminal sequence consistent with the addition of a glycosylphosphatidylinositol (GPI) anchor. DGPI (J. Kronegg and D. Buloz; URL no longer active), PredGPI (42), and bigPI Predictor (8) all indicated the presence of a GPI anchor addition signal but differed in the predicted locations of the ω GPI anchor attachment site. Highly charged amino acids made up 32% of the predicted BmSA1 protein sequence, and the predicted pI was 5.1.
Purified recombinant BmSA1 protein that was cleaved with thrombin to remove the GST fusion partner was used to immunize mice for monoclonal antibody production. The apparent molecular mass of the cleaved product was approximately 45 kDa on a 12% polyacrylamide gel (data not shown). We obtained one IgG1 subclass monoclonal antibody, designated 3A6D, and used it to probe a Western blot of Triton X-114 detergent extracts from uninfected and B. microti-infected red blood cell membranes. Both the monoclonal antibody and a babesiosis patient serum reacted with an antigen band found only in the infected RBC detergent extract (indicated by the arrow in Fig. 3). The apparent molecular mass of the native antigen on the Western blot of an 8 to 16% polyacrylamide gel was 44 kDa.
Fig 3.
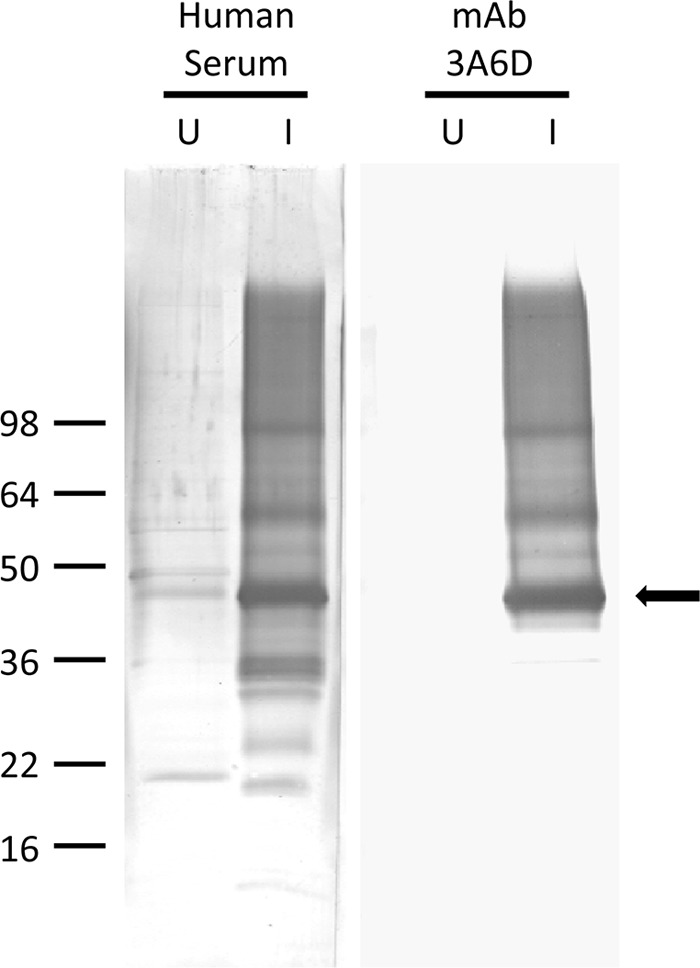
The immunodominant membrane-associated antigen is found only in B. microti-infected red blood cells. Triton X-114-extracted proteins (3.5 μg/lane) from uninfected (U) and B. microti-infected (I) red blood cell membranes were resolved on an 8 to 16% polyacrylamide gel and transferred to PVDF. PVDF sections were incubated with either serum from a confirmed-babesiosis patient (Human Serum) or a mouse monoclonal antibody raised against rBmSA1 protein (mAb 3A6D). Bound IgG antibodies were visualized as described for Fig. 1. The positions of molecular mass markers (in thousands) are indicated on the left. Monoclonal antibody reactivity confirmed the identity of the immunodominant protein band in the parasitized red blood cell detergent extract as BMN1-9/BmSA1 (indicated by the arrow).
We cloned and expressed a fragment of the antigen previously reported to have the highest signal/noise ratio (S/N) with human sera, BMN1-17 (35), in addition to the BmSA1 protein. Because the protein coding sequence reported by Lodes et al. was shared between two overlapping clones from a gene family (35), we designed a forward primer using the BMN1-20 sequence that deleted the predicted 22-amino-acid signal peptide and a reverse primer using the BMN1-17 sequence that deleted a stretch of 22 hydrophobic amino acids from the carboxy terminus. The 1,221-bp PCR product should have resulted in a GST fusion protein with a predicted size of 72 kDa. In fact, we obtained two types of clones, each with an insert size of ∼1,200 bp. One clone, Bm17, weakly expressed a GST fusion protein doublet in the 105-kDa molecular mass range that was not easily purified to homogeneity, while the other clone, Bm17N, abundantly expressed a single GST fusion protein in the 79-kDa molecular mass range that was soluble and easily purified on a glutathione Sepharose 4B column (data not shown). The expressed proteins reacted strongly with babesiosis patient sera by ELISA and yielded the expected 26-kDa GST fragments when cleaved with thrombin (data not shown).
The nucleic acid sequence of the Bm17N clone (GenBank accession number JX112362) differed from published sequences at 4 base positions and contained a 96-bp deletion compared to the sequence of BMN1-17 (35). One of the base changes, a G-to-T base substitution located in the third repeat, resulted in the introduction of an in-frame stop codon (GGA to TGA) that shortened the expressed protein by an additional 167 amino acids. The predicted truncated Bm17N translation product is a 26.8-kDa protein with a pI of 4.3 (53.1-kDa GST fusion protein; fusion protein pI of 4.7). The predicted protein sequence (Fig. 4) includes two of the 32-amino-acid repeats identified by Lodes et al. (35) and two copies (100% and 85% sequence identity) of the 26-residue immunodominant peptide sequence identified by Houghton et al. (22). Because of the ease of GST fusion protein production and because two-thirds (67%) of the deleted protein sequence was made up of repeated sequence (Fig. 4), the truncated rBm17N/GST protein was used in our work.
Fig 4.

Deduced amino acid sequence of the Bm17N clone. The deduced amino acid sequence of clone Bm17N is presented in a manner to highlight the repeat structure of the protein. The sequence contains two 47-amino-acid repeats and a substructure of six 32-amino-acid repeats. Compared to the published BMN1-17 and BMN1-20 sequences (35), the Bm17N sequence contained four base substitutions that resulted in three amino acid substitutions (indicated in the sequence by boldface italic letters) and the introduction of an in-frame stop codon (#, position 239) in the third repeat. The immunodominant 26-amino-acid peptide sequence identified by Houghton et al. (22) is indicated in boldface with an underline.
Human immune responses to B. microti antigens.
IgG antibody responses to both recombinant B. microti antigens were assessed using the MBA. MFI−BG responses to GST alone (GST-coupled beads were included in each assay well as a control) ranged from 2 to 677 (median = 21; mean = 39) and were always below the cutoff values determined for the B. microti antigens. Figure 5 shows the distributions of MBA responses for rBmSA1/GST (Fig. 5A) and rBm17N/GST (Fig. 5B) assays for presumed negative (control) sera and sera confirmed to be B. microti positive. Using receiver-operating characteristic (ROC) analysis (58), a cutoff value for rBmSA1/GST (1,122 MFI−BG units) that resulted in a sensitivity of 98.7% and a specificity of 100% (J-index = 0.987; 99.4% correct) was identified. The optimal cutoff value for rBm17N/GST, determined to be 5,849 MFI−BG units, resulted in a sensitivity of 97.4% and a specificity of 97.6% (J index = 0.95; 97.5% correct). Using these cutoff values, one B. microti-positive sample was misclassified as negative by both assays (IFA titer = 1:256), while a second positive sample was misclassified only by the rBm17N/GST assay (IFA titer = 1:64) (Table 1). Two presumed-negative samples had strongly positive responses in the rBm17N/GST assay but were negative in the rBmSA1/GST assay (Table 1). Both of these samples were tested using the routine diagnostic B. microti IFA (3) and were determined to be truely negative (titer, <1:8). Given that the maximum value of each assay was approximately 29,500 MFI−BG units, the rBmSA1/GST assay had a wider dynamic range and a higher signal/noise ratio (geomean-positive signal/geomean-negative signal; S/N = 994) than did the rBm17N/GST assay (S/N = 262).
Fig 5.
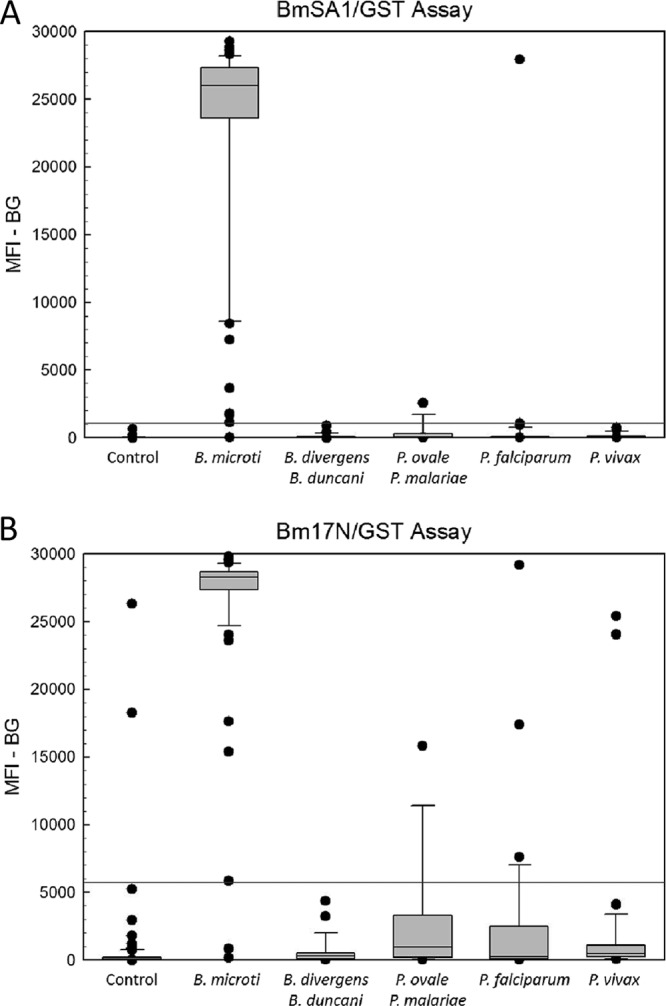
MBA detection of IgG antibodies among presumed-negative cases and confirmed-babesiosis and malaria cases. MBA was conducted as described in Materials and Methods using beads coated with rBmSA1/GST (A) or rBm17N/GST (B). Bound IgG antibodies were detected using a biotinylated mouse monoclonal anti-human IgG and R-phycoerythrin-labeled streptavidin. Distributions for the presumed-negative sample set (Control; n = 82), B. microti-positive samples (n = 78), B. duncani- and B. divergens-positive samples (n = 25), P. ovale- and P. malariae-positive samples (n = 13), P. falciparum-positive samples (n = 33), and P. vivax-positive samples (n = 35) are presented. Boxes include values between the 25th and 75th percentiles, whiskers include values between the 10th and 90th percentiles, and outliers are indicated by data points. The median values are indicated within the boxes by a line. The respective cutoff values determined by ROC analysis for rBmSA1/GST (MFI−BG = 1,122) and rBm17N/GST (MFI−BG = 5,849) are indicated by horizontal lines in panel A and panel B, respectively.
Table 1.
MBA results for specimens tested
| Test | Patient group | No. of specimens | No. (%) positive by MBA |
||
|---|---|---|---|---|---|
| rBmSA1 | rBm17N | Botha | |||
| Sensitivity | B. microti infectedb,c | 78 | 77 (98.7) | 76 (97.4) | 76 (97.4) |
| Specificity | B. duncani infected | 10 | 0 (0) | 0 (0) | 0 (0) |
| B. divergens infected | 15 | 0 (0) | 0 (0) | 0 (0) | |
| Plasmodium species infectedd | 81 | 2 (2.5) | 7 (8.6) | 1 (1.2) | |
| Presumed B. microti negativec,e | 82 | 0 (0) | 2 (2.4) | 0 (0) | |
Positive by MBA for antibodies to both rBmSA1 and rBm17N.
IFA-positive (titer, >1:64) and blood smear-positive patients.
Used in the ROC analysis for determination of cutoff values.
Includes patients infected with P. falciparum (n = 33), P. vivax (n = 35), P. ovale (n = 7), and P. malariae (n = 6).
Includes C. parvum outbreak patients who are residents of Texas (n = 44) and Kansas (n = 8) and healthy Haitian blood donors (n = 30).
To assess the species specificity of the recombinant-protein MBA, sera from B. divergens group (n = 15) and B. duncani group (n = 10) babesiosis cases and sera from 81 Plasmodium species cases were assayed for antibody reactivity to the two recombinant B. microti antigens by MBA. As expected, none of the non-B. microti babesiosis sera reacted with the rBmSA1/GST or rBm17N/GST protein (Fig. 5). In contrast, one P. falciparum serum specimen was strongly positive by MBA (MFI−BG > 27,000) for both recombinant antigens. A second serum sample, from a P. ovale patient, was weakly positive only for antibodies to rBmSA1/GST (MFI−BG = 2552), while positive rBm17N/GST responses were detected for P. falciparum (n = 3), P. vivax (n = 2), and P. malariae (n = 1) patient sera (Fig. 5). Overall, 2/81 (2.5%) malaria sera had positive responses to rBmSA1/GST, and 7/81 (8.6%) had positive responses to rBm17N/GST (Table 1). Because demographic data were not linked to the serum aliquots, we were unable to determine which of the cases positive for malaria lived in regions of the United States where babesiosis is endemic.
Even though the majority of malaria MBA values were below the positive cutoff threshold, P. falciparum and P. vivax MBA values for both recombinant B. microti antigens were statistically greater than those of presumed-negative controls (Kruskal-Wallis test with multiple comparisons; P < 0.05). The P. ovale/P. malariae MBA values were significantly greater than control values only in the rBm17N/GST assay.
IgG MBA performance.
To address concerns about the stability of the antibody-antigen-reporter molecule complex and about the reproducibility between instruments, B. microti-positive sample assay plates were read consecutively using two different instruments. As shown in Fig. 6A, MBA values (including controls, n = 84) obtained during primary rBmSA1/GST analysis on one instrument were stable upon secondary analysis of the same assay plates with a different Bio-Plex instrument (Spearman correlation coefficient = 0.968). No significant differences were observed by Wilcoxon analysis of the paired values for either MBA (rBmSA1/GST assay, P = 0.088; rBm17N/GST assay, P = 0.323). The Babesia and Plasmodium specificity study samples (including controls, n = 115) were used to examine the interassay stability of the rBmSA1/GST MBA (Fig. 6B). The interassay correlation was quite good (Spearman correlation coefficient = 0.986), and although two borderline-negative values were borderline positive in the repeat analysis, the overall differences observed among the MBA results were not statistically significant (Wilcoxon analysis; P = 0.633).
Fig 6.
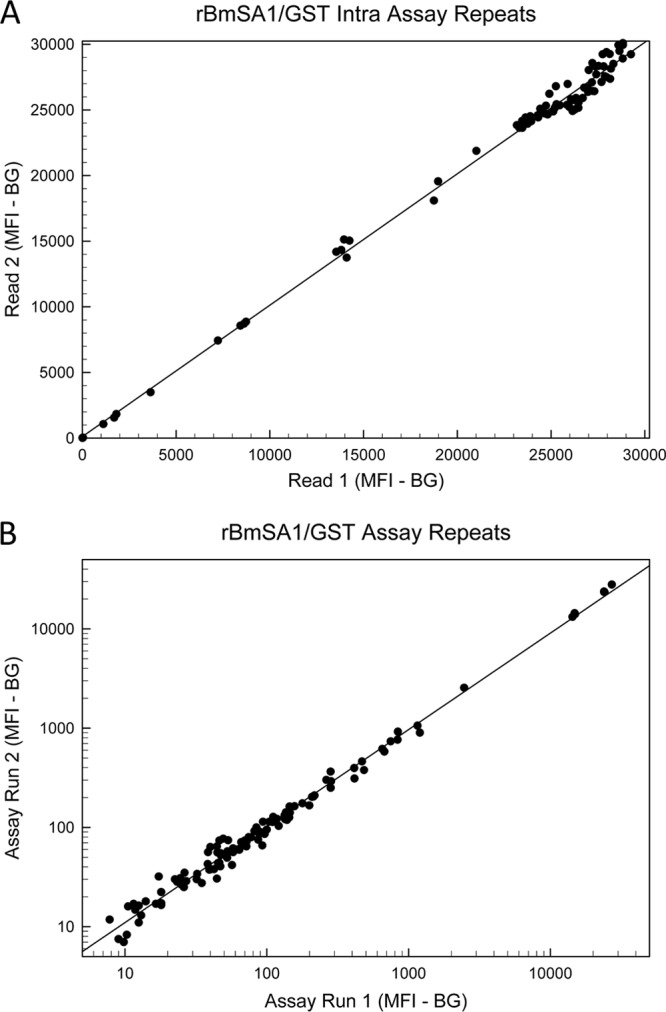
Reproducibility of the rBmSA1/GST MBA assay. (A) MBA values (MFI−BG) for assay plates that were run consecutively on two different Bio-Plex instruments on the same day; (B) MBA values (MFI−BG) for samples that were independently assayed approximately 2 weeks apart. Note that the values in panel B are plotted on a logarithmic scale to allow visualization of a wider range of values. Least-squares regression lines are given in both plots. The positive cutoff value for the rBmSA1/GST MBA was determined to be 1,122 MFI−BG units.
The MBA results described above were collected over the course of 8 months. During that time, the rBmSA1/GST beads were stable at 4°C in the presence of protease inhibitors. With one exception (deleted from the analysis; likely a dilution error, as the two assays were equally affected), individual positive-control sample values fell within ±7% of their overall study mean values (positive-control means, 23,989 and 14,202; standard deviations, 431 and 510, respectively). In contrast, the rBm17N/GST beads degraded sometime between the third month and the eighth month of storage. The reason for the degradation is unclear, but we were able to recouple beads with rBm17N/GST and complete the study with control value ranges comparable to those obtained for the rBmSA1/GST assay (positive-control means, 25,842 and 12,902; standard deviations, 523 and 449, respectively).
DISCUSSION
The risk of acquiring human B. microti infection is likely increasing due to increasing vector tick populations in areas of endemicity, increasing levels of recreational exposure to potentially infected ticks, and the lack of a sensitive and specific high-throughput screening assay for donated blood and blood products (14, 19, 25, 27, 32). In addition to traditional blood smear microscopy, Western blotting, ELISA, IFA, and PCR assays have been developed by various laboratories for diagnosis. Unfortunately, none of these assays are 100% sensitive and specific in every phase of infection; PCR and microscopy are superior to IFA in the acute infection phase before an immune response has developed, but antibody-based assays are more likely to detect low-level infections in the late or chronic phases, when parasite loads are extremely low (14, 29, 30, 34, 50). These low-level, largely asymptomatic infections are a particular concern for transfusion-transmitted babesiosis (19, 20, 23, 24, 53).
In an effort to develop an improved antibody detection technique, we surveyed B. microti total antigen Western blots for reactive bands and identified an immunodominant antigen using detergent extraction and protein sequencing techniques. The expressed recombinant protein, originally named BMN1-9 by Lodes et al. (35) but more recently called BmSA1 by Luo et al. (36), migrated on polyacrylamide gels with an apparent molecular mass greater than the predicted molecular mass (45 kDa versus 32.7 kDa). The published polyacrylamide gels of Luo et al. for the same BmSA1/GST construct that we used also suggest an apparent molecular mass significantly greater than 33 kDa (36). The apparent molecular mass of the Triton X-114-extracted native protein was 40 to 46 kDa and appeared to vary with polyacrylamide gel conditions. While some of the size variability of the native antigen may be the result of posttranslational modifications (such as a GPI anchor), the majority of the unexpected size differentials of both native and recombinant proteins may result from the high proportion of charged amino acids (32%) present in the sequence. We have previously documented similar aberrant gel migration patterns with another highly charged, GPI-anchored parasite surface protein, the 17-kDa Cryptosporidium parvum antigen (43).
Host reactivity to the BmSA1 protein has been characterized using animal models, and the early appearance and long duration of the specific immune response may make this antigen a good target for an antibody detection assay. Hamster antibodies to BmSA1 were apparent by day 4 to 5 of experimental B. microti infection, and an indirect ELISA using recombinant BmSA1 as the antigen was 100% sensitive compared to blood film microscopy (36, 37). Sera from pathogen-free animals or from animals infected with pathogens other than B. microti (including other Babesia spp.) were uniformly negative by indirect ELISA (36, 37). Data from human serum assays using this antigen, however, are limited (35). In our study, we incorporated recombinant BmSA1/GST into a multiplex bead-based serologic assay for the detection of specific IgG antibodies and, using human samples with a wide range of IFA titers, determined that the assay was both sensitive (98.7% sensitive using sera from microscopy- and IFA-positive B. microti patients) and specific (100% specific using presumed-negative sera and 97.5% using malaria-positive sera). Because information on the relative timing of the antibody response and the appearance of parasitemia in humans naturally infected by tick inoculation is lacking, additional studies will be needed to assess the utility of the MBA in the early phases of infection.
The true power of the multiplex assay is that it allows for the simultaneous detection of multiple antibody responses in a single assay well. As a benchmark for sensitivity and specificity comparisons, we expressed and multiplexed a sequence related to the BMN1-17/BMN1-20 protein family previously identified as immunodominant antigens by Lodes et al. (35). The BMN1-17 protein contains 7 copies of a 32-amino-acid repeat and has been reported to migrate on polyacrylamide gels as a doublet with an apparent molecular mass greater than the predicted molecular mass (81 kDa versus 49.4kDa) (35). Even though we used a different expression system (GST versus 6×His), our results with the Bm17 clone confirm the presence of a doublet and the aberrant gel migration of the full-length protein (data not shown). We have some limited evidence that points to the single 90-kDa protein band on our Western blots being the Bm1-17 antigen (data not shown) and suggest that the observed recombinant protein doublet is an artifact of the expression system. Our Bm17N clone contained only 6 repeats (like the BMN1-20 clone of Lodes et al. [35]) and expressed a truncated recombinant protein because of an in-frame stop codon within the third repeat. We have not yet determined whether the stop codon is a PCR artifact. It is possible that the truncated protein coding sequence is a previously unrecognized member of the gene family represented by BMN1-17 and BMN1-20 that may be expressed in lower abundance. Compared to assays using the rBmSA1/GST protein, assays using the truncated rBm17N/GST protein were slightly less sensitive (97.4%) and less specific with presumed-negative sera (97.6%) and malaria-positive sera (91.3%). Although we did not extensively test the full-length recombinant protein from our Bm17 clone, we think it unlikely that the inclusion of the additional repeat sequences downstream of the internal Bm17N stop codon would markedly improve the specificity of the assay. The issue of cross-reactivity with malaria-positive sera (perhaps as high as 30%) has been previously documented for the full-length BMN1-17 antigen (22).
If, however, a definition of “positive for two specific antigens” was adopted for the MBA, the result would be a relatively sensitive (97.4%) and highly specific (100% for presumed-negative sera, 100% for sera infected with other Babesia spp., and 98.8% for malaria-positive sera) assay. The one malaria-infected donor who was positive for antibodies to both B. microti antigens had very high responses (>27,000). Although IFA cross-reactivity between sera from malaria-infected patients and antigens from B. microti has been previously reported (4), the magnitude of the responses to both recombinant antigens and the fact that some of the malaria samples were submitted from regions of the United States where babesiosis is endemic suggest the possibility that this individual may have been infected with both malaria and B. microti. We hypothesize that the use of a two-antigen MBA for screening of low-prevalence populations would minimize the number of false positives and, in the case of blood donor screening, would detect the majority of babesiosis-positive donors while all but eliminating unnecessary donor deferrals. Studies with a well-defined, longitudinal set of sera from a babesiosis patient or from experimentally infected animals are needed to confirm the timing of the IgG response and the utility of the assays in all phases of Babesia infection.
The MBA results were reproducible on different days and using different instruments. Even values well below the positive cutoff were reproduced upon repeated assay. Beads coupled with the BmSA1 antigen were stable for at least 8 months, but beads coupled with the Bm17N antigen appeared to degrade during long-term storage. The fact that we were able to recouple fresh rBm17N/GST to beads and reproduce our MBA control values suggests that there is some robustness to the process. We are currently exploring low-temperature storage conditions for the Bm17N/GST-coupled beads in an attempt to prevent degradation in future work.
Unfortunately, the strength of a highly specific MBA is also a weakness in terms of the detection of individuals who are infected with other Babesia species. Cross-reacting antibody responses to B. microti antigens have not been reported for sera from other humans and animals infected with Babesia species, and our results for sera from B. duncani- and B. divergens-infected patients support those findings (14, 17, 18, 36, 37, 41, 47). Given the apparent species specificity of the response, one or more antigens from both B. duncani and B. divergens would be required to cover all of the potential Babesia infections that might be found in the U.S. blood supply. With an additional investment in antigen discovery, cloning, and multiplex assay development, this goal is certainly achievable.
ACKNOWLEDGMENTS
Use of trade names is for identification only and does not imply endorsement by the Public Health Service or by the U.S. Department of Health and Human Services. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Footnotes
Published ahead of print 1 August 2012
REFERENCES
- 1. Asad S, Sweeney J, Mermel LA. 2009. Transfusion-transmitted babesiosis in Rhode Island. Transfusion 49:2564–2573 [DOI] [PubMed] [Google Scholar]
- 2. Beattie JF, Michelson ML, Holman PJ. 2002. Acute babesiosis caused by Babesia divergens in a resident of Kentucky. N. Engl. J. Med. 347:697–698 [DOI] [PubMed] [Google Scholar]
- 3. Chisholm ES, Ruebush TK, II, Sulzer AJ, Healy GR. 1978. Babesia microti infection in man: evaluation of an indirect immunofluorescent antibody test. Am. J. Trop. Med. Hyg. 27:14–19 [DOI] [PubMed] [Google Scholar]
- 4. Chisholm ES, Sulzer AJ, Ruebush TK., II 1986. Indirect immunofluorescence test for human Babesia microti infection: antigenic specificity. Am. J. Trop. Med. Hyg. 35:921–925 [DOI] [PubMed] [Google Scholar]
- 5. Chittum HS, et al. 1998. Rabbit beta-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading gaps. Biochemistry 37:10866–10870 [DOI] [PubMed] [Google Scholar]
- 6. Conrad PA, et al. 2006. Description of Babesia duncani n. sp. (Apicomplexa: Babesiidae) from humans and its differentiation from other piroplasms. Int. J. Parasitol. 36:779–789 [DOI] [PubMed] [Google Scholar]
- 7. Duh D, Jelovsek M, Avsic-Zupanc T. 2007. Evaluation of an indirect fluorescence immunoassay for the detection of serum antibodies against Babesia divergens in humans. Parasitology 134:179–185 [DOI] [PubMed] [Google Scholar]
- 8. Eisenhaber B, Bork P, Eisenhaber F. 1998. Sequence properties of GPI-anchored proteins near the omega-site: constraints for the polypeptide binding site of the putative transamidase. Protein Eng. 11:1155–1161 [DOI] [PubMed] [Google Scholar]
- 9. Eng JK, McCormack AL, Yates JR., III 1994. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom. 5:976–989 [DOI] [PubMed] [Google Scholar]
- 10. Filstein MR, et al. 1980. Serosurvey for human babesiosis in New York. J. Infect. Dis. 141:518–521 [DOI] [PubMed] [Google Scholar]
- 11. Fitzpatrick JE, et al. 1968. Human case of piroplasmosis (babesiosis). Nature 217:861–862 [DOI] [PubMed] [Google Scholar]
- 12. Gerber MA, et al. 1994. The risk of acquiring Lyme disease or babesiosis from a blood transfusion. J. Infect. Dis. 170:231–234 [DOI] [PubMed] [Google Scholar]
- 13. Gubernot DM, et al. 2009. Babesia infection through blood transfusions: reports received by the US Food and Drug Administration, 1997–2007. Clin. Infect. Dis. 48:25–30 [DOI] [PubMed] [Google Scholar]
- 14. Gubernot DM, et al. 2009. Transfusion-transmitted babesiosis in the United States: summary of a workshop. Transfusion 49:2759–2771 [DOI] [PubMed] [Google Scholar]
- 15. Hatcher JC, Greenberg PD, Antique J, Jimenez-Lucho VE. 2001. Severe babesiosis in Long Island: review of 34 cases and their complications. Clin. Infect. Dis. 32:1117–1125 [DOI] [PubMed] [Google Scholar]
- 16. Healy GR, Speilman A, Gleason N. 1976. Human babesiosis: reservoir in infection on Nantucket Island. Science 192:479–480 [DOI] [PubMed] [Google Scholar]
- 17. Herwaldt B, et al. 1996. A fatal case of babesiosis in Missouri: identification of another piroplasm that infects humans. Ann. Intern. Med. 124:643–650 [DOI] [PubMed] [Google Scholar]
- 18. Herwaldt BL, et al. 2004. Babesia divergens-like infection, Washington State. Emerg. Infect. Dis. 10:622–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herwaldt BL, et al. 2011. Transfusion-associated babesiosis in the United States: a description of cases. Ann. Intern. Med. 155:509–519 [DOI] [PubMed] [Google Scholar]
- 20. Herwaldt BL, et al. 2002. Transmission of Babesia microti in Minnesota through four blood donations from the same donor over a 6-month period. Transfusion 42:1154–1158 [DOI] [PubMed] [Google Scholar]
- 21. Homer MJ, Aguilar-Delfin I, Telford SR, III, Krause PJ, Persing DH. 2000. Babesiosis. Clin. Microbiol. Rev. 13:451–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Houghton RL, et al. 2002. Identification of Babesia microti-specific immunodominant epitopes and development of a peptide EIA for detection of antibodies in serum. Transfusion 42:1488–1496 [DOI] [PubMed] [Google Scholar]
- 23. Johnson ST, Cable RG, Leiby DA. 2012. Lookback investigations of Babesia microti-seropositive blood donors: seven-year experience in a Babesia-endemic area. Transfusion 52:1509–1516 [DOI] [PubMed] [Google Scholar]
- 24. Johnson ST, et al. 2009. Seroprevalence of Babesia microti in blood donors from Babesia-endemic areas of the northeastern United States: 2000 through 2007. Transfusion 49:2574–2582 [DOI] [PubMed] [Google Scholar]
- 25. Kjemtrup AM, Conrad PA. 2000. Human babesiosis: an emerging tick-borne disease. Int. J. Parasitol. 30:1323–1337 [DOI] [PubMed] [Google Scholar]
- 26. Krause PJ, et al. 2008. Persistent and relapsing babesiosis in immunocompromised patients. Clin. Infect. Dis. 46:370–376 [DOI] [PubMed] [Google Scholar]
- 27. Krause PJ, et al. 2003. Increasing health burden of human babesiosis in endemic sites. Am. J. Trop. Med. Hyg. 68:431–436 [PubMed] [Google Scholar]
- 28. Krause PJ, et al. 1998. Persistent parasitemia after acute babesiosis. N. Engl. J. Med. 339:160–165 [DOI] [PubMed] [Google Scholar]
- 29. Krause PJ, et al. 1996. Comparison of PCR with blood smear and inoculation of small animals for diagnosis of Babesia microti parasitemia. J. Clin. Microbiol. 34:2791–2794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Krause PJ, et al. 1994. Diagnosis of babesiosis: evaluation of a serologic test for the detection of Babesia microti antibody. J. Infect. Dis. 169:923–926 [DOI] [PubMed] [Google Scholar]
- 31. Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685 [DOI] [PubMed] [Google Scholar]
- 32. Leiby DA. 2006. Babesiosis and blood transfusion: flying under the radar. Vox Sang. 90:157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leiby DA, et al. 2005. Demonstrable parasitemia among Connecticut blood donors with antibodies to Babesia microti. Transfusion 45:1804–1810 [DOI] [PubMed] [Google Scholar]
- 34. Loa CC, Adelson ME, Mordechai E, Raphaelli I, Tilton RC. 2004. Serological diagnosis of human babesiosis by IgG enzyme-linked immunosorbent assay. Curr. Microbiol. 49:385–389 [DOI] [PubMed] [Google Scholar]
- 35. Lodes MJ, et al. 2000. Serological expression cloning of novel immunoreactive antigens of Babesia microti. Infect. Immun. 68:2783–2790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Luo Y, et al. 2011. Identification and characterization of a novel secreted antigen 1 of Babesia microti and evaluation of its potential use in enzyme-linked immunosorbent assay and immunochromatographic test. Parasitol. Int. 60:119–125 [DOI] [PubMed] [Google Scholar]
- 37. Luo Y, et al. 2012. A double antibody sandwich enzyme-linked immunosorbent assay for detection of secreted antigen 1 of Babesia microti using hamster model. Exp. Parasitol. 130:178–182 [DOI] [PubMed] [Google Scholar]
- 38. Maniatis T, Fritsch EF, Sambrook J. 1982. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 39. Moss DM, Montgomery JM, Newland SV, Priest JW, Lammie PJ. 2004. Detection of Cryptosporidium antibodies in sera and oral fluids using multiplex bead assay. J. Parasitol. 90:397–404 [DOI] [PubMed] [Google Scholar]
- 40. Moss DM, et al. 2011. Multiplex bead assay for serum samples from children in Haiti enrolled in a drug study for the treatment of lymphatic filariasis. Am. J. Trop. Med. Hyg. 85:229–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Persing DH, et al. 1995. Infection with a Babesia-like organism in northern California. N. Engl. J. Med. 332:298–303 [DOI] [PubMed] [Google Scholar]
- 42. Pierleoni A, Martelli PL, Casadio R. 2008. PredGPI: a GPI-anchor predictor. BMC Bioinformatics 9:392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Priest JW, Kwon JP, Arrowood MJ, Lammie PJ. 2000. Cloning of the immunodominant 17-kDa antigen from Cryptosporidium parvum. Mol. Biochem. Parasitol. 106:261–271 [DOI] [PubMed] [Google Scholar]
- 44. Priest JW, et al. 1999. Detection by enzyme immunoassay of serum immunoglobulin G antibodies that recognize specific Cryptosporidium parvum antigens. J. Clin. Microbiol. 37:1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Priest JW, Mehlert A, Arrowood MJ, Riggs MW, Ferguson MA. 2003. Characterization of a low molecular weight glycolipid antigen from Cryptosporidium parvum. J. Biol. Chem. 278:52212–52222 [DOI] [PubMed] [Google Scholar]
- 46. Priest JW, et al. 2010. Multiplex assay detection of immunoglobulin G antibodies that recognize Giardia intestinalis and Cryptosporidium parvum antigens. Clin. Vaccine Immunol. 17:1695–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Prince HE, Lape-Nixon M, Patel H, Yeh C. 2010. Comparison of the Babesia duncani (WA1) IgG detection rates among clinical sera submitted to a reference laboratory for WA1 IgG testing and blood donor specimens from diverse geographic areas of the United States. Clin. Vaccine Immunol. 17:1729–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rosner F, Zarrabi MH, Benach JL, Habicht GS. 1984. Babesiosis in splenectomized adults. Review of 22 reported cases. Am. J. Med. 76:696–701 [DOI] [PubMed] [Google Scholar]
- 49. Ruebush TK, II, et al. 1977. Human babesiosis on Nantucket Island. Evidence for self-limited and subclinical infections. N. Engl. J. Med. 297:825–827 [DOI] [PubMed] [Google Scholar]
- 50. Ryan R, et al. 2001. Diagnosis of babesiosis using an immunoblot serologic test. Clin. Diagn. Lab. Immunol. 8:1177–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Skrabalo Z, Deanovic Z. 1957. Piroplasmosis in man; report of a case. Doc. Med. Geogr. Trop. 9:11–16 [PubMed] [Google Scholar]
- 52. Spielman A, Clifford CM, Piesman J, Corwin MD. 1979. Human babesiosis on Nantucket Island, USA: description of the vector, Ixodes (Ixodes) dammini, n. sp. (Acarina: Ixodidae). J. Med. Entomol. 15:218–234 [DOI] [PubMed] [Google Scholar]
- 53. Tonnetti L, et al. 2009. Transfusion-transmitted Babesia microti identified through hemovigilance. Transfusion 49:2557–2563 [DOI] [PubMed] [Google Scholar]
- 54. Vannier E, Gewurz BE, Krause PJ. 2008. Human babesiosis. Infect. Dis. Clin. N. Am. 22:viii–ix, 469–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Western KA, Benson GD, Gleason NN, Healy GR, Schultz MG. 1970. Babesiosis in a Massachusetts resident. N. Engl. J. Med. 283:854–856 [DOI] [PubMed] [Google Scholar]
- 56. White DJ, et al. 1998. Human babesiosis in New York State: review of 139 hospitalized cases and analysis of prognostic factors. Arch. Intern. Med. 158:2149–2154 [DOI] [PubMed] [Google Scholar]
- 57. Young C, et al. 2012. Preventing transfusion-transmitted babesiosis: preliminary experience of the first laboratory-based blood donor screening program. Transfusion 52:1523–1529 [DOI] [PubMed] [Google Scholar]
- 58. Zweig MH, Campbell G. 1993. Receiver-operating characteristic (ROC) plots: a fundamental evaluation tool in clinical medicine. Clin. Chem. 39:561–577 [PubMed] [Google Scholar]