

Abstract
Recent studies have found that cannabinoids may improve neuropsychological performance, ameliorate negative symptoms, and have antipsychotic properties for a subgroup of the schizophrenia population. These findings are in contrast to the longstanding history of adverse consequences of cannabis use, predominantly on the positive symptoms, and a balanced neurochemical basis for these opposing views is lacking. This paper details a review of the neurobiological substrates of schizophrenia and the neurochemical effects of cannabis use in the normal population, in both cortical (in particular prefrontal) and subcortical brain regions. The aim of this paper is to provide a holistic neurochemical framework in which to understand how cannabinoids may impair, or indeed, serve to ameliorate the positive and negative symptoms as well as cognitive impairment. Directions in which future research can proceed to resolve the discrepancies are briefly discussed.
1. Introduction
A body of literature over the past three decades has established cannabis use to be a significant risk factor for the onset and development of schizophrenia [1–5], with early onset of use (by age 15) believed to confer greater risk than later onset of use [6]. In a comprehensive meta-analysis of prospective and population-based studies by Moore et al. [7], an increased risk of psychosis was found in individuals who had ever used cannabis, and a dose-response effect was yielded in which greater risk applied to people who used cannabis most frequently. The authors could not, however, draw a definitive causal link between cannabis use and psychosis on grounds that the studies were observational in nature, and no robust evidence supports the view that early-age onset of cannabis use might be more harmful than later-age onset of use. The increased risk of psychosis in people using cannabis from a younger age could indicate greater cumulative exposure to cannabis rather than a sensitive period of exposure.
The high prevalence of cannabis use in schizophrenia combined with observations that cannabis exacerbates the positive symptoms has driven research to investigate why cannabis is used so widely in this population. According to the self-medication hypothesis, there are beneficial effects of cannabis and cannabinoids on the symptoms of schizophrenia. Cross-sectional studies comparing users and nonusers have shown that cannabis use is associated with a reduction in negative symptoms [8–14]. Animal and human models have also suggested that cannabinoids possess antipsychotic properties [15–17]. One of these studies [15] involved a randomised, double-blind controlled trial in 42 patients with acute schizophrenia and found antipsychotic properties of cannabidiol (CBD) that were comparable to amisulpride. Other studies in which CBD has been administered to rats as well as healthy human volunteers have also demonstrated antipsychotic properties of CBD [16, 17].
These mixed findings on how cannabis or cannabinoids impact on the positive and negative symptoms of schizophrenia may in part be attributable to differences in methodology and research design. For example, studies have used different methods to measure cannabis exposure and assess outcome [7], and results are likely to be influenced by whether the studies were observational (prospective versus cross sectional) or experimental (direct drug administration by way of randomised controlled trials or other means). A range of other potential confounding factors are also likely to have contributed to the inconsistent results, particularly where observational designs were employed.
From a neuropsychological perspective, a paucity of research exists. Cannabis use, in some studies of schizophrenia, has been found to be associated with worse neuropsychological performance [18–20]. Conversely, cannabis has been suggested to improve neuropsychological functioning [21–25]. Only one of these studies [18] involved a randomised, double-blind, placebo-controlled trial, in which the effects of i.v. ∆9-tetrahydrocannabinol (THC) on cognition were studied in 13 stabilised schizophrenia patients and 22 healthy controls, 30 minutes after drug administration. All participants administered THC relative to their placebo baseline performance demonstrated cognitive impairments in domains such as memory and attention, and the schizophrenia group performed more poorly than the control group. Five of the remaining studies [19, 21–25] were cross sectional in nature in which current or past cannabis users with schizophrenia were compared to non current users or those without a past history of use; and one study [20] performed a correlational analysis examining the relationship between level of cannabis use over the preceding year and cognition in persons with a psychotic illness.
Again, the inconsistencies in the findings between these neuropsychological studies are likely to be at least, in part, attributable to methodological variability between the studies, as well as methodological limitations within each study (for full review, see Coulston et al. [26]). For example, different methods were used to measure cannabis exposure, as per the studies detailed earlier which assessed the clinical symptoms. All seven neuropsychological studies used only a single index to classify cannabis use, which does not adequately reflect the variable and fluctuating level of cannabis use that may occur over time in the schizophrenia population. In three instances [19, 22, 23], cannabis use was defined solely with respect to abuse/dependence according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV) [27]. In one study [18], only the acute effects of cannabis were examined, whilst in another study [21], only the residual, longer-term effects of cannabis were examined in participants who had been abstinent for at least 28 days. Such indices of cannabis use fail to consider factors such as recency and frequency of use (beyond the acute intoxication phase), which have been found to impact on cognition in the normal population [28].
Other limitations in several of these neuropsychological studies, some of which are also relevant to those studies mentioned earlier that focused on the associations between cannabis use and the positive/negative symptoms, include absence of drug screening procedures prior to assessment, restricted range of cognitive functions assessed, minimal or no control of important confounding variables such as current and past history of other substance use (including caffeine and nicotine), and a range of demographic and medical/psychiatric variables (e.g., gender, level of education, premorbid IQ, early-age onset of psychosis, duration of mental illness, medications). Furthermore, several of these studies did not include a control group which limits interpretation of their findings, because it cannot be determined if the cognitive performance of the cannabis users was frankly impaired (i.e., fell below the normal range of performance) or, was in fact within the normal range of performance.
One of the most comprehensive cross-sectional neuropsychological studies to date [29] was conducted by considering all the limitations and range of potential confounds detailed above. The investigators found that cannabis use was associated with enhanced cognitive functioning, predominantly in the areas of attention, processing speed, and executive functioning, domains which rely heavily on prefrontal cortical networks.
Additional reasons for the discrepancies across the literature on the effects of cannabis use on symptomatology and cognition may pertain to the fact that separate and specialised groups of people with schizophrenia react differently to cannabis and other substances. Empirical literature indicates that systematic research on schizophrenia is difficult due to the disorder's heterogeneity and different diagnostic conceptualisations [30], which would imply that the neurochemical imbalances in different brain regions are not equal in all persons. As such, the schizophrenia population is deemed pharmacologically heterogenous [31], in which case the interaction between cannabinoids, neurotransmitters, and antipsychotic treatments is likely to vary between individuals. Subsequently, a balanced neurochemical framework to conceptualise both the adverse and potential therapeutic use of cannabinoids for respective groups of the population would be useful for two reasons; first, in light of the new emerging literature supporting a therapeutic benefit for all symptoms, and second, in the context of relatively minimal research that has been conducted in the field of neuropsychology. Further, positing a more balanced neurochemical framework that perhaps underpins an aspect of the core pathophysiology of schizophrenia allows a reconceptualisation of aetiology and treatment of this complex disorder. In particular, a balanced framework promotes awareness that there may not be a unidirectional method of treatment for all subgroups of the population, thereby providing a foundation for novel approaches to investigating the basis of this neuropsychiatric condition.
This paper briefly outlines the neurochemical processes associated with schizophrenia and cannabis use in the normal population, with implications for the effects of cannabinoids on the positive and negative symptoms, as well as cognitive impairment.
2. Symptomatology in Schizophrenia and Neurotransmitter Systems
The effectiveness of conventional antipsychotic medications in alleviating positive symptoms is largely attributed to the blockade of dopamine transmission, especially in the mesolimbic system [32, 33]. Atypical antipsychotics are also believed to exert their therapeutic benefits on psychotic symptoms (in the same cerebral regions) via their antagonistic actions on serotonin, acetylcholine, and noradrenaline receptors [34–37], and via agonistic actions on GABA [35] and glutamate transmission [38]. This suggests that in the unmedicated state, the positive symptoms of schizophrenia are, at least in part, attributable to hyperactivation of dopamine, serotonin, acetylcholine, and noradrenaline, and hypoactivity of GABA and glutamate in the mesolimbic and other subcortical regions.
Moreover, reduced prefrontal dopamine activity is associated with exaggerated striatal dopaminergic activity [39], which is thought to account for the negative symptoms and cognitive impairment. Negative symptoms and cognitive impairments have also been associated with decreased prefrontal acetylcholine, serotonin, noradrenaline, and glutamate [39–47]. The therapeutic action of atypical antipsychotics involves facilitating prefrontal neurotransmission across all of these neurotransmitter systems, except GABA [48–53]. This suggests that in the unmedicated state, the negative symptoms and cognitive impairments of schizophrenia are, at least in part, attributable to hypoactivity of dopamine, serotonin, acetylcholine, noradrenaline, and glutamate in the prefrontal region.
With respect to GABA, although direct enhancement of prefrontal activity would be a potential treatment for schizophrenia [54], atypical antipsychotics tend to exert their effect in the prefrontal cortex by inhibiting release of GABA from interneurons [55]. This subsequently enhances prefrontal neurotransmitter activity. Dopamine and serotonin signalling, associated with antipsychotics, may therefore be responsible for the inhibition of GABAergic currents, and subsequent increase in prefrontal activity [56, 57].
3. The Endocannabinoid System
Cannabinoids exert their effect by binding to specific endogenous cannabinoid receptors [58] known as CB1 and CB2, which are located in the CNS and peripherally in the spleen and immune cells [59, 60]. The CB1 receptors are widely distributed in the brain including the cerebral cortex (especially the frontal and medial temporal lobes), limbic areas, basal ganglia, ventral tegmental area, thalamus, hypothalamus, cerebellum, and brainstem [58, 61–65].
CB1 receptors reside within the lipid membrane of the presynaptic neuron terminals, are modulated by the postsynaptic release of endocannabinoids (namely anandamide and 2-arachidonoyl glycerol), and are also influenced by exogenously consumed cannabinoids. CB1 receptors act as neuromodulators through coupling with intracellular G-proteins controlling cyclic adenosine monophosphate (c-AMP) formation and Ca2+ and K+ transport. In this respect, the cannabinoid system has important interactions with several neurotransmitter systems.
Cannabinoids augment potassium stimulated striatal dopamine efflux and subsequently dopamine release in the mesolimbic system (especially the nucleus accumbens), medial prefrontal cortex, midbrain regions, and substantia nigra [58, 66–74]. THC has been shown to facilitate stimulation of dopamine release in the medial forebrain bundle of the mesolimbic system at dose ranges pharmacologically relevant to human recreational use [68]. As the positive symptoms of schizophrenia are related to elevated dopamine release, particularly in the mesolimbic system, cannabinoids have long been implicated in worsening the positive symptoms of schizophrenia [73, 75, 76].
In contrast to the effect of cannabinoids on dopamine release in the mesolimbic system, significant evidence exists for the CB1 receptor mechanism to play an important role in “dampening” neuroexcitability [59, 60, 77]. It has been demonstrated that in the cerebral cortex, serotonin activity is inhibited by cannabinoid receptor agonists (e.g., WIN 55,212 and CP-55,940) [78]. Conversely, studies employing CB1 receptor antagonists (e.g., SR 141716A) have demonstrated increases in serotonin in various brain regions including the medial prefrontal cortex and nucleus accumbens [79].
Cannabinoids have been demonstrated to decrease acetylcholine release in the medial prefrontal cortex, hippocampus, and striatum [80–82]. Likewise, the CB1 receptor antagonist (SR 141716A) has resulted in an increase in acetylcholine in the medial prefrontal cortex and hippocampus [79, 83].
Noradrenaline activity in various areas of the brain including the hippocampus, cerebellum, hypothalamus, and cerebral cortex, has been inhibited by the cannabinoid receptor agonist (WIN 55,212) [84]. This inhibition was subsequently attenuated by the cannabinoid receptor antagonist (SR 141716). Cannabinoid-inhibited release of noradrenaline has also been demonstrated in the hypothalamus and striatum [60, 85–87].
The CB1 receptor has been demonstrated to be present in high concentration at presynaptic terminals of glutaminergic synapses in the hippocampus and other areas of the brain [88]. In general, activation of the CB1 receptor has been shown to inhibit glutamate release in the cerebellum, hippocampus, prefrontal cortex, and substantia nigra [77, 78, 89–91].
CB1 receptor mediated decreases in GABA has also been demonstrated in brain regions including the prefrontal cortex [78, 88, 92, 93], hippocampus [90, 94, 95], and subcortical regions including the basal ganglia [96–100].
4. Cannabinoids and Prefrontal Neurotransmission
Despite the general consensus that cannabinoids inhibit all major neurotransmitters (except dopamine) in various brain regions including the prefrontal cortex, a number of studies have supported the converse notion that cannabinoids in fact stimulate neurotransmission release in the prefrontal cortex [88, 92, 93].
An example of how neurotransmission of dopamine could be theoretically enhanced by cannabinoids in the prefrontal cortex is related to CB1 receptors being preferentially located on GABAergic interneurons in areas including the cerebral cortex, ventral tegmental area, and hippocampus [92]. Cannabinoids such as THC decrease the excitability of GABA-ergic interneurons, and therefore, given GABA is the major inhibitory neurotransmitter in the CNS, disinhibition of dopamine and glutamate follows. Consequently, increases in the outflow of neurotransmission parallel the decrease in extracellular GABA.
As another example of how neurotransmission is theoretically enhanced by cannabinoids in the prefrontal cortex, studies have demonstrated that increased dopamine neurotransmission in the mesolimbic projections from the nucleus accumbens triggers an inhibition of GABA-ergic efferents to the basal forebrain. The process of inhibiting the release of GABA, in turn, decreases the inhibition of other neurotransmitters such as dopamine, acetylcholine, noradrenaline, and glutamate. A specific example of this process has been postulated where dopamine's direct inhibitory effect on both glutamate and GABA-ergic activity leads to an increased excitability of cholinergic flow [46, 101].
Proceeding from these lines of reasoning, studies have demonstrated that in the prefrontal region, cannabinoids mediate increases in noradrenaline [102], acetylcholine [103, 104], and glutamate [92, 105]. With respect to serotonin and GABA, studies have demonstrated that cannabinoids mediate the inhibition of reuptake in cortical areas, which leads to their increase [106].
5. The Effects of Cannabis on Cognition and the Positive/Negative Symptoms of Schizophrenia
Table 1 presents a summary of the information discussed thus far. The grey arrows in the “Schizophrenia” columns represent the directions of neurotransmitter release in the unmedicated state of the disorder in both the prefrontal cortex and subcortical regions. As discussed above, the negative symptoms and cognitive impairments are generally conceptualised in terms of decreased neurotransmission of all six major systems in the prefrontal cortex, whilst the positive symptoms are generally thought to arise because of increased neurotransmission of dopamine, serotonin, acetylcholine and noradrenaline systems, and decreased neurotransmission of glutamate and GABA systems in subcortical regions.
Table 1.
Directions of neurotransmitter release in the prefrontal cortex and subcortical regions.
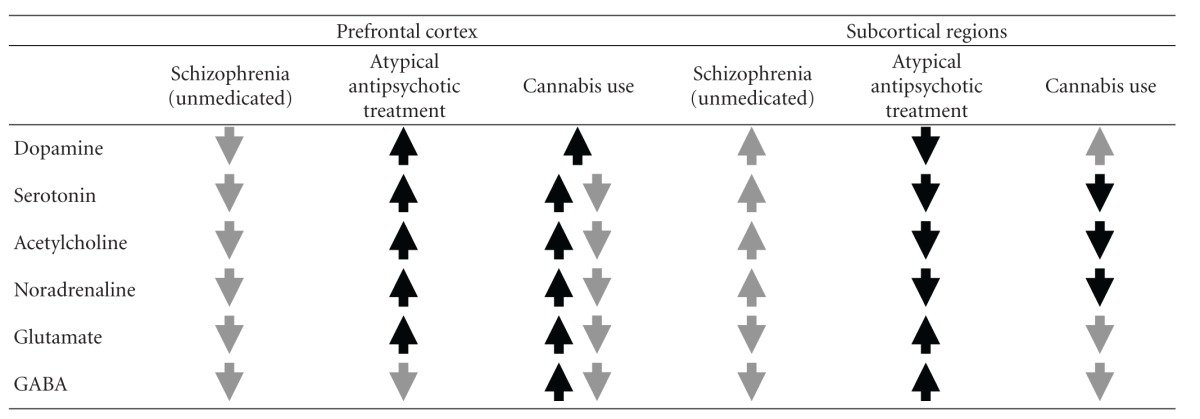
|
The black arrows in the “Atypical antipsychotic treatment” columns represent how these medications are purported to ameliorate the symptoms and cognitive impairments of schizophrenia. Specifically, atypical antipsychotics are postulated to ameliorate the negative symptoms and cognitive impairment by stimulating neurotransmission in the prefrontal cortex (of all neurotransmitters except GABA), and are thought to ameliorate the positive symptoms by inhibiting dopamine, serotonin, acetylcholine, and noradrenaline, and by increasing glutamate and GABA in subcortical regions.
Of pertinent interest to this review is the theoretical amelioration and/or exacerbation of the symptoms and cognitive impairments of schizophrenia that cannabis use may produce. As Table 1 shows, cannabis in the normal population leads to excitation of dopamine in the prefrontal cortex. However, what remains relatively less certain is whether cannabis leads to excitation or inhibition of the other five major neurotransmitters in the prefrontal cortex (i.e., serotonin, acetylcholine, noradrenaline, glutamate, and GABA), given that evidence for both modes of action have been presented. Hence, the postulated effect of cannabis on these five systems is indicated by both grey and black arrows.
Consequently, looking at the “Schizophrenia (unmedicated)” and “Cannabis use” columns in conjunction, Table 1 shows that although cannabis has the potential to exacerbate the negative symptoms and cognitive impairments of schizophrenia via contributing further to the “hypofrontal” nature of the disorder, cannabis also has a therapeutic potential by facilitating neurotransmission (at least in the dopaminergic system) in the prefrontal cortex. In line with the latter perspective, research has demonstrated enhanced cognitive performance in schizophrenia with comorbid cannabis use [21–25, 29], which may be attributable to stimulation of prefrontal neurotransmission.
In subcortical regions, Table 1 shows that cannabis use is associated with excitation of dopamine and inhibition of all the other five major neurotransmitters (i.e., serotonin, noradrenaline, acetylcholine, glutamate, and GABA). Hence, cannabis has the potential to counteract increased levels of serotonin, acetylcholine, and noradrenaline that occur in subcortical regions in untreated schizophrenia and thus, provide an antipsychotic therapeutic effect. However, excitation of dopamine and inhibition of glutamate and GABA in subcortical regions produced by cannabis use may augment the dysregulation that occurs in these three systems in untreated schizophrenia, and possibly exacerbate the positive symptoms.
6. Summary and Conclusion
Cannabis use in the schizophrenia population has been shown to worsen the prognosis and increase the burden of the disorder. However, evidence exists for a subgroup of the population to suggest that cannabinoids have therapeutic effects on the negative and positive symptoms, as well as cognitive impairments. Although this evidence is not conclusive and requires further research and replication, a more comprehensive and broad-based neurochemical framework has been presented in this paper, offering an explanation for the potentially therapeutic effects of cannabinoids in addition to its adverse effects.
Whilst the neurochemical effects of cannabinoids are complex, cannabinoids appear to have at least in part, a “restorative” effect on neurotransmitter dysfunctions in schizophrenia, which may underpin the biological substrate of the therapeutic effects cannabis has been demonstrated to have in recent studies.
In the context of recent research by Coulston et al. [29], future studies need to establish which subgroups of schizophrenia most benefit from cannabinoids as a putative “treatment”; which subgroups demonstrate nil or minimal exacerbation of positive symptoms in the context of cannabis use, or indeed, which subgroups may experience antipsychotic effects of cannabis; how such a treatment may complement or interact with standard antipsychotics and other psychiatric treatments; what constitutes an effective quantity or dose of cannabinoids to exert a beneficial impact on cognition; what precise frequency of administration would be required; how long the effects of cannabinoids last on cognition following a recent dose; how such a treatment may generalise to other domains of real-life functioning such as employment and social settings (and in particular, cognitive processes required to function well in these settings); and whether such a treatment can be generalised to older age groups. Methods by which future research could proceed to address these questions include randomised, double-blind, controlled drug trials.
Clearly there are many questions that need to be addressed and the use of further randomised, double-blind studies is necessary to establish the effects of cannabis across the domains of cognition, mood, and mentation. However, given the prevalence of cannabis use and its integral role in the clinical manifestation of psychosis, this is an area for future research, and reconceptualising our neurochemical understanding is perhaps a useful first step. In this context, the consideration, appraisal, and rigorous testing of novel models are necessary, and are likely to advance our understanding.
References
- 1.Andréasson S, Allebeck P, Engström A, Rydberg U. Cannabis and schizophrenia. A longitudinal study of Swedish conscripts. Lancet. 1987;2(8574):1483–1486. doi: 10.1016/s0140-6736(87)92620-1. [DOI] [PubMed] [Google Scholar]
- 2.Fergusson DM, Horwood LJ, Swain-Campbell NR. Cannabis dependence and psychotic symptoms in young people. Psychological Medicine. 2003;33(1):15–21. doi: 10.1017/s0033291702006402. [DOI] [PubMed] [Google Scholar]
- 3.Silva PA, Stanton WR. From Child to Adult: The Dunedin Multidisciplinary Health and Development Study. Auckland, UK: Oxford University Press; 1996. [Google Scholar]
- 4.Van Os J, Bak M, Hanssen M, Bijl RV, De Graaf R, Verdoux H. Cannabis use and psychosis: a longitudinal population-based study. American Journal of Epidemiology. 2002;156(4):319–327. doi: 10.1093/aje/kwf043. [DOI] [PubMed] [Google Scholar]
- 5.Zammit S, Allebeck P, Andréasson S, Lundberg I, Lewis G. Self reported cannabis use as a risk factor for schizophrenia in Swedish conscripts of 1969: historical cohort study. British Medical Journal. 2002;325(7374):1199–1201. doi: 10.1136/bmj.325.7374.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arseneault L, Cannon M, Poulton R, Murray R, Caspi A, Moffitt TE. Cannabis use in adolescence and risk for adult psychosis: longitudinal prospective study. British Medical Journal. 2002;325(7374):1212–1213. doi: 10.1136/bmj.325.7374.1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore TH, Zammit S, Lingford-Hughes A, et al. Cannabis use and risk of psychotic or affective mental health outcomes: a systematic review. Lancet. 2007;370(9584):319–328. doi: 10.1016/S0140-6736(07)61162-3. [DOI] [PubMed] [Google Scholar]
- 8.Bersani G, Orlandi V, Gherardelli S, Pancheri P. Cannabis and neurological soft signs in schizophrenia: absence of relationship and influence on psychopathology. Psychopathology. 2002;35(5):289–295. doi: 10.1159/000067064. [DOI] [PubMed] [Google Scholar]
- 9.Bersani G, Orlandi V, Kotzalidis GD, Pancheri P. Cannabis and schizophrenia: impact on onset, course, psychopathology and outcomes. European Archives of Psychiatry and Clinical Neuroscience. 2002;252(2):86–92. doi: 10.1007/s00406-002-0366-5. [DOI] [PubMed] [Google Scholar]
- 10.Compton MT, Furman AC, Kaslow NJ. Lower negative symptom scores among cannabis-dependent patients with schizophrenia-spectrum disorders: preliminary evidence from an African American first-episode sample. Schizophrenia Research. 2004;71(1):61–64. doi: 10.1016/j.schres.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 11.Compton MT, Whicker NE, Hochman KM. Alcohol and cannabis use in urban, African American, first-episode schizophrenia-spectrum patients: associations with positive and negative symptoms. Journal of Clinical Psychiatry. 2007;68(12):1939–1945. doi: 10.4088/jcp.v68n1215. [DOI] [PubMed] [Google Scholar]
- 12.Dubertret C, Bidard I, Adès J, Gorwood P. Lifetime positive symptoms in patients with schizophrenia and cannabis abuse are partially explained by co-morbid addiction. Schizophrenia Research. 2006;86(1–3):284–290. doi: 10.1016/j.schres.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Peralta V, Cuesta MJ. Influence of cannabis abuse on schizophrenic psychopathology. Acta Psychiatrica Scandinavica. 1992;85(2):127–130. doi: 10.1111/j.1600-0447.1992.tb01456.x. [DOI] [PubMed] [Google Scholar]
- 14.Skosnik PD, Park S, Dobbs L, Gardner WL. Affect processing and positive syndrome schizotypy in cannabis users. Psychiatry Research. 2008;157(1–3):279–282. doi: 10.1016/j.psychres.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 15.Leweke FM, Koethe D, Gerth CW, et al. Cannabidiol as an antipsychotic agent. European Psychiatry. 2007;22(supplement 1):p. S21. [Google Scholar]
- 16.Zuardi AW, Crippa JAS, Hallak JEC, Moreira FA, Guimarães FS. Cannabidiol, a Cannabis sativa constituent, as an antipsychotic drug. Brazilian Journal of Medical and Biological Research. 2006;39(4):421–429. doi: 10.1590/s0100-879x2006000400001. [DOI] [PubMed] [Google Scholar]
- 17.Zuardi AW, Rodrigues JA, Cunha JM. Effects of cannabidiol in animal models predictive of antipsychotic activity. Psychopharmacology. 1991;104(2):260–264. doi: 10.1007/BF02244189. [DOI] [PubMed] [Google Scholar]
- 18.D’Souza DC, Abi-Saab WM, Madonick S, et al. Delta-9-tetrahydrocannabinol effects in schizophrenia: implications for cognition, psychosis, and addiction. Biological Psychiatry. 2005;57(6):594–608. doi: 10.1016/j.biopsych.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 19.Liraud F, Verdoux H. Effect of comorbid substance use on neuropsychological performance in subjects with psychotic or mood disorders. Encephale. 2002;28(2):160–168. [PubMed] [Google Scholar]
- 20.Pencer A, Addington J. Substance use and cognition in early psychosis. Journal of Psychiatry and Neuroscience. 2003;28(1):48–54. [PMC free article] [PubMed] [Google Scholar]
- 21.Jockers-Scherübl MC, Wolf T, Radzei N, et al. Cannabis induces different cognitive changes in schizophrenic patients and in healthy controls. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2007;31(5):1054–1063. doi: 10.1016/j.pnpbp.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Kumra S, Thaden E, DeThomas C, Kranzler H. Correlates of substance abuse in adolescents with treatment-refractory schizophrenia and schizoaffective disorder. Schizophrenia Research. 2005;73(2-3):369–371. doi: 10.1016/j.schres.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Sevy S, Burdick KE, Visweswaraiah H, et al. Iowa Gambling Task in schizophrenia: a review and new data in patients with schizophrenia and co-occurring cannabis use disorders. Schizophrenia Research. 2007;92(1–3):74–84. doi: 10.1016/j.schres.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stirling J, White C, Lewis S, et al. Neurocognitive function and outcome in first-episode schizophrenia: a 10-year follow-up of an epidemiological cohort. Schizophrenia Research. 2003;65(2-3):75–86. doi: 10.1016/s0920-9964(03)00014-8. [DOI] [PubMed] [Google Scholar]
- 25.Stirling J, Lewis S, Hopkins R, White C. Cannabis use prior to first onset psychosis predicts spared neurocognition at 10-year follow-up. Schizophrenia Research. 2005;75(1):135–137. doi: 10.1016/j.schres.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 26.Coulston CM, Perdices M, Tennant CT. The Neuropsychology of cannabis and other substance use in schizophrenia: review of the literature and critical evaluation of methodological issues. Australian and New Zealand Journal of Psychiatry. 2007;41(11):869–884. doi: 10.1080/00048670701634952. [DOI] [PubMed] [Google Scholar]
- 27.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4th edition. Washington, DC, USA: APA; 1994. [Google Scholar]
- 28.Pope HG, Jr., Gruber AJ, Hudson JI, Huestis MA, Yurgelun-Todd D. Neuropsychological performance in long-term cannabis users. Archives of General Psychiatry. 2001;58(10):909–915. doi: 10.1001/archpsyc.58.10.909. [DOI] [PubMed] [Google Scholar]
- 29.Coulston CM, Perdices M, Tennant CC. The neuropsychological correlates of cannabis use in schizophrenia: lifetime abuse/dependence, frequency of use, and recency of use. Schizophrenia Research. 2007;96(1–3):169–184. doi: 10.1016/j.schres.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 30.Keyes M. Assessing schizophrenia with the MMPI-2. Dissertation Abstracts International. Section B. 2004;64:p. 5788. [Google Scholar]
- 31.Ban TA. Neuropsychopharmacology and the genetics of schizophrenia: a history of the diagnosis of schizophrenia. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2004;28(5):753–762. doi: 10.1016/j.pnpbp.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 32.Binder EB, Kinkead B, Owens MJ, Nemeroff CB. The role of neurotensin in the pathophysiology of schizophrenia and the mechanism of action of antipsychotic drugs. Biological Psychiatry. 2001;50(11):856–872. doi: 10.1016/s0006-3223(01)01211-2. [DOI] [PubMed] [Google Scholar]
- 33.Farde L. Brain imaging of schizophrenia—the dopamine hypothesis. Schizophrenia Research. 1997;28(2-3):157–162. doi: 10.1016/s0920-9964(97)00121-7. [DOI] [PubMed] [Google Scholar]
- 34.Bymaster FP, Nelson DL, DeLapp NW, et al. Antagonism by olanzapine of dopamine D1, serotonin2, muscarinic, histamine H1 and α1-adrenergic receptors in vitro. Schizophrenia Research. 1999;37(1):107–122. doi: 10.1016/s0920-9964(98)00146-7. [DOI] [PubMed] [Google Scholar]
- 35.Ma J, Ye N, Lange N, Cohen BM. Dynorphinergic GABA neurons are a target of both typical and atypical antipsychotic drugs in the nucleus accumbens shell, central amygdaloid nucleus and thalamic central medial nucleus. Neuroscience. 2003;121(4):991–998. doi: 10.1016/s0306-4522(03)00397-x. [DOI] [PubMed] [Google Scholar]
- 36.Tarsy D, Baldessarini RJ, Tarazi FI. Effects of newer antipsychotics on extrapyramidal function. CNS Drugs. 2002;16(1):23–45. doi: 10.2165/00023210-200216010-00003. [DOI] [PubMed] [Google Scholar]
- 37.Waddington JL, Scully PJ, O’Callaghan E. The new antipsychotics, and their potential for early intervention in schizophrenia. Schizophrenia Research. 1997;28(2-3):207–222. doi: 10.1016/s0920-9964(97)00115-1. [DOI] [PubMed] [Google Scholar]
- 38.Goff DC, Coyle JT. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. American Journal of Psychiatry. 2001;158(9):1367–1377. doi: 10.1176/appi.ajp.158.9.1367. [DOI] [PubMed] [Google Scholar]
- 39.Meyer-Lindenberg A, Miletich RS, Kohn PD, et al. Reduced prefrontal activity predicts exaggerated striatal dopaminergic function in schizophrenia. Nature Neuroscience. 2002;5(3):267–271. doi: 10.1038/nn804. [DOI] [PubMed] [Google Scholar]
- 40.Benes FM, Vincent SL, Marie A, Khan Y. Up-regulation of GABA(A) receptor binding on neurons of the prefrontal cortex in schizophrenic subjects. Neuroscience. 1996;75(4):1021–1031. doi: 10.1016/0306-4522(96)00328-4. [DOI] [PubMed] [Google Scholar]
- 41.Clarke HF, Dalley JW, Crofts HS, Robbins TW, Roberts AC. Cognitive inflexibility after prefrontal serotonin depletion. Science. 2004;304(5672):878–880. doi: 10.1126/science.1094987. [DOI] [PubMed] [Google Scholar]
- 42.Coyle JT. The GABA-glutamate connection in schizophrenia: which is the proximate cause? Biochemical Pharmacology. 2004;68(8):1507–1514. doi: 10.1016/j.bcp.2004.07.034. [DOI] [PubMed] [Google Scholar]
- 43.Dalley JW, Theobald DE, Pereira EAC, Li PMMC, Robbins TW. Specific abnormalities in serotonin release in the prefrontal cortex of isolation-reared rats measured during behavioural performance of a task assessing visuospatial attention and impulsivity. Psychopharmacology. 2002;164(3):329–340. doi: 10.1007/s00213-002-1215-y. [DOI] [PubMed] [Google Scholar]
- 44.Friedman JI, Temporini H, Davis KL. Pharmacologic strategies for augmenting cognitive performance in schizophrenia. Biological Psychiatry. 1999;45(1):1–16. doi: 10.1016/s0006-3223(98)00287-x. [DOI] [PubMed] [Google Scholar]
- 45.Ichikawa J, Dai J, O’Laughlin IA, Fowler WL, Meltzer HY. Atypical, but not typical, antipsychotic drugs increase cortical acetylcholine release without an effect in the nucleus accumbens or striatum. Neuropsychopharmacology. 2002;26(3):325–339. doi: 10.1016/S0893-133X(01)00312-8. [DOI] [PubMed] [Google Scholar]
- 46.Sarter M, Bruno JP. Abnormal regulation of corticopetal cholinergic neurons and impaired information processing in neuropsychiatric disorders. Trends in Neurosciences. 1999;22(2):67–74. doi: 10.1016/s0166-2236(98)01289-2. [DOI] [PubMed] [Google Scholar]
- 47.Zavitsanou K, Wards PB, Huang X-F. Selective alterations in inotropic glutamate receptors in the anterior cingulated cortex in schizophrenia. Neuropsychopharmacology. 2002;27:826–833. doi: 10.1016/S0893-133X(02)00347-0. [DOI] [PubMed] [Google Scholar]
- 48.Hertel P, Nomikos GG, Schilström B, Arborelius L, Svensson TH. Risperidone dose-dependently increases extracellular concentrations of serotonin in the rat frontal cortex: role of α2-adrenoceptor antagonism. Neuropsychopharmacology. 1997;17(1):44–55. doi: 10.1016/S0893-133X(97)00002-X. [DOI] [PubMed] [Google Scholar]
- 49.Ichikawa J, Kuroki T, Dai J, Meltzer HY. Effect of antipsychotic drugs on extracellular serotonin levels in rat medial prefrontal cortex and nucleus accumbens. European Journal of Pharmacology. 1998;351(2):163–171. doi: 10.1016/s0014-2999(98)00308-2. [DOI] [PubMed] [Google Scholar]
- 50.Jardemark KE, Ninan I, Liang X, Wang RY. Protein kinase C is involved in clozapine’s facilitation of N-methyl-D-aspartate- and electrically evoked responses in pyramidal cells of the medial prefrontal cortex. Neuroscience. 2003;118(2):501–512. doi: 10.1016/s0306-4522(02)00976-4. [DOI] [PubMed] [Google Scholar]
- 51.Lacroix LP, Hows ME, Shah AJ, Hagan JJ, Heidbreder CA. Selective antagonism at dopamine D3 receptors enhances monoaminergic and cholinergic neurotransmission in the rat anterior cingulate cortex. Neuropsychopharmacology. 2003;28(5):839–849. doi: 10.1038/sj.npp.1300114. [DOI] [PubMed] [Google Scholar]
- 52.Potvin S, Stip E, Roy J-Y. Clozapine, quetiapine and olanzapine among addicted schizophrenic patients: towards testable hypotheses. International Clinical Psychopharmacology. 2003;18(3):121–132. doi: 10.1097/01.yic.0000063501.97247.38. [DOI] [PubMed] [Google Scholar]
- 53.Zhang W, Perry KW, Wong DT, et al. Synergistic effects of olanzapine and other antipsychotic agents in combination with fluoxetine on norepinephrine and dopamine release in rat prefrontal cortex. Neuropsychopharmacology. 2000;23(3):250–262. doi: 10.1016/S0893-133X(00)00119-6. [DOI] [PubMed] [Google Scholar]
- 54.Costa E, Davis JM, Dong E, et al. A GABAergic cortical deficit dominates schizophrenia pathophysiology. Critical Reviews in Neurobiology. 2004;16(1-2):1–23. doi: 10.1615/critrevneurobiol.v16.i12.10. [DOI] [PubMed] [Google Scholar]
- 55.Bourdelais AJ, Deutch AY. The effects of haloperidol and clozapine on extracellular GABA levels in the prefrontal cortex of the rat: an in vivo microdialysis study. Cerebral Cortex. 1994;4(1):69–77. doi: 10.1093/cercor/4.1.69. [DOI] [PubMed] [Google Scholar]
- 56.Wang X, Zhong P, Yan Z. Dopamine D4 receptors modulate GABAergic signaling in pyramidal neurons of prefrontal cortex. Journal of Neuroscience. 2002;22(21):9185–9193. doi: 10.1523/JNEUROSCI.22-21-09185.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yan Z. Regulation of GABAergic inhibition by serotonin signaling in prefrontal cortex: molecular mechanisms and functional implications. Molecular Neurobiology. 2002;26(2-3):203–216. doi: 10.1385/MN:26:2-3:203. [DOI] [PubMed] [Google Scholar]
- 58.Ashton CH. Pharmacology and effects of cannabis: a brief review. British Journal of Psychiatry. 2001;178:101–106. doi: 10.1192/bjp.178.2.101. [DOI] [PubMed] [Google Scholar]
- 59.Grant I, Gonzalez R, Carey CL, Natarajan L, Wolfson T. Non-acute (residual) neurocognitive effects of cannabis use: a meta-analytic study. Journal of the International Neuropsychological Society. 2003;9(5):679–689. doi: 10.1017/S1355617703950016. [DOI] [PubMed] [Google Scholar]
- 60.Kathmann M, Bauer U, Schlicker E, Göthert M. Cannabinoid CB1 receptor-mediated inhibition of NMDA- and kainate- stimulated noradrenaline and dopamine release in the brain. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1999;359(6):466–470. doi: 10.1007/pl00005377. [DOI] [PubMed] [Google Scholar]
- 61.Breivogel CS, Childers SR. The functional neuroanatomy of brain cannabinoid receptors. Neurobiology of Disease. 1998;5(6):417–431. doi: 10.1006/nbdi.1998.0229. [DOI] [PubMed] [Google Scholar]
- 62.Gifford AN, Makriyannis A, Volkow ND, Gatley SJ. In vivo imaging of the brain cannabinoid receptor. Chemistry and Physics of Lipids. 2002;121(1-2):65–72. doi: 10.1016/s0009-3084(02)00148-2. [DOI] [PubMed] [Google Scholar]
- 63.Ilan AB, Smith ME, Gevins A. Effects of marijuana on neurophysiological signals of working and episodic memory. Psychopharmacology. 2004;176(2):214–222. doi: 10.1007/s00213-004-1868-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Shah YB, Prior MJW, Dixon AL, Morris PG, Marsden CA. Detection of cannabinoid agonist evoked increase in BOLD contrast in rats using functional magnetic resonance imaging. Neuropharmacology. 2004;46(3):379–387. doi: 10.1016/j.neuropharm.2003.09.023. [DOI] [PubMed] [Google Scholar]
- 65.Vik PW, Cellucci T, Jarchow A, Hedt J. Cognitive impairment in substance abuse. Psychiatric Clinics of North America. 2004;27(1):97–109. doi: 10.1016/S0193-953X(03)00110-2. [DOI] [PubMed] [Google Scholar]
- 66.Diana M, Melis M, Muntoni AL, Gessa GL. Mesolimbic dopaminergic decline after cannabinoid withdrawal. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(17):10269–10273. doi: 10.1073/pnas.95.17.10269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.French ED, Dillon K, Wu X. Cannabinoids excite dopamine neurons in the ventral tegmentum and substantia nigra. NeuroReport. 1997;8(3):649–652. doi: 10.1097/00001756-199702100-00014. [DOI] [PubMed] [Google Scholar]
- 68.Gessa G, Melis M, Muntoni A, Diana M. Cannabinoids activate mesolimbic dopamine neurons by an action on cannabinoid CB1 receptors. European Journal of Pharmacology. 1998;341(1):39–44. doi: 10.1016/s0014-2999(97)01442-8. [DOI] [PubMed] [Google Scholar]
- 69.Hiroi N. Dependence, tolerance, and alteration in gene expression. In: Nahas GG, Sutin KM, Harvey D, editors. Marihuana and Medicine. Totowa, NJ, USA: Humana Press; 1999. pp. 207–211. [Google Scholar]
- 70.Patel V, Borysenko M, Kumar MSA. Effect of Δ9-THC on brain and plasma catecholamine levels as measured by HPLC. Brain Research Bulletin. 1985;14(1):85–90. doi: 10.1016/0361-9230(85)90179-0. [DOI] [PubMed] [Google Scholar]
- 71.Stahl SM. Getting stoned without inhaling: anandamide is the brain’s natural marijuana. Journal of Clinical Psychiatry. 1998;59(11):566–567. doi: 10.4088/jcp.v59n1101. [DOI] [PubMed] [Google Scholar]
- 72.Valjent E, Pagès C, Rogard M, Besson M-J, Maldonado R, Caboche J. δ9-tetrahydrocannabinol-induced MAPK/ERK and Elk-1 activation in vivo depends on dopaminergic transmission. European Journal of Neuroscience. 2001;14(2):342–352. doi: 10.1046/j.0953-816x.2001.01652.x. [DOI] [PubMed] [Google Scholar]
- 73.Voruganti LNP, Slomka P, Zabel P, Mattar A, Awad AG. Cannabis induced dopamine release: an in-vivo SPECT study. Psychiatry Research: Neuroimaging. 2001;107(3):173–177. doi: 10.1016/s0925-4927(01)00104-4. [DOI] [PubMed] [Google Scholar]
- 74.Wu X, French ED. Effects of chronic Δ9-tetrahydrocannabinol on rat midbrain dopamine neurons: an electrophysiological assessment. Neuropharmacology. 2000;39(3):391–398. doi: 10.1016/s0028-3908(99)00140-9. [DOI] [PubMed] [Google Scholar]
- 75.Degenhardt L, Hall W, Lynskey M. Testing hypotheses about the relationship between cannabis use and psychosis. Drug and Alcohol Dependence. 2003;71(1):37–48. doi: 10.1016/s0376-8716(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 76.Hall W. Reducing the harms caused by cannabis use: the policy debate in Australia. Drug and Alcohol Dependence. 2001;62(3):163–174. doi: 10.1016/s0376-8716(00)00171-x. [DOI] [PubMed] [Google Scholar]
- 77.Akinshola BE, Taylor RE, Ogunseitan AB, Onaivi ES. Anandamide inhibition of recombinant AMPA receptor subunits in Xenopus oocytes is increased by forskolin and 8-bromo-cyclic AMP. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1999;360(3):242–248. doi: 10.1007/s002109900078. [DOI] [PubMed] [Google Scholar]
- 78.Nakazi M, Bauer U, Nickel T, Kathmann M, Schlicker E. Inhibition of serotonin release in the mouse brain via presynaptic cannabinoid CB1 receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2000;361(1):19–24. doi: 10.1007/s002109900147. [DOI] [PubMed] [Google Scholar]
- 79.Tzavara ET, Davis RJ, Perry KW, et al. The CB1 receptor antagonist SR141716A selectively increases monoaminergic neurotransmission in the medial prefrontal cortex: implications for therapeutic actions. British Journal of Pharmacology. 2003;138(4):544–553. doi: 10.1038/sj.bjp.0705100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Carta G, Nava F, Gessa GL. Inhibition of hippocampal acetylcholine release after acute and repeated delta-9-tetrahydrocannabinoid administration in rats. Brain Research. 1998;809:1–4. doi: 10.1016/s0006-8993(98)00738-0. [DOI] [PubMed] [Google Scholar]
- 81.Domino EF. Cannabinoids and the cholinergic system. Journal of Clinical Pharmacology. 1981;21(supplement 8-9):2495–2555. doi: 10.1002/j.1552-4604.1981.tb02602.x. [DOI] [PubMed] [Google Scholar]
- 82.Gessa GL, Mascia MS, Casu MA, Carta G. Inhibition of hippocampal acetylcholine release by cannabinoids: reversal by SR 141716A. European Journal of Pharmacology. 1997;327(1):R1–R2. doi: 10.1016/s0014-2999(97)89683-5. [DOI] [PubMed] [Google Scholar]
- 83.Gessa GL, Casu MA, Carta G, Mascia MS. Cannabinoids decrease acetylcholine release in the medial-prefrontal cortex and hippocampus, reversal by SR 141716A. European Journal of Pharmacology. 1998;355(2-3):119–124. doi: 10.1016/s0014-2999(98)00486-5. [DOI] [PubMed] [Google Scholar]
- 84.Schlicker E, Timm J, Zentner J, Göthert M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn-Schmiedeberg’s Archives of Pharmacology. 1997;356(5):583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- 85.Göbel I, Trendelenburg AU, Cox SL, Meyer A, Starke K. Electrically evoked release of [3H]noradrenaline from mouse cultured sympathetic neurons: release-modulating heteroreceptors. Journal of Neurochemistry. 2000;75(5):2087–2094. doi: 10.1046/j.1471-4159.2000.0752087.x. [DOI] [PubMed] [Google Scholar]
- 86.Tzavara ET, Perry KW, Rodriguez DE, Bymaster FP, Nomikos GG. The cannabinoid CB1 receptor antagonist SR141716A increases norepinephrine outflow in the rat anterior hypothalamus. European Journal of Pharmacology. 2001;426(3):R3–R4. doi: 10.1016/s0014-2999(01)01228-6. [DOI] [PubMed] [Google Scholar]
- 87.Vizi ES, Katona I, Freund TF. Evidence for presynaptic cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the guinea pig lung. European Journal of Pharmacology. 2001;431(2):237–244. doi: 10.1016/s0014-2999(01)01413-3. [DOI] [PubMed] [Google Scholar]
- 88.Ferraro L, Tomasini MC, Cassano T, et al. Cannabinoid receptor agonist WIN 55,212-2 inhibits rat cortical dialysate γ-aminobutyric acid levels. Journal of Neuroscience Research. 2001;66(2):298–302. doi: 10.1002/jnr.1224. [DOI] [PubMed] [Google Scholar]
- 89.Auclair N, Otani S, Soubrie P, Crepel F. Cannabinoids modulate synaptic strength and plasticity at glutamatergic synapses of rat prefrontal cortex pyramidal neurons. Journal of Neurophysiology. 2000;83(6):3287–3293. doi: 10.1152/jn.2000.83.6.3287. [DOI] [PubMed] [Google Scholar]
- 90.Hoffman AF, Lupica CR. Mechanisms of cannabinoid inhibition of GABA(A) synaptic transmission in the hippocampus. Journal of Neuroscience. 2000;20(7):2470–2479. doi: 10.1523/JNEUROSCI.20-07-02470.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. Journal of Neurophysiology. 1999;82(3):1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 92.Pistis M, Ferraro L, Pira L, et al. Δ9-tetrahydrocannabinol decreases extracellular GABA and increases extracellular glutamate and dopamine levels in the rat prefrontal cortex: an in vivo microdialysis study. Brain Research. 2002;948(1-2):155–158. doi: 10.1016/s0006-8993(02)03055-x. [DOI] [PubMed] [Google Scholar]
- 93.Trettel J, Levine ES. Cannabinoids depress inhibitory synaptic inputs received by layer 2/3 pyramidal neurons of the neocortex. Journal of Neurophysiology. 2002;88(1):534–539. doi: 10.1152/jn.2002.88.1.534. [DOI] [PubMed] [Google Scholar]
- 94.Hájos N, Katona I, Naiem SS, et al. Cannabinoids inhibit hippocampal GABAergic transmission and network oscillations. European Journal of Neuroscience. 2000;12(9):3239–3249. doi: 10.1046/j.1460-9568.2000.00217.x. [DOI] [PubMed] [Google Scholar]
- 95.Hampson RE, Deadwyler SA. Cannabinoids, hippocampal function and memory. Life Sciences. 1999;65(6-7):715–723. doi: 10.1016/s0024-3205(99)00294-5. [DOI] [PubMed] [Google Scholar]
- 96.Chan PK, Chan SC, Yung WH. Presynaptic inhibition of GABAergic inputs into rat substantia nigra pars reticulate neurons by cannabinoid agonists. Neuroreport. 1998;9:671–675. doi: 10.1097/00001756-199803090-00020. [DOI] [PubMed] [Google Scholar]
- 97.Hoffman AF, Lupica CR. Direct actions of cannabinoids on synaptic transmission in the nucleus accumbens: a comparison with opioids. Journal of Neurophysiology. 2001;85(1):72–83. doi: 10.1152/jn.2001.85.1.72. [DOI] [PubMed] [Google Scholar]
- 98.Manzoni OJ, Bockaert J. Cannabinoids inhibit GABAergic synaptic transmission in mice nucleus accumbens. European Journal of Pharmacology. 2001;412(2):R3–R5. doi: 10.1016/s0014-2999(01)00723-3. [DOI] [PubMed] [Google Scholar]
- 99.Szabo B, Siemes S, Wallmichrath I. Inhibition of GABAergic neurotransmission in the ventral tegmental area by cannabinoids. European Journal of Neuroscience. 2002;15(12):2057–2061. doi: 10.1046/j.1460-9568.2002.02041.x. [DOI] [PubMed] [Google Scholar]
- 100.Wallmichrath I, Szabo B. Cannabinoids inhibit striatonigral GABAergic neurotransmission in the mouse. Neuroscience. 2002;113(3):671–682. doi: 10.1016/s0306-4522(02)00109-4. [DOI] [PubMed] [Google Scholar]
- 101.Crook JM, Tomaskovic-Crook E, Copolov DL, Dean B. Low muscarinic receptor binding in prefrontal cortex from subjects with schizophrenia: a study of brodmann’s areas 8, 9, 10, and 46 and the effects of neuroleptic drug treatment. American Journal of Psychiatry. 2001;158(6):918–925. doi: 10.1176/appi.ajp.158.6.918. [DOI] [PubMed] [Google Scholar]
- 102.Jentsch JD, Andrusiak E, Tran A, Bowers MB, Jr., Roth RH. δ9-tetrahydrocannabinol increases prefrontal cortical catecholaminergic utilization and impairs spatial working memory in the rat: blockade of dopaminergic effects with HA966. Neuropsychopharmacology. 1997;16(6):426–432. doi: 10.1016/S0893-133X(97)00018-3. [DOI] [PubMed] [Google Scholar]
- 103.Acquas E, Pisanu A, Marrocu P, Goldberg SR, Di Chiara G. Δ9-tetrahydrocannabinol enhances cortical and hippocampal acetylcholine release in vivo: a microdialysis study. European Journal of Pharmacology. 2001;419(2-3):155–161. doi: 10.1016/s0014-2999(01)00967-0. [DOI] [PubMed] [Google Scholar]
- 104.Verrico CD, Jentsch JD, Dazzi L, Roth RH. Systemic, but not local, administration of cannabinoid CB1 receptor agonists modulate prefrontal cortical acetylcholine efflux in the rat. Synapse. 2003;48(4):178–183. doi: 10.1002/syn.10202. [DOI] [PubMed] [Google Scholar]
- 105.Ferraro L, Tomasini MC, Gessa GL, Bebe BW, Tanganelli S, Antonelli T. The cannabinoid receptor agonist WIN 55,212-2 regulates glutamate transmission in rat cerebral cortex: an in vivo and in vitro study. Cerebral Cortex. 2001;11(8):728–733. doi: 10.1093/cercor/11.8.728. [DOI] [PubMed] [Google Scholar]
- 106.Steffens M, Feuerstein TJ. Receptor-independent depression of DA and 5-HT uptake by cannabinoids in rat neocortex—involvement of Na+/K+-ATPase. Neurochemistry International. 2004;44(7):529–538. doi: 10.1016/j.neuint.2003.08.009. [DOI] [PubMed] [Google Scholar]