

Abstract
Despite much research, there remains controversy over the role of gap junctions in seizure processes. Many studies report anticonvulsant effects of gap junction blockade, but contradictory results have also been reported. The aim of this study was to clarify the role of connexin36 (Cx36) gap junctions in neocortical seizures. We used the mouse neocortical slice preparation to investigate the effect of pharmacological (mefloquine) and genetic (Cx36 knockout mice (Cx36KO)) manipulation of Cx36 gap junctions on two seizure models: low-magnesium artificial cerebrospinal fluid (ACSF) and aconitine perfusion in low-magnesium ACSF. Low-magnesium- (nominally zero) and aconitine- (230 nM) induced seizure-like event (SLE) population activity was recorded extracellularly. The results were consistent in showing that neither mefloquine (25 μM) nor genetic knockdown of Cx36 expression had anticonvulsant effects on SLE activity generated by either method. These findings call into question the widely held idea that open Cx36 gap junctions promote seizure activity.
1. Introduction
The role of gap junctions in seizurogenesis has been extensively investigated in the last 10 years, but remains an area of controversy. It seems intuitively reasonable that open gap junctions, acting as electrical synapses, should enhance synchrony between cortical neurons and promote seizure activity. Experimental studies, however, have reported conflicting results. While a number of in vivo and in vitro studies have shown anticonvulsant effects of pharmacological gap junction blockade [1–5], proconvulsant effects have also been reported [6]. Furthermore, transgenic mice lacking connexin36 (Cx36) gap junctions, the predominant neuronal gap junction subtype in adult animals, are seizure prone [7]. These discrepancies in the literature probably reflect two main complicating factors. Firstly, all known gap-junction-blocking drugs have off-target effects which could confound the experiments that were done with these agents [8]. Secondly, the effect of gap junction blockade on seizure activity will almost certainly depend on the details of exactly which gap junction subtype is manipulated. For example, Cx36 gap junctions are found almost exclusively between inhibitory interneurons, and a disruption to inhibitory timing as a result of their blockade could have a disinhibitory effect on pyramidal cell activity and hence an increase in seizures. Conversely, closure of putative pyramidal cell axonal gap junctions should suppress seizure activity [9].
Given the aforementioned inconsistencies, the aim of this study was to clarify the role of Cx36 gap junctions in the generation of neocortical seizure activity. We utilized the mouse neocortical slice preparation to investigate the effect of pharmacological and genetic manipulation of Cx36 gap junctions on two seizure models: low-magnesium artificial cerebrospinal fluid (ACSF) and aconitine perfusion in low-magnesium ACSF. The low-magnesium ACSF model of seizure activity has been extensively studied in the cortical slice preparation [10, 11] and is thought to be primarily of glutamatergic origin [12]. Aconitine is a potent neurotoxin that increases neuronal excitability by increasing intracellular calcium independent of NMDA and non-NMDA receptors [13]. It is thought to prevent synaptic voltage-gated sodium channel inactivation, causing cell depolarization and a prolonged increase in intracellular calcium [14]. A nano-molar concentration of aconitine induces a unique pattern of persistent seizure-like activity in the cortical slice preparation [15]. Neocortical seizure-like activity of both low magnesium and aconitine is sensitive to the anticonvulsant effects of phenytoin (data not shown) and some anaesthetics [16], and therefore they both represent valid models for investigating seizure mechanisms.
The seizure-like event (SLE) activity generated by these two methods were compared across cortical slices from wild-type (WT)—with and without pretreatment with the Cx36 gap junction blocker mefloquine—and connexin36 knockout (Cx36 KO) animals. The results clearly show that in the cerebrocortical seizure models tested, Cx36 gap junction blockade does not have a measurable anticonvulsant effect.
2. Materials and Methods
2.1. Slice Preparation
The study procedures were approved by the Waikato Ethics Committee. Neocortical slices were prepared from mixed sex C57/Bl6/129SV adult wild-type (WT) and Connexin36 knockout (Cx36 KO) mice. The latter were gifted by professor David Paul, Harvard University. The mice were decapitated while anesthetised with carbon dioxide, in accordance with local animal ethics guidelines. The brain was removed and immediately placed in ice-cold artificial cerebrospinal fluid (ACSF), modified for neuronal protection [17], with the following composition (in mM): NaCl 92.7; NaHCO3 24; NaH2PO4 1.2; KCl 3; MgCl2 19; CaCl2 0; D-glucose 25; bubbled with carbogen (95% O2, 5% CO2). Slices (400 μm) were cut in the coronal plane between bregma −2 mm to −5 mm on a vibratome (Campden Instruments, UK) in ice-cold ACSF as above and transferred to a holding chamber containing carbogenated, low-magnesium ACSF of the following composition: NaCl 124; NaHCO3 26; NaH2PO4 1.25; KCl 5; MgCl2 0; CaCl2 2; D-glucose 10, with or without added mefloquine (25 μm). The holding chamber was maintained at room temperature (18–20°C) where the slices were held for at least one hour before being transferred in pairs to an immersion-style recording chamber. Submerged slices were perfused with carbogenated ACSF at a flow rate of 5 mL/min using a peristaltic pump that recirculated the ACSF through the recording bath. All solutions were replaced after no more than 1 week of storage at 1–4°C.
2.2. Electrical Recording
Spontaneous local field potential activity was recorded from the neocortex using a 50 μm teflon-coated tungsten electrode. No specific neocortical layer or region was targeted. A silver/silver chloride disc electrode served as a common reference/bath ground. The signal was amplified (1000x, A-M Systems, USA) and bandpass filtered (1 and 1000 Hz) before analog-digital conversion (Power 1401, CED, UK) and recording on computer for later analysis (Spike2, CED, UK). The recording setup was enclosed within a grounded Faraday cage to reduce electrical noise.
2.3. Experimental Protocols
Three different exposures were investigated and compared.
(1) Control WT Slices —
Once transferred to the recording chamber, a stable pattern of seizure-like activity was established and recorded for at least 10 minutes in low-magnesium ACSF. Thereafter, aconitine (230 nM) was added to the low-magnesium perfusate and run continuously until the full effect of aconitine had been established, or in the absence of an obvious effect, for one hour. The aconitine-induced activity in low-magnesium ACSF is the same as when delivered in “normal” ACSF [15], except that the sequence of changes occurs somewhat more quickly when magnesium is absent from the solution. Thus aconitine is a unique seizure model with effects independent of the neuronal excitability induced by low magnesium.
(2) Mefloquine Pretreatment in WT Slices —
Slices were mefloquine pretreated by adding 25 μM mefloquine (in 0.1% DMSO) to the low-magnesium ACSF solution in which slices were held immediately after cutting. The dose of DMSO used has no effect on cortical SLE activity (results not shown). Because this holding period was at least one hour, the minimum mefloquine exposure time was one hour for recordings made from the initial pair of slices and up to 3 hours for slices recorded from subsequently. This length of exposure is sufficient to block the majority of Cx36 gap junctions in 400 μm thick brain slices [18]. Mefloquine perfusion was continued at 25 μM throughout the recording period. Thereafter, aconitine (230 nM) was added to the low-magnesium perfusate and run continuously, as above.
(3) Cx36 KO Slices —
Once transferred to the recording chamber, a stable pattern of seizure-like activity was established and recorded for at least 10 minutes in low-magnesium ACSF. Thereafter, aconitine (230 nM) was added to the low-magnesium perfusate and run continuously, as above.
2.4. Statistical Analysis
To investigate the effect of Cx36 gap junction blockade on low-magnesium SLE activity, baseline recordings were compared across the three groups: control WT, mefloquine-pretreated WT, and Cx36 KO slices. The amplitude (maximum peak-to-peak), frequency, and length of SLEs were quantified and compared using the Kruskal Wallis test (nonparametric ANOVA). The effect of Cx36 gap junction blockade on aconitine SLE activity was investigated by quantifying: SLE amplitude and frequency approximately 60 minutes after beginning aconitine perfusion: SLE length at approximately 30 and 60 minutes after beginning aconitine perfusion, and the time to the maximum increase in SLE frequency. These parameters were compared across groups using the Kruskal Wallis test. Unless otherwise stated, the data are expressed as median (95% confidence interval), and P < .05 was considered statistically significant.
3. Results
3.1. Low-Magnesium SLEs
Low-magnesium SLE activity was recorded from 12 control WT slices (from 3 animals), 12 mefloquine-pretreated WT slices (from 3 animals), and 14 Cx36 KO slices (from 3 animals). In the control WT group, each SLE consisted of a large-population depolarization, followed by a 4–7 Hz oscillation of lower amplitude than the initial component (Figure 1). The median SLE amplitude was 129(99–172) μV, SLE frequency 3.0(2.6–3.6) events/min, and SLE length 1.2(0.9–1.6) s. There was no significant difference in any of these parameters between the three groups, although there was a tendency toward a higher frequency in the mefloquine-pretreated slices (Table 1).
Figure 1.
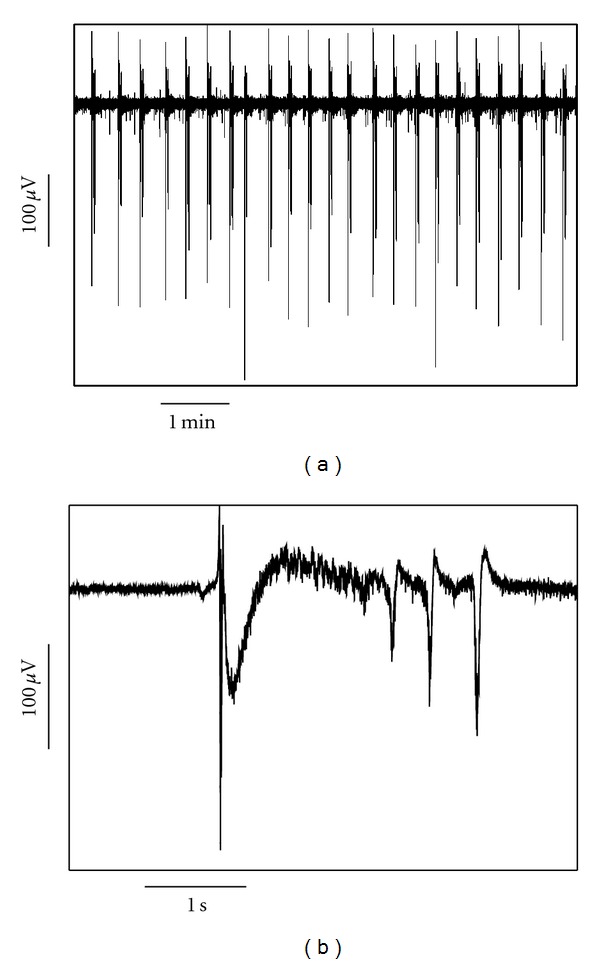
An example from a control WT slice showing the pattern of low-magnesium seizure-like event (SLE) activity. (a) shows a compressed time view and (b) an enlarged view of one SLE.
Table 1.
Low-magnesium seizure-like event amplitude, frequency, and length for the three groups. Data are median (95% confidence interval).
| WT (n = 12, 3 animals) |
WT + mefloquine (n = 12, 3 animals) |
Cx36 KO (n = 14, 3 animals) |
P value | |
|---|---|---|---|---|
| Amplitude (μV) | 129 (99–172) | 134 (98–179) | 146 (108–217) | .72 |
| Frequency (/min) | 3.0 (2.6–3.6) | 4.3 (3.3–6.9) | 3.3 (2.3–5.8) | .06 |
| Length (s) | 1.2 (0.9–1.6) | 0.9 (0.8–1.5) | 0.9 (0.3–2.8) | .69 |
WT: wild-type.
Cx36 KO, connexin36 knockout.
P values from Kruskal-Wallis test (nonparametric ANOVA) comparing the values across all three groups.
3.2. Aconitine SLEs
Acontine induced a consistent series of changes in the pattern of SLE activity in the WT slices, as illustrated in Figure 2. The most noticeable effects were a dramatic increase in event frequency (900(657–1272)%, P < .001, Wilcoxon test) and reduction in SLE amplitude (−37(−54–−26)%, P = .002, Wilcoxon test). Event length showed a biphasic pattern, with an increase (11(−10–24)%, P = .002, Wilcoxon test) followed by a decrease (−28(−50–1)%, P = .03, Wilcoxon test) compared to baseline. All of these effects were similar across all three groups (Table 2). The time to maximum change in event frequency was also similar across groups (P = .14, Kruskal-Wallis test). The only difference between the groups was evident in a trend toward a biphasic amplitude peak in the Cx36 KO recordings (18(8–59)% increase, P = .009, Wilcoxon test), similar to that observed for event length in all three groups.
Figure 2.
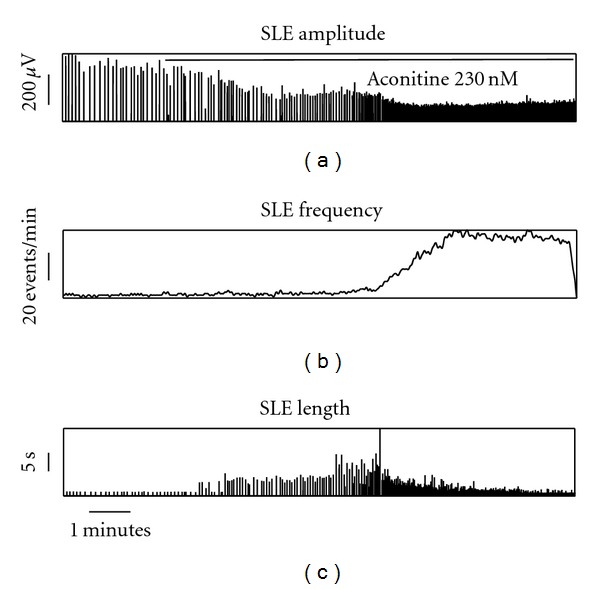
An example from a control WT slice showing the effect of aconitine on seizure-like event (SLE) activity. Each vertical line in (a) and (c) represents a single SLE.
Table 2.
Change in seizure-like event amplitude, frequency, and length (% change) for the three groups after 60 minutes of aconitine perfusion. Data are median (95% confidence interval).
| WT (n = 12, 3 animals) |
WT + mefloquine (n = 12, 3 animals) |
Cx36 KO (n = 14, 3 animals) |
P value | |
|---|---|---|---|---|
| Amplitude (% change) | −37 (−54–−26) | −48 (−64–−25) | −49 (−61–5) | .76 |
| Length (% change) | −28 (−50–1) | −37 (−61–−27) | −46 (−66–9) | .50 |
| Frequency (% change) | 900 (657–1272) | 557 (322–737) | 640 (416–1284) | .13 |
WT: wild-type.
Cx36 KO, connexin36 knockout.
P-values from Kruskal-Wallis test (nonparametric ANOVA) comparing the % change across all three groups.
4. Discussion
In this study we sought to clarify the role of Cx36 gap junctions in regulating neocortical seizure activity. The study was motivated by conflicting data in the literature showing both anti- and proepileptiform effects of gap junction blockade. The important finding was that Cx36 gap junction blockade in the adult cerebral cortex did not significantly alter two different models of neocortical seizure-like activity. Certainly, no anticonvulsant effect was observed, calling into question the widely held idea that open Cx36 gap junctions promote seizure activity.
4.1. Cerebral Cortex Connexin Expression
Only a limited number of connexins have been shown to be unequivocally expressed in the mature cerebral cortex. Cx30, 32, 43, 45, and 47 gap junctions are expressed by cortical glial cells [19–22]. The most common of these are Cx30 and Cx43, which are expressed by astrocytes [19, 21]. Central nervous system neurons express Cx36, Cx45, and Cx57, with the latter restricted to horizontal cells of the retina [23]. Cx36 is the most common neuronal gap junction in the mature cerebral cortex, and its expression appears to be restricted almost exclusively to inhibitory interneurons [24]. Thus, in the mature cerebral cortex, the most prominent gap junctions are those between inhibitory neurons (Cx36) and those between astrocytes (Cx30 and Cx43).
4.2. Targeted Effect of Cx36 Blockade
In this study we focused on Cx36 gap junctions using pharmacological (mefloquine) and genetic knock-down (Cx36 KO mice) methods. The dose (25 μM) and application time (at least 1 hour) of mefloquine used in this study was chosen to ensure penetration of the drug into the full depth of the slice [18]. The specificity of this dose for Cx36 gaps in brain slices is not known, but testing in cell cultures indicates that the dose required to significantly block Cx36 has little effect on other cortical gap junction subtypes [18]. It is reasonable to assume, therefore, that gap junction blockade by mefloquine in our study was targeted to Cx36. Like its related antimalarial compounds (quinine and quinidine), mefloquine is not void of off-target effects [18, 25, 26]. However, despite the possibility of some modest side effects, it is worth reiterating that the important result from this study was the absence of an anticonvulsant effect with a drug that demonstrably blocks Cx36 gap junctions. This, coupled with a similar lack of effect in the genetically modified animals, is a strong evidence that Cx36 gap junctions do not play a major role in preventing seizures in the cerebral cortex.
4.3. Possible Role of Astrocytic and Axoaxonal Gap Junctions in Seizure Activity
The fact that Cx36 blockade had no measurable anticonvulsant effect in our study implicates astrocytic gap junctions (Cx30 and Cx43) as the major players in the anticonvulsant effects reported in previous gap junction blockade studies [2, 4, 5]. Many of these studies used broad-spectrum gap junction blockers such as carbenoxolone and octanol, which would effect blockade of both neuronal and astrocytic gap junctions. The importance of astrocytic gap junctions in seizure mechanisms is further supported by a recent study showing anticonvulsant effects of mimetic peptides directed towards astrocytic gap junctions in hippocampal slice cultures [27]. Interestingly, in one study in which a more specific Cx36 gap junction blocker was used (quinine), the frequency of seizure events increased [28], in keeping with the present study and our previous findings [6]. Whether these proepileptiform observations reflect gap junction or off-target drug effects remains unclear.
Gap-junction-linked astrocytic networks could contribute to seizure genesis by facilitating the spread of neuronal activity via propagating calcium waves [29]. Astrocytes produce spontaneous slow calcium transients during seizure-like activity in many in vitro models of epilepsy [30–32]; these events occur independent of neuronal activity [33, 34] and are causally linked to astrocytic release of glutamate [32, 35] and an increase in neuronal excitability [36] and synchronicity [37].
Pyramidal cell axoaxonal gap junctions have been implicated in epileptogenesis in modeling studies [9]; blockade is theorised to have an anticonvulsant effect. Interestingly, there is evidence that axoaxonal gap junctions identified in the hippocampus are composed of Cx36 [38]. If this is correct, the results of our study suggest that either these gap junctions are not present in the cortex or are so few as to have minimal impact on seizure processes.
4.4. Genetic Knockout Compensation
One of the recognised disadvantages of using genetic knockout animals is the likelihood of compensatory effects during development, which could mask or confound investigation into the gene in question. Importantly, compensatory up- or downregulation in other connexin subtypes is not seen in Cx36 KO mice [7], suggesting that the functional consequence of Cx36 removal in these animals may be minimal. Looking beyond direct gap junction effects, compensatory changes to GABAergic modulation in the cerebral cortex have been reported in Cx36 KO animals [39]. While the exact nature of these changes is unclear, they are reflected functionally as a more robust GABAergic system. This would seem to run counter to the finding that these animals express seizure-like activity at least as robustly as WT mice. Further compensatory effects in Cx36 mice have been reported in other systems [40, 41]. However, these studies report effects at central nervous system sites outside the cerebral cortex, so their relevance to the present study is uncertain. While we cannot completely rule out undiscovered compensatory effects in Cx36 KO mice, current knowledge suggests that compensation to Cx36 ablation does not explain the lack of effect in the KO animals in our study.
4.5. Subcortical Effects
This study does not address the possibility that Cx36 gap junctions may regulate cortical seizure activity indirectly via expression at subcortical sites. Compared to WTs, Cx36 mice are highly prone to the convulsant effects of pentylenetetrazol (PTZ) in vivo [7]. However, a comparison of WT and Cx36 KO cortical slices shows no difference in PTZ sensitivity (unpublished observations), implicating a subcortical seizure-promoting effect in the Cx36 KO animals.
5. Conclusion
The findings of this study indicate that cerebral Cx36 gap junctions do not promote seizure activity and implicate astrocytic gap junctions as the main source of gap-junction-mediated regulation of seizure activity in the mature cerebral cortex.
References
- 1.Bostanci MO, Bagirici F. Anticonvulsive effects of quinine on penicillin-induced epileptiform activity: an in vivo study. Seizure. 2007;16(2):166–172. doi: 10.1016/j.seizure.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 2.Bostanci MO, Bağirici F. Anticonvulsive effects of carbenoxolone on penicillin-induced epileptiform activity: an in vivo study. Neuropharmacology. 2007;52(2):362–367. doi: 10.1016/j.neuropharm.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 3.Nassiri-Asl M, Zamansoltani F, Torabinejad B. Antiepileptic effects of quinine in the pentylenetetrazole model of seizure. Seizure. 2009;18(2):129–132. doi: 10.1016/j.seizure.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 4.Köhling R, Gladwell SJ, Bracci E, Vreugdenhil M, Jefferys JGR. Prolonged epileptiform bursting induced by 0-Mg in rat hippocampal slices depends on gap junctional coupling. Neuroscience. 2001;105(3):579–587. doi: 10.1016/s0306-4522(01)00222-6. [DOI] [PubMed] [Google Scholar]
- 5.Medina-Ceja L, Cordero-Romero A, Morales-Villagrán A. Antiepileptic effect of carbenoxolone on seizures induced by 4-aminopyridine: a study in the rat hippocampus and entorhinal cortex. Brain Research. 2008;1187(1):74–81. doi: 10.1016/j.brainres.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 6.Voss LJ, Jacobson G, Sleigh JW, Steyn-Ross A, Steyn-Ross M. Excitatory effects of gap junction blockers on cerebral cortex seizure-like activity in rats and mice. Epilepsia. 2009;50(8):1971–1978. doi: 10.1111/j.1528-1167.2009.02087.x. [DOI] [PubMed] [Google Scholar]
- 7.Jacobson GM, Voss LJ, Melin SM, et al. Connexin36 knockout mice display increased sensitivity to pentylenetetrazol-induced seizure-like behaviors. Brain Research. 2010;1360:198–204. doi: 10.1016/j.brainres.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 8.Juszczak GR, Swiergiel AH. Properties of gap junction blockers and their behavioural, cognitive and electrophysiological effects: animal and human studies. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33(2):181–198. doi: 10.1016/j.pnpbp.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 9.Traub RD, Draguhn A, Whittington MA, et al. Axonal gap junctions between principal neurons: a novel source of network oscillations, and perhaps epileptogenesis. Reviews in the Neurosciences. 2002;13(1):1–30. doi: 10.1515/revneuro.2002.13.1.1. [DOI] [PubMed] [Google Scholar]
- 10.Tsau Y, Guan LI, Wu JY. Initiation of spontaneous epileptiform activity in the neocortical slice. Journal of Neurophysiology. 1998;80(2):978–982. doi: 10.1152/jn.1998.80.2.978. [DOI] [PubMed] [Google Scholar]
- 11.Golomb D, Shedmi A, Curtu R, Ermentrout GB. Persistent synchronized bursting activity in cortical tissues with low magnesium concentration: a modeling study. Journal of Neurophysiology. 2006;95(2):1049–1067. doi: 10.1152/jn.00932.2005. [DOI] [PubMed] [Google Scholar]
- 12.Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251(4992):432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- 13.Ameri A, Peters T. Calcium-dependent, sustained enhancement of excitability during washout of aconitine in rat hippocampal slices. Experimental Brain Research. 1997;114(3):518–524. doi: 10.1007/pl00005661. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt H, Schmitt O. Effect of aconitine on the sodium permeability of the node of Ranvier. Pflugers Archiv European Journal of Physiology. 1974;349(2):133–148. doi: 10.1007/BF00586624. [DOI] [PubMed] [Google Scholar]
- 15.Voss LJ, Voss JM, McLeay L, Sleigh JW. Aconitine induces prolonged seizure-like events in rat neocortical brain slices. European Journal of Pharmacology. 2008;584(2-3):291–296. doi: 10.1016/j.ejphar.2008.02.024. [DOI] [PubMed] [Google Scholar]
- 16.Voss LJ, Sleigh JW. Enhancement of cortical GABAergic function does not account for the anticonvulsant effects of midazolam, isoflurane or etomidate. The Open Anesthesiology Journal. 2010;4:5–12. [Google Scholar]
- 17.Nowak LG, Bullier J. Spread of stimulating current in the cortical grey matter of rat visual cortex studied on a new in vitro slice preparation. Journal of Neuroscience Methods. 1996;67(2):237–248. [PubMed] [Google Scholar]
- 18.Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC, Srinivas M. Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(33):12364–12369. doi: 10.1073/pnas.0402044101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Condorelli DF, Mudò G, Trovato-Salinaro A, Mirone MB, Amato G, Belluardo N. Connexin-30 mRNA is up-regulated in astrocytes and expressed in apoptotic neuronal cells of rat brain following kainate-induced seizures. Molecular and Cellular Neuroscience. 2002;21(1):94–113. doi: 10.1006/mcne.2002.1155. [DOI] [PubMed] [Google Scholar]
- 20.Dermietzel R, Farooq M, Kessler JA, Althaus H, Hertzberg EL, Spray DC. Oligodendrocytes express gap junction proteins connexin32 and connexin45. GLIA. 1997;20(2):101–114. [PubMed] [Google Scholar]
- 21.Dermietzel R, Hertzberg EL, Kessler JA, Spray DC. Gap junctions between cultured astrocytes: immunocytochemical, molecular, and electrophysiological analysis. Journal of Neuroscience. 1991;11(5):1421–1432. doi: 10.1523/JNEUROSCI.11-05-01421.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Condorelli DF, Trovato-Salinaro A, Mudò G, Mirone MB, Belluardo N. Cellular expression of connexins in the rat brain: neuronal localization, effects of kainate-induced seizures and expression in apoptotic neuronal cells. European Journal of Neuroscience. 2003;18(7):1807–1827. doi: 10.1046/j.1460-9568.2003.02910.x. [DOI] [PubMed] [Google Scholar]
- 23.Hombach S, Janssen-Bienhold U, Söhl G, et al. Functional expression of connexin57 in horizontal cells of the mouse retina. European Journal of Neuroscience. 2004;19(10):2633–2640. doi: 10.1111/j.0953-816X.2004.03360.x. [DOI] [PubMed] [Google Scholar]
- 24.Deans MR, Gibson JR, Sellitto C, Connors BW, Paul DL. Synchronous activity of inhibitory networks in neocortex requires electrical synapses containing connexin36. Neuron. 2001;31(3):477–485. doi: 10.1016/s0896-6273(01)00373-7. [DOI] [PubMed] [Google Scholar]
- 25.Caridha D, Yourick D, Cabezas M, Wolf L, Hudson TH, Dow GS. Mefloquine-induced disruption of calcium homeostasis in mammalian cells is similar to that induced by ionomycin. Antimicrobial Agents and Chemotherapy. 2008;52(2):684–693. doi: 10.1128/AAC.00874-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou C, Xiao C, McArdle JJ, Ye JH. Mefloquine enhances nigral γ-aminobutyric acid release via inhibition of cholinesterase. Journal of Pharmacology and Experimental Therapeutics. 2006;317(3):1155–1160. doi: 10.1124/jpet.106.101923. [DOI] [PubMed] [Google Scholar]
- 27.Samoilova M, Wentlandt K, Adamchik Y, Velumian AA, Carlen PL. Connexin 43 mimetic peptides inhibit spontaneous epileptiform activity in organotypic hippocampal slice cultures. Experimental Neurology. 2008;210(2):762–775. doi: 10.1016/j.expneurol.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 28.Gajda Z, Szupera Z, Blazsó G, Szente M. Quinine, a blocker of neuronal Cx36 channels, suppresses seizure activity in rat neocortex in vivo. Epilepsia. 2005;46(10):1581–1591. doi: 10.1111/j.1528-1167.2005.00254.x. [DOI] [PubMed] [Google Scholar]
- 29.Nedergaard M. Direct signaling from astrocytes to neurons in cultures of mammalian brain cells. Science. 1994;263(5154):1768–1771. doi: 10.1126/science.8134839. [DOI] [PubMed] [Google Scholar]
- 30.Stout C, Charles A. Modulation of intercellular calcium signaling in astrocytes by extracellular calcium and magnesium. GLIA. 2003;43(3):265–273. doi: 10.1002/glia.10257. [DOI] [PubMed] [Google Scholar]
- 31.Tashiro A, Goldberg J, Yuste R. Calcium oscillations in neocortical astrocytes under epileptiform conditions. Journal of Neurobiology. 2002;50(1):45–55. doi: 10.1002/neu.10019. [DOI] [PubMed] [Google Scholar]
- 32.Tian GF, Azmi H, Takano T, et al. An astrocytic basis of epilepsy. Nature Medicine. 2005;11(9):973–981. doi: 10.1038/nm1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parri HR, Gould TM, Crunelli V. Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nature Neuroscience. 2001;4(8):803–812. doi: 10.1038/90507. [DOI] [PubMed] [Google Scholar]
- 34.Wang TF, Zhou C, Tang AH, Wang SQ, Chai Z. Cellular mechanism for spontaneous calcium oscillations in astrocytes. Acta Pharmacologica Sinica. 2006;27(7):861–868. doi: 10.1111/j.1745-7254.2006.00397.x. [DOI] [PubMed] [Google Scholar]
- 35.Parpura V, Basarsky TA, Liu F, Jeftinija K, Jeftinija S, Haydon PG. Glutamate-mediated astrocyte-neuron signalling. Nature. 1994;369(6483):744–747. doi: 10.1038/369744a0. [DOI] [PubMed] [Google Scholar]
- 36.Fellin T, Gomez-Gonzalo M, Gobbo S, Carmignoto G, Haydon PG. Astrocytic glutamate is not necessary for the generation of epileptiform neuronal activity in hippocampal slices. Journal of Neuroscience. 2006;26(36):9312–9322. doi: 10.1523/JNEUROSCI.2836-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fellin T, Pascual O, Gobbo S, Pozzan T, Haydon PG, Carmignoto G. Neuronal synchrony mediated by astrocytic glutamate through activation of extrasynaptic NMDA receptors. Neuron. 2004;43(5):729–743. doi: 10.1016/j.neuron.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 38.Hamzei-Sichani F, Kamasawa N, Janssen WGM, et al. Gap junctions on hippocampal mossy fiber axons demonstrated by thin-section electron microscopy and freeze-fracture replica immunogold labeling. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(30):12548–12553. doi: 10.1073/pnas.0705281104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Voss LJ, Melin S, Jacobson G, Sleigh JW. GABAergic compensation in connexin36 knock-out mice evident during low-magnesium seizure-like event activity. Brain Research. 2010;1360:49–55. doi: 10.1016/j.brainres.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 40.De Zeeuw CI, Chorev E, Devor A, et al. Deformation of network connectivity in the inferior olive of connexin 36-deficient mice is compensated by morphological and electrophysiological changes at the single neuron level. Journal of Neuroscience. 2003;23(11):4700–4711. doi: 10.1523/JNEUROSCI.23-11-04700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cummings DM, Yamazaki I, Cepeda C, Paul DL, Levine MS. Neuronal coupling via connexin36 contributes to spontaneous synaptic currents of striatal medium-sized spiny neurons. Journal of Neuroscience Research. 2008;86(10):2147–2158. doi: 10.1002/jnr.21674. [DOI] [PubMed] [Google Scholar]