

Abstract
The mucosal surfaces of the gastrointestinal tract are continually exposed to an enormous antigenic load of microbial and dietary origin, yet homeostasis is maintained. Pattern recognition molecules (PRMs) have a key role in maintaining the integrity of the epithelial barrier and in promoting maturation of the mucosal immune system. Commensal bacteria modulate the expression of a broad range of genes involved in maintaining epithelial integrity, inflammatory responses, and production of antimicrobial peptides. Mice deficient in PRMs can develop intestinal inflammation, which is dependent on the microbiota, and in humans, PRM polymorphisms are associated with exacerbated inflammatory bowel disease. Innate immune responses and epithelial barrier function are regulated by PRM-induced signaling at multiple levels, from the selective expression of receptors on mucosal cells or compartments to the expression of negative regulators. Here, we describe recent advances in our understanding of innate signaling pathways, particularly by Toll-like receptors and nucleotide-binding domain and leucine-rich repeat containing receptors at mucosal surfaces.
INTRODUCTION
Activation of the innate immune system is triggered when pathogen-associated molecular patterns or endogenous damage-associated molecular patterns engage pattern recognition molecules (PRMs) on cells including epithelial cells, macrophages, and dendritic cells (DCs). These molecules can be grouped into Toll-like receptors (TLRs), nucleotide-binding domain and leucine-rich repeat containing receptors (NLRs), and retinoic acid-inducible gene-I (RIG-)-like receptors (RLRs).1 Engagement of PRMs on mucosal cells and consequent activation of signaling cascades including NF-κB, interferon (IFN) response factors (IRFs), activator protein 1 (AP1), and mitogen-activated protein kinases can promote the production of proinflammatory cytokines and antimicrobial peptides, as well as the maintenance of epithelial barrier function and epithelial cell proliferation. TLR signaling, particularly through TLR2, has an important role in maintaining intestinal epithelial homeostasis and protection from epithelial injury.2 Commensal bacteria including Bacteroides thetaiotaomicron induce the modulation of many genes involved in intestinal barrier function and nutrient absorption.3 The uptake of soluble and particulate antigens across the intestinal epithelium by both transcellular and paracellular routes is influenced by microbial and inflammatory factors and is regulated by PRMs. The microbiota are localized to the intestinal lumen and the mucosal immune compartment, which comprises the lamina propria, Peyer’s patches, and mesenteric lymph nodes.4 The ability to contain the microbiota in this region is dependent on PRMs, as a deficiency in TLR signaling results in the increased uptake of bacteria into the spleens of mice.5,6
The expression and localization of PRMs on intestinal epithelial cells (IECs) and DCs in the intestinal mucosa differ significantly from those of other tissues. In particular, TLR expression on IECs is generally low and many of the receptors are expressed basolaterally, preventing interaction with pathogen-associated molecular patterns in the lumen. Nevertheless, epithelial cells are responsive to certain TLR ligands and recognition of commensal bacteria by TLRs on IEC under steady-state conditions induces tissue protective factors including interleukin (IL)-6 and the chemokine KC-1, suggesting that low-level recognition of commensals by TLRs enhances protection from intestinal epithelial injury.2 Microbiota-induced TLR signaling triggers innate immune responses that are required to prevent exaggerated adaptive immunity to these organisms.6 In this context, deficiency in TLR signaling resulted in high titer serum antibody responses against the intestinal microbiota that were essential to maintain host-commensal mutualism in the deficient animals. Furthermore, a deficiency in TLR or NLR signaling can result in intestinal inflammatory responses, and in murine models these responses depend on the microbiota.7 Adaptive immune responses in the intestine are characterized by high numbers of lgA producing plasma cells, regulatory T cells and IL-17-producing T cells whose development is closely linked to factors produced by PRMs expressing IECs, DCs, and macrophages. Thus, commensal ligands for PRMs are involved in the positive and negative regulation of both innate and adaptive immunity in the intestine and elucidating the molecular basis of this regulation will be of great value in the design of both vaccines and therapeutic anti-inflammatory strategies.
TOLL-LIKE RECEPTORS
The TLRs are a family of transmembrane glycoproteins, which contains 10 members in humans and 13 in mice. They are located on the cell surface or on endosomes and recognize the presence of microbes through pathogen-associated molecular patterns and damage-associated molecular patterns.1 The TLRs contain 16–28 leucine-rich repeats (LRRs) that mediate ligand binding.8 The LRRs are 20–30 amino acids long, contain a conserved “LxxLxLxxN” motif,9,10 and share a common horseshoe-like shape, with the “‘LxxLxLxxN” motifs located on the inner concave surfaces.11 On ligand binding, the C termini of the extracellular domains come together, bringing the intracellular Toll/IL-1R (TIR) domains into close proximity,12 facilitating the docking of adapter molecules. For example, the TLR1/2 heterodimer and the TLR3 homodimers are induced by binding agonistic ligands, forming a common “m”-shaped architecture. There are four activating adapters: Myeloid differentiating factor-88 (MyD88), MyD88 adapter-like (Mal), TIR domain-containing adapter-inducing IFN-β (TRIF), and TRIF-related adapter molecule (TRAM), whereas sterile-α and Armadillo repeat-containing molecule (SARM) negatively regulates TRIF signaling.13 Binding of the activating adapters through their TIR domains results in the recruitment of the IL-1R-associated kinases (IRAKs) and downstream activation of transcription factors including NF-κB and IFN regulatory factor 3 (IRF3), which induces proinflammatory cytokines and type I IFNs.13 MyD88 is required for signaling by all TLRs with the exception of TLR3 which uses TRIF. Lipopolysaccharide (LPS)-induced IRF3 production is also MyD88 independent. TLR4 recruits TRAM–TRIF through their TIR domains and the TLR4-TRAM complex moves into early endosomes where it signals through TRIF to activate IRF3 and IRF7 through TRAF3, TBK1, and IKKε.14 Mal was originally found to be required for both TLR2 and TLR4 signaling,15 but it is now thought that Mal may be dispensable for TLR2 signaling at high ligand concentrations in macrophages and DCs.16 Furthermore, Mal was dispensable for IL-6 production at high levels of infection with the enteric pathogen, Salmonella typhimurium.16
Primary human IECs express constitutive TLR3 and TLR5 and low levels of TLR2 and TLR4.17 The location of TLRs is crucial to their function as TLRs 1, 2, 4, 5, and 6 expressed on the cell surface recognize extracellular microbes, whereas TLR3, 7, 8, and 9 are thought to detect the presence of viral particles and are located on early endosomal membranes. TLR3 recognizes double-stranded RNA, TLR7 and TLR8 recognize single-stranded RNA, whereas TLR9 recognizes unmethylated CpG DNA. Both TLR7 and TLR9 are highly expressed on plasmacytoid DCs (pDCs). On viral infection, pDCs secrete high concentrations of type I IFN18 and a population of CCR9+ pDCs are present in the murine small intestine.19 It will be important to resolve the role of these cells in the recognition of viral nucleic acids and induction of antiviral immunity in the intestine.
In contrast to control tissue, the expression of TLR4 is increased on IECs in ulcerative colitis (UC) and Crohn’s disease (CD), and expression of both TLR2 and TLR4 was selectively increased on intestinal macrophages.17,20 Intestinal monocytes from inflamed tissue also responded to LPS stimulation with increased expression of IL-1β and a higher percentage of intestinal DCs expressed TLR4 (and TLR2) in inflammatory bowel disease compared with control patients.20,21 Thus, the expression of specific PRMs and the consequences of PRM engagement by cognate ligands differ greatly between normal and pathological conditions. This may depend in part on whether intestinal injury occurs, as although resident mucosal cells may be hyporesponsive to specific ligands such as LPS, naive cells may be recruited from other tissues that respond strongly to these ligands. In addition, injury may allow the paracellular uptake of TLR ligands that engage basolateral TLRs on IEC.
Although the injection of TLR agonists is a potent means of inducing adaptive immune responses, commensal ligands do not generally elicit strong adaptive immunity. Challenge of MyD88/TRIF-deficient mice with the commensal bacterium, Escherichia coli K-12, resulted in the recovery of more live bacteria from the spleens than in the case of control mice.6 Thus, the ability to localize the microbiota in the lumen and mucosal immune compartment is dependent on intact TLR signaling. This effect was not a result of compromised intestinal barrier function or intestinal pathology and deficiency in TLR signaling resulted in the spontaneous induction of serum antibody responses against the microbiota, which restored host-commensal mutualism. The induction of a protective humoral immune response against bacteria in mice deficient in TLR signaling indicates an important role for other PRMs including NLRs or RLRs or both in triggering adaptive immunity by intestinal bacteria.
TLR SIGNALING MAINTAINS THE INTEGRITY AND FUNCTION OF THE INTESTINAL EPITHELIUM
Disruption of the epithelial layer in mice promotes inflammation in the intestine, proving that maintaining epithelial barrier integrity is vital. The intestinal microbiota have a key role in promoting mucosal homeostasis and the underlying mechanisms are currently being addressed.3
TLR2
Intestinal epithelial cells express TLR2 and commensal ligand-induced activation of TLR2 is thought to have an important role in maintaining the integrity of the intestinal epithelial barrier.22 TLR2 interacts with ligands including bacterial lipopeptides, lipoteichoic acid and yeast zymosan, and forms heterodimers with TLR1 or TLR 6.1 Repeated stimulation of TLR2 causes IECs to become unresponsive to lipopeptide ligands.23 The uptake of molecules by paracellular and transcellular pathways is regulated by tight and gap junctions between IECs. Tight junctions are regulated by protein kinase C (PKC) and the tight junction protein, zonula occludens-1, which may be a specific PKC target. Ligand-induced TLR2 activation led to the specific activation of PKC-α and PKC-δ,22 which correlated with enhanced transepithelial resistance to invading bacteria and increased apical tightening and sealing of the tight junctions. The TLR2-induced phosphorylation of PKC was blocked by transfection with a TLR2 deletion mutant. TLR2 signaling has recently been shown to promote gap junctional intercellular communication,24 a process that coordinates the cell–cell passage of ions and small metabolites ( < 1 kDa) on acute IEC injury. Treatment of IECs with the TLR2 agonist Pam3CSK4 increased gap junctional intercellular communication through the connexin, C×43, during IEC injury24 and knockdown of C×43 using siRNA inhibited TLR2-induced IEC barrier restitution and delayed wound closure in the acute (dextran sodium sulfate, DSS) colitis model.24 The protective effects of TLR2 stimulation on tight junction-associated barrier assembly have been shown to be MyD88 dependent and to require PI3K/Akt-mediated cell survival.25 Prophylaxis with Pam3CSK4 also prevented spontaneous disease onset in a chronic colitis model. Among Caucasians, the TLR2-R753Q polymorphism correlates with the incidence of sepsis26 and is associated with a more severe disease phenotype in UC patients,27 which was recently associated with a defect in gap junctional intercellular communication.24 Furthermore, in a neonatal rat model of necrotizing enterocolitis-induced mucosal injury, there was increased expression of both TLR2 and TLR4, suggesting that TLR2 may promote intestinal inflammation under circumstances where the epithelial barrier has been compromised.28 Combined, these findings highlight the importance of TLR2 in intestinal epithelial barrier function. Further characterization of TLR2 signaling effects in this context are required, however, as pharmacological targeting of TLR2 in IECs to improve barrier integrity may be a potential therapeutic approach.
TLR4
Ligand binding induces aggregation of a TLR4–MD2 complex, which initiates signaling.12 However, in addition to MD2, TLR signaling also requires two additional accessory proteins, LPS-binding protein and CD14. LPS-binding protein extracts LPS from the outer membrane of Gram-negative bacteria and transfers it to CD14. As CD14 has no intracellular signaling domain, LPS must be transferred to TLR4–MD2 to initiate signaling. Under normal conditions, expression of the TLR4–MD2–CD14 receptor complex is low and the intestinal tissue does not respond strongly to LPS.29 However, cells from the murine intestinal crypt epithelium express TLR4 internally and may be more responsive to LPS than epithelial cells from other sites.30 A recent study found that intestinal DCs from rats were responsive to the TLR agonists Pam3CSK4 (TLR2), poly I: C (TLR3), flagellin (TLR5), R848 (TLR7/8), and CpG (TLR9), but not to LPS.31 Poor responsiveness to LPS stimulation has also been associated with low TLR4 expression in rat intestinal DCs31 and murine lamina propria CD11c+ cells.32 However, mucosal DCs can respond to other TLR stimuli and intestinal DCs are not generally hyporesponsive to TLR stimulation, suggesting that other mechanisms must act to prevent their activation. As there is great variation in TLR expression in IEC and DC populations in the intestine, it is difficult to fully ascertain the responsiveness to TLR stimuli from in vitro studies. Delivery of TLR ligands by oral gavage and challenge studies with enteric pathogens or commensals may provide a better indication of the consequences of intestinal exposure to TLR ligands on intestinal innate and adaptive immunity.
Stimulation of IECs with TLR4 induced proliferation, NF-κB activation and release of proinflammatory cytokines,33,34 and the inflammatory cytokines IFN-γ and TNF-α enhanced IEC expression of TLR4 and MD2 on IECs.35 Although short-term exposure of IECs to LPS induced proinflammatory signaling cascades, persistent exposure led to reduced cell surface-expressed TLR4 and increased the inhibitory protein Toll-interacting protein (TOLLIP) leading to reduced IRAK activity.33 The migration of enterocytes into areas of damaged intestine is important for wound healing and this was inhibited by LPS-induced activation of Rho-GTPase and increased focal adhesions.36 Treatment of mice with a TLR4 antagonist inhibited the development of disease in the acute DSS colitis model and a spontaneous (MDR1α–/–) chronic colitis model.37 It was proposed that this was a result of blocking the interaction between enteric bacteria and the innate immune system. Necrotizing enterocolitis is associated with increased expression of TLR4 in the intestinal mucosa and exposure to LPS and hypoxia sensitized the intestinal epithelium to LPS through TLR4 upregulation. TLR4-mutant C3H/HeJ mice, which are hyporesponsive to LPS, were protected from the development of necrotizing enterocolitis and exhibited enhanced enterocyte restitution and proliferation after mucosal injury.38 However, in a further study, it was shown that although a TLR4-blocking antibody could limit acute inflammation in the DSS model of colitis, subsequent tissue repair was also inhibited.39 This indicates an important role for TLR4 in the resolution of inflammation and restoration of tissue integrity, highlighting the complexity of TLR4-induced effects in the mucosa.
In addition to the uptake of molecules between cells by paracellular routes and transcellular uptake across IECs, microfold (M) cells have an important role in the uptake of both soluble and particulate antigens and their transport to the underlying lymphoid tissue.40 M cells are found in the follicle-associated epithelium overlying intestinal Peyer’s patches and isolated lymphoid follicles. MyD88 and TLR4 were found to have a role in the development of Peyer’s patches in the early postnatal period in mice.41 Challenge with Streptococcus pneumoniae upregulated the M-cell-mediated uptake of microparticles across the follicle-associated epithelium of the rabbit peyer’s patches.42 This effect was due to the increased intraluminal concentrations of macrophage migration inhibitory factor (MIF), which induced the appearance of cells with the functional attributes of antigen sampling M cells.40 Furthermore, increased numbers of M cells were documented in the inflamed ileal mucosa in humans.43 Thus, M-cell-mediated uptake of antigens and particulates is regulated by exposure to microbes and it will be important to determine the PRMs involved in these processes. In addition to the vital role of peyer’s patches in the initiation of mucosal immune responses, isolated lymphoid follicles are also implicated in the induction of adaptive immunity in the intestine, including the T-cell-independent development of IgA+ cells that principally occurs at these sites.44 The development of isolated lymphoid follicles is compromised in germ-free mice, suggesting an important role for the commensal flora in promoting their development. Furthermore, the microbiota is altered in the absence of isolated lymphoid follicles. This promoting effect is mediated through peptidoglycan-induced nucleotide-binding oligomerization domain 1 (NOD1) signaling.44,45 The identification of PRMs that regulate M-cell transport of particulates can have potential in the development of improved oral vaccines by enhancing vaccine uptake and induction of innate immune responses.
ROLES FOR OTHER TLRs IN REGULATING INTESTINAL INFLAMMATORY RESPONSES
Many bacterial species that colonize the intestine express flagellin,46 including E. coli, which is normally a commensal,47 but when the gastrointestinal barrier is compromised, it can become pathogenic. Ligation of TLR5 on IECs by flagellin leads to increased expression of antiapoptotic genes,48 which may correlate with the observed protective effects of TLR5 signaling in epithelial homeostasis. This suggests that under normal conditions, ligation of TLR5 by flagellin in IECs does not exert inflammatory effects and may be cytoprotective. In contrast, flagellated bacteria including E. coli can induce proinflammatory responses by interacting with basolateral TLR5 in epithelial cell lines49 and the intact human colonic mucosa.50 Exposure of the injured mouse colon to flagellin resulted in aggravated colonic inflammation, whereas the normal colon was not responsive to flagellin.50 TLR5-deficient mice develop spontaneous colitis46 and this was abrogated in mice that were also deficient in TLR4, indicating a key role for TLR4 ligands in promoting the inflammatory response. The inflammatory pathology associated with Salmonella infection, which occurs in peyer’s patches, is dependent on TLR551 and the migration of DCs into the gut lumen in response to S. typhimurium was dependent on flagellin and MyD88.52 Thus, the consequences of TLR5 ligation with flagellin depend on whether the interaction is with IECs under noninflammatory conditions, where the outcome may be cytoprotective, or with nonepithelial cells, where the outcome may be amplified inflammatory responses.
An additional role for TLR5 has been revealed in promoting IgA + B-cell differentiation. A population of TLR5+ CD11c+ CD11b+ DCs in the small intestine promotes IgA+ cell differentiation by a retinoic acid-dependent process.53 Flagellin-induced expression of retinal dehydrogenase 2 in these cells converts vitamin A into retinoic acid, which acts on naive B cells to promote their differentiation into IgA+ plasma cells. These cells can also promote antigen-specific Th1 and Th17 responses and their efficacy in promoting Th17 cells is associated with the production of retinoic acid.53 Flagellin may therefore have potential in mucosal vaccines as a means to promote vaccine-specific T-cell and IgA responses. However, delivery systems will be required to transport the flagellin across the epithelium to enable interaction with basolateral TLR5 on epithelial cells and TLR5-expressing DCs and macrophages.
A human IEC line expressed TLR9 and responded to CpG DNA stimulation with enhanced activation of NF-κB and production of IL-8. However, fully differentiated human colonic epithelial cells were unresponsive when challenged with CpG.54 However, polarity-dependent discrimination was recently shown between the responsiveness and nonresponsiveness of IECs to TLR9 activation.55 Although localized exclusively to the endosome in immune cells, TLR9 was detected on the basolateral (and apical) surface of polarized (and nonpolarized) IECs. Activation of basolateral TLR9 resulted in IκBα degradation and NF-κB activation, whereas stimulation of apical TLR9 led to the accumulation of poly-ubiquitinated IκBα in the cytoplasm, preventing NF-κB activation. This effect was associated with intracellular tolerance to subsequent TLR challenges. IECs in TLR9-deficient mice showed a lower NF-κB activation threshold and greater susceptibility to experimental colitis. In addition to the role of TLR9 in IECs, Paneth cells express TLR9 close to the antimicrobial peptide-containing secretory granules and injection of CpG resulted in a reduction in the numbers of secretory granules, consistent with Paneth cell degranulation and defensin release.56 Furthermore, the delivery of CpG increased the resistance of mice to oral challenge with S. typhimurium . The use of probiotics has been proposed as a therapeutic strategy for inflammatory bowel diseases. TLR9 signaling was required to mediate the anti-inflammatory effect of probiotics or probiotic DNA in the DSS colitis model57 and the anti-inflammatory effects of TLR9 agonists in colitis are mediated by type I IFN.58 Recent evidence was reported for genetic interactions between the single-nucleotide TLR9 polymorphisms, 1237T/C and 2848A/G, and CD-associated variants in NOD2, IL-23R, and DLG5 in modulating susceptibility to CD.59 The stimulation of human cells with the NOD2 agonist muramyl dipeptide (MDP) enhanced CpG-induced TLR9 responses.60 This indicates that PRMs do not act in isolation but that complex synergistic interactions operate to amplify the inflammatory responses. Regulatory T cells in the intestine maintain immunological tolerance to self-antigens and have an important role in suppressing damaging inflammatory responses.61 However, in contrast to the anti-inflammatory roles for TLR9 described above, DNA from commensal bacteria limits the induction of inducible regulatory T cells through TLR9 signaling.62
REGULATION OF IgA RESPONSES
The microbiota has an essential role in promoting the development of IgA-producing plasma cells and there is evidence that this enhancing effect is driven by specific bacterial species.63 Mice deficient in activation-induced cytidine deaminase, which leads to a loss of IgA-producing plasma cells, exhibit expansion of anaerobic, particularly segmented filamentous bacteria in the small intestine.64 The commensal bacterium B. thetaiotaomicron induces strong innate immune responses in the absence of IgA.65 TLR agonists can act directly on B cells in the presence of TGF-β to promote IgA class switching in B cells.66 A population of TNF-α/iNOS-producing DCs in the intestinal lamina propria produce nitric oxide and this leads to the induction of BAFF and APRIL, which promote activation-induced cytidine deaminase expression and IgA class switching.67 The development of TNF-α/iNOS-producing DCs requires TLR signaling as the numbers of these cells in the lamina propria are reduced in MyD88-deficient and TLR2−\− TLR4−\−\TLR9−\− mice.67 As IgA can have protective roles in immunity against pathogens, targeting cells including TNF-α/iNOS-producing DCs and TLR5-expressing DCs may have potential in vaccine development.
NEGATIVE REGULATION OF TLR SIGNALING IN THE INTESTINE
In addition to regulation mediated by tissue and cell-type-specific expression of PRMs in the gastrointestinal tract, there is also selective expression of negative innate signaling regulators in intestinal cells (Table 1). Mice deficient in IL-10 develop spontaneous commensal-dependent colitis, characterized by the presence of proinflammatory Th1 cells, and this is MyD88 dependent.68 IL-10 signaling pathways are dependent on signal transducer and activator of transcription (STAT) 3 and a myeloid cell-specific deletion of STAT3 resulted in enterocolitis with enhanced Th1 responses.69 This was dependent on IL-12 p40 and was reduced in mice that were also deficient in TLR4, suggesting that the commensal TLR4 ligands induced IL-12 p40. As it is now known that IL-12 p40 is a component of IL-23 as well as IL-12, the proinflammatory phenotype in the STAT3-deficient mice may reflect contributions from both cytokines.
Table 1.
Regulation of innate and adaptive intestinal responses by PRM signaling
| Deficient Immune molecule |
Disease pathology | Reference |
|---|---|---|
| MyD88 | Impaired development of Peyer’s patches Spontaneous intestinal tumors |
2,136 |
| TRIF | More commensal bacteria in spleen when challenged with E. coli |
6 |
| TLR5 | Spontaneous colitis | 46 |
| TLR9 | Lower NF-κB activation, greater susceptibility to DSS colitis |
55 |
| TLR2 | Compromised integrity of epithelial barrier |
137 |
| TLR4– (blocking with antagonist) |
Inhibits DSS-induced colitis and in spontaneous chronic colitis model |
39 |
| NOD2 | Abnormal development and function of Peyer’s patches, increased bacteria and yeast translocation |
90 |
| Increased susceptibility to oral infection with L. monocy -togenes |
92 | |
| NOD1 | Increased susceptibility to H. pylori |
91 |
| STAT3 | Develop chronic enterocolitis | 69 |
| SOCS1 | Increased susceptibility to DSS-induced colitis |
70 |
| SIGIRR | Increased susceptibility to DSS-induced colitis |
74 |
| IRAK-M | Amplified inflammatory responses to S. typhimurium |
75 |
DSS, dextran sodium sulfate.
Suppressor of cytokine signaling 1 (SOCS1) deficient mice exhibit more severe DSS-induced colitis than do wild-type mice,70 which is dependent on the microbiota.71 The TLR adapter protein, Mal, contains a PEST domain that allows it to be degraded by SOCS1.72 Thus, the absence of SOCS1 may result in amplified Mal signaling in response to TLR4 and TLR2 agonists.
Single immunoglobulin IL-1 receptor-related molecule (SIGIRR)/TIR8 is a negative regulator for Toll-IL-1R signaling.73 Mice deficient in SIGIRR were more susceptible to DSS-induced colitis and commensal bacteria induced higher levels of cytokines by epithelial cells from SIGIRR-deficient cells than from wild-type cells.74 SIGIRR functions as an intracellular decoy for TRAF6 and IRAK1 in both IECs and intestinal DCs. SIGIRR-deficient colonic epithelial cells exhibited commensal-dependent upregulation of inflammatory genes, whereas gut epithelium-specific expression of the SIGIRR transgene in the SIGIRR-deficient mice abrogated the hypersensitivity to DSS-induced colitis.73
IRAK-M is an inactive kinase, restricted to monocytes and macrophages whose expression is increased by TLR stimulation. IRAK-M prevented the dissociation of IRAK-1 and IRAK-4 from MyD88 and the formation of IRAK–TRAF6 complexes and IRAK-M-deficient mice exhibited amplified inflammatory responses to S. typhimurium.75 However, it is not clear whether IRAK-M has a role in regulating colitis. The cytoplasmic ubiquitin-modifying enzyme, A20, removes ubiquitin molecules from TRAF6, abrogating TLR-dependent responses.76 Deficiency in A20 led to spontaneous intestinal inflammation, which was eliminated if mice were also deficient in MyD88, and was also reduced by treatment of mice with antibiotics.77 Thus, a number of regulatory pathways, particularly directed at MyD88-dependent signaling, operate in the intestine to limit inflammatory responses to the microbiota.
THE ROLE OF NLRs IN MUCOSAL IMMUNITY
To date, 23 NLR proteins have been identified in humans and 34 NLR genes are present in mice.78 These intracellular receptors are structurally composed of an N-terminal effector domain that can comprise caspase recruitment domains (CARDs) as in the case of the NODs, a pryin domain as in the case of NLRP, or a baculovirus-inhibitor-of-apoptosis repeats domain for NAIP.1 NLRs comprise a central oligomerization domain and a C-terminal LRR domain79,80 and mediate signaling cascades through homophilic and heterophilic protein–protein interactions. In addition to their roles in PRM recognition, NLR family members are increasingly implicated in the proinflammatory forms of cell death, pyroptosis, and pyronecrosis.81 Thus, in inflammatory conditions, where significant mucosal cell death is ongoing, it will be important to assess the contribution of NLR family members, including NLRP1 and NLRP3, to the inflammatory process.
NOD 1 AND NOD 2
Nucleotide-binding oligomerization domains 1 and 2 (CARD15) are intracellular sensors of bacterial peptidoglycan components. NOD2 recognizes the presence of MDP, whereas NOD1 senses γ-D-meso-DAP (iE-DAP).82–84 NOD1 and NOD2 activating ligands are thought to enter cells by endocytosis through clathrin-coated pits.85 The internalization of the peptides is pH dependent and the peptides are enzymatically processed in endosomes before transport into the cytosol. NOD1 is ubiquitously expressed in adult human tissues, whereas NOD2 is expressed in leukocytes, DCs, and epithelial cells.86,87 NOD1 expression is upregulated by IFN-γ in IECs and NOD2 expression is increased by inflammatory stimuli such as LPS, TNF-α, or IFN-γ.87,88 NOD1 and NOD2 interact with the inhibitor of apoptosis proteins (cIAP1 and cIAP2) that possess C-terminal RING-finger domains with E3 ligase activity (Figure 1).89 Although cIAP proteins have CARD domains, the NOD1 and NOD2 interactions occur independently of these, indicating that the interaction between the NODs, cIAPs, and receptorinteracting protein 2 (RIP2) is independent of their CARD domains. Following recruitment to NOD1 and NOD2, the cIAP proteins function as E3 ligases and mediate the binding of lysine (K) 63-linked ubiquitin to RIP2. Ubiquitination of RIP2 causes its activation and subsequent binding and activation of TGF-β-associated kinase 1 (TAK1). The kinase activity of TAK1 activates both IKKγ and MKK, which culminates in the translocation of NF-κB and mitogen-activated protein kinase into the nucleus to upregulate the transcription of proinflammatory genes and mediate antibacterial effects by the upregulation of defensins.
Figure 1.

Nucleotide-binding domain and Leucine-rich repeat containing receptor (NLR)-and Toll-like receptor (TLR)-mediated signaling cascades in the gut epithelia. NLRs are composed of N-terminal effector domains that mediate protein–protein interactions with their binding partners. NOD1 and NLRP4 (IPAF) are composed of one caspase recruitment domain (CARD), whereas NOD2 is composed of two. All NLRs have a central oligomerization domain and C-terminal LRR regions. Cytosolic L. monocytogenes are recognized by NOD1 and NOD2, H. pylori is identified by NOD1, and S. typhimurium is recognized by NOD2. Extracellular flagellated S. typhimurium is recognized by TLR5, whereas NLRP4 identifies flagellin, which reaches the cytosol. NOD1 and NOD2 interact with the inhibitor of apoptosis proteins (cIAP1 and cIAP2), which have been found to possess C-terminal RING-finger domains with E3 ligase activity. cIAP1 or cIAP2 bind and ubiquitinate RIP2, adding K63-linked ubiquitin units to the kinase protein. Ubiquitinated RIP2 binds and activates TAK1. The kinase activity of TAK1 activates both IKKγ and MKK, which culminates in the translocation of NF-κB and mitogen-activated protein kinase into the nucleus, upregulating the transcription of proinflammatory genes and mediating the antibacterial effects by the upregulation of defensins. The bacterial translocation complex SipB mediates the translocation of flagellin across the plasma membrane, which is recognized by NLRP4, which in turn recruits apoptosis speck-like protein (ASC) through its N-terminal CARD domain. ASC then recruits and activates caspase-1, forming the NLRP4 inflammasome. TLRs contain an N-terminal LRR domain, a transmembrane domain, and a C-terminal cytoplasmic TIR domain. TLR5 is activated on ligation to extracellular flagellin, and TLR5 recruits adaptor MyD88 to upregulate a signaling cascade that activates the proinflammatory transcription factors NF-κB and AP1. Basolaterally polarized TLR5 is found in the colon and is upregulated by invasive bacteria that have breached the transepithelial barrier. TLR4 and TLR2 are found on the epithelial surface at a relatively low level; however, the expression of TLR4 and TLR2 is increased by proinflammatory cytokines. Basolateral TLR9 has been found to activate NF-κB, whereas apical TLR9 has been shown to cause the accumulation of ubiquitinated IκB preventing the activation of NF-κB.
NOD2 deficiency resulted in the abnormal development and function of peyer’s patches in mice.90 This phenotype was peyer’s patch specific and was not apparent after birth but progressed with time, being observed for up to 52 weeks. peyer’s patches in NOD2-deficient mice were larger than controls with increased numbers of both M cells and CD4+ T cells. The knockout mice exhibited increased translocation of yeast and bacteria across peyer’s patches and higher concentrations of the cytokines TNF-α, IFN-γ, IL-12, and IL-4.
NOD1-deficient mice are highly susceptible to infection with Helicobacter pylori,91 whereas NOD2-deficient mice are more susceptible to oral infection with Listeria monocytogenes.92 The uptake of L. monocytogenes into phagolysosomes generates bacterial peptides that enter the cytosol and interact with NOD2. This interaction is thought to specifically trigger type I IFN responses.93 NOD2 is expressed in ileal Paneth cells, and production of the cryptidin subgroup of antimicrobial peptides is reduced in NOD2-deficient cells.92 Furthermore, the expression of α-defensins is reduced in CD patients with NOD2 mutations.94
Crohn’s disease, which is characterized by chronic granulomatous intestinal inflammation and ulcers of the mucosa, has a complex pathology associated with a combination of genetic factors, deregulated interaction with the microbiota, and immune-mediated tissue injury. Three polymorphisms of NOD2 are associated with CD (Arg702Trp, Gly908Arg, and Leu1007fsinsC). These missense polymorphisms are located within the LRR domain of NOD2, involved in sensing MDP, and are associated with decreased MDP-induced NF-κB activity and impaired bacterial clearance.83,95,96 Individuals with the Leu1007fsinsC frameshift mutation have an inability to signal in response to MDP, and an impaired response to intracellular Salmonella, indicating that NOD2 is required for protection against intracellular bacteria.97 A NOD2 gain-of-function mutation is also associated with a chronic systemic inflammatory disorder known as “Blau syndrome,” in which NOD2 2932iC is constitutively active and can cause ligand-independent activation of NF-κB. In mice, this mutation enhanced NF-κB activation and increased apoptosis and secretion of IL-1β, which was linked to increased susceptibility to bacteria-induced intestinal inflammation.98 The 3020insC (SNP13) mutation in NOD2, found in Paneth cells, is associated with reduced levels of human defensins 5 and 6.94,99,100 A genetic association was identified between the Wnt-signaling pathway transcription factor TCF-4, which regulates Paneth cell differentiation and α-defensin expression, and ileal CD, indicating that deficiency in Paneth cell α-defensins is a primary factor in the pathogenesis of ileal CD.101 In addition to these findings on the importance of NOD2 in Paneth cells, NOD2-sufficient mast cells were significantly increased in the colonic lamina propria of CD patients in comparison with those of UC and control individuals.102 It was suggested that NOD2+ mast cells have a role in pathogenicity, associated with the recruitment of inflammatory cells in individuals with CD. In addition to the established link between NOD2 mutations and CD, NOD1 gene polymorphisms are also associated with susceptibility to CD and UC.103
INFLAMMASOMES
Inflammasomes are cytoplasmic multiprotein complexes containing an NLR and caspase-1, which assemble on activation, leading to the multimerization of the adapter molecule, apoptosis speck-like protein (ASC). Procaspase-1 is recruited to ASC through CARD domain interactions and autocleavage of caspase-1 is triggered.104 The cleaved caspase-1 p10 and p20 subunits assemble into active caspase-1, which cleaves not only pro-IL-1β into active IL-1β but also IL-18 into its active form. A number of NLR family members have now been shown to form inflammasomes including NLRP1, NLRP2, NLRP3, and NLRP4 (IPAF).105 NLRP4 responds to the presence of flagellated bacteria (Figure 1) and Salmonella uses a type III secretion system to deliver flagellin into the cytosol,106 resulting in NLRP4 inflammasome activation. Salmonella-induced activation of caspase-1 and secretion of IL-1β in macrophages is NLRP4 dependent and TLR5-independent.106,107
NLRP3
NLRP3 forms an inflammasome complex with ASC, cardinal, and procaspase-1.108–110 The NLPR3 inflammasome has attracted increasing interest with growing recognition of its role in innate responses induced by stimuli including adenosine triphosphate (ATP), pore-forming toxins, and uric acid.105 Our recent work suggests that the combination of particulates and TLR agonists is a strong stimulus for NLRP3 inflammasome activation.111 Furthermore, the enteric pathogens L. monocytogenes and Shigella flexneri have been found to signal through NLRP3 and S. flexneri induces a form of pyronecrosis in human monocytes, which is dependent on ASC, NLRP3, and cathepsin B.110 Extracellular ATP is a potent stimulus for NLRP3 inflammasome activation and is produced in high concentrations by commensal bacteria.112 In vitro, the addition of ATP to macrophages and DCs following TLR stimulation potently promotes NLRP3-mediated secretion of active IL-1β and IL-18. This process is mediated by ATP binding to P2X7 receptors and requires potassium efflux from cells.113 In addition, ATP binds specifically to NLRP3, which exerts its effect as an ATPase, and it was proposed that this nucleotide hydrolysis was required for NLRP3 self-association, interaction with ASC, activation of caspase-1, and release of IL-1β.114
IL-17-producing CD4+T cells (Th17 cells) are a subset of T helper cells that are implicated in protection against a number of extracellular pathogens, including the enteric pathogen Citrobacter rodentium,115 and also in autoimmune conditions.116 These cells are characterized by the production of IL-17A, IL-17F, IL-21, and IL-22 and require the STAT3-dependent expression of the transcription factor ROR-γt.117 Th17 cells are highly enriched and constitutively present in the intestinal lamina propria,118 and this requires the microbiota as the numbers of Th17 cells are reduced in germ-free mice.112 Remarkably, the development of Th17 cells in the lamina propria was independent of TLR signaling as it was intact in MyD88/TRIF-deficient mice. ATP activates a population of CD70(high) CD11c(low) cells in the intestinal lamina propria leading to the differentiation of Th17 cells.112 There were lower concentrations of luminal ATP and Th17 cells in germ-free mice, implicating the microbiota in both processes. Furthermore, the administration of ATP to germ-free mice resulted in a large increase in the numbers of Th17 cells in the lamina propria and delivery of ATP-exacerbated pathology in a T-cell-mediated colitis model. The CD70(high) CD11c(low) cells produced the Th17 polarizing factors IL-6, IL-23, and integrin-αV and -β8, which are involved in activating latent complexes of TGF-β. We have reported an essential role for IL-1 in the induction of Th17 cells in mice.119 As ATP is a potent stimulus for NLRP3 inflammasome activation, it is likely that ATP produced by the microbiota would promote NLRP3 activation in intestinal DCs and macrophages. This may particularly be the case during epithelial injury where PRM ligands can access their receptors on epithelial and innate cells. This would induce pro-IL-1β production and ATP activation could activate the NLRP3 inflammasome with consequent activation of caspase-1 and cleavage and secretion of IL-1β and IL-18. Thus, both MyD88-dependent and -independent mechanisms for Th17 cell development probably exist, as NLRP3 may be a mediator of commensal bacteria-induced Th17 responses in the intestine (Figure 2 ). In addition, γδT cells, which are abundant in the intestinal epithelium,120 respond to stimulation with IL-1 and IL-23 by secreting IL-17 and act to amplify Th17 cell responses.121 Thus, NLRP3 activation in the intestine may promote IL-1 secretion in response to ATP, which acts in synergy with other PRM-induced polarizing cytokines to generate an environment particularly conducive to the differentiation of Th17 cells in the lamina propria (Figure 2). As ATP-induced Th17 cell differentiation was independent of TLR signaling, this suggests that the induction of IL-23 and other Th17 cell-polarizing cytokines must be driven through signaling through other PRMs.112 In this regard, NOD2 and Dectin 1 have been implicated in the induction of IL-23.122
Figure 2.
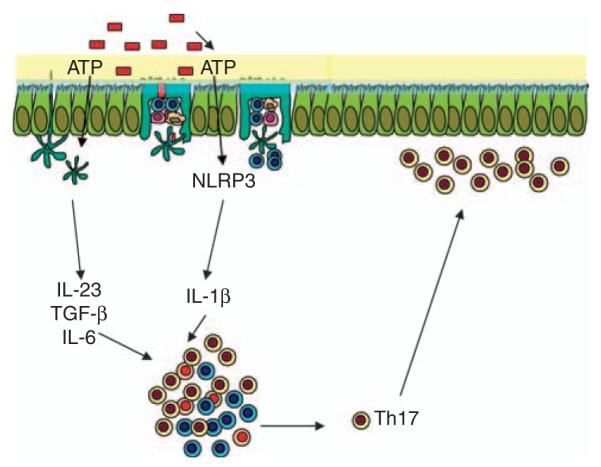
Hypothetical model for ATP-driven NLRP3 activation in promoting Th17 responses in the intestine. ATP produced by commensal bacteria (red boxes) acts on CD70(high)CD11c(low) cells to enhance the Toll-like receptor (TLR)-independent secretion of the Th17 cell-polarizing cytokines, IL-6, IL-23, and TGF-β.112 In parallel, ATP may act on intestinal dendritic cells (DCs) and macrophages through P2X7 receptors to enhance NLRP3 inflammasome activation, promoting caspase-1 activity and secretion of IL-1β. These Th17-polarizing cytokines act in synergy to drive Th17 responses in the lamina propria.
Despite evidence from the studies above, indicating that the promoting effects of the microbiota on Th17 cells are TLR independent, TLR5-expressing DCs can promote the differentiation of Th17 cells on stimulation with flagellin53 and there were reduced numbers of lamina propria Th17 cells in TLR9-deficient mice.62 Thus, the role of TLR signaling in promoting Th17 cells may be context dependent. The Th17-promoting cytokine IL-23 mediates intestinal inflammation123,124 and an IL-23 receptor variant is associated with inflammatory bowel disease.125
RLRs
The detection of viruses can be largely attributed to the presence of intracellular TLRs and RLRs.126 The RLRs, RIG-I, and Mda-5 are cytoplasmic RNA helicases containing a DExD/H box RNA helicase domain and two CARD-like domains.126 These recognize viral RNA and signal downstream to NF-κB, mitogen-activated protein kinases, and IRFs, which transcribe genes encoding type I IFN and proinflammatory cytokines. RIG-I specifically recognizes 5′-triphosphate-containing viral RNA and upregulates downstream signaling cascades that cause the phosphorylation and dimerization of IRF3 through a TBK1–IKKε–DDX3 complex. Activation of the IRF3 complex causes its translocation into the nucleus to upregulate the transcription of IFN-β, protecting the host from viral infection.
Downstream of RIG-I and Mda5 is an adapter molecule known as mitochondrial antiviral signaling adapter, also known as IPS-1. It has an N-terminal CARD domain and a C-terminal transmembrane domain that localizes it to the mitochondrial membrane. Mitochondrial antiviral signaling adapter functions to activate IFN-α, IFN-β, and NF-κB promoters in a TBK1-dependent manner.127 Recently, RIG-I was also shown to sense AT-rich double-stranded DNA through the induction of an RNA polymerase III-transcribed RNA intermediate.128
Signaling through RLRs, similar to TLRs, induces the phosphorylation and homodimerization of constitutively expressed IRF3, triggering the translation of type I IFN (IFN-β). IRF3 is present in cells in an inactive state, its activation requiring the phosphorylation of multiple serine/threonine residues at its C terminus.129 The phosphorylation of IRF3 on serine clusters is carried out by TBK-1, and IKKε, which allows IRF3 dimerization to occur, followed by its nuclear accumulation. The IRF3 dimer forms a complex with the transcription factors ATF-2/c-Jun (AP-1) and NF-κB (p65 and p50 heterodimer) and the co-activator CBP/p300, allowing it to bind to DNA in a sequence-specific manner.129–132 RIG-I disruption in mice resulted in reduced body weight, which was caused by severe damage and inflammatory infiltration into the colonic mucosa.133,134 Given the recent findings on the ability of RIG-I to recognize DNA as well as RNA, it will be important to identify the ligand promoting this process. In addition to these inflammatory effects, the number of peyer’s patches was significantly reduced in RIG-I-deficient mice and RIGI was found to control the transcriptional activity of Gαi2, a negative regulator of T-cell responses, which may mediate the modulatory effects of RIG-I.134
CONCLUSIONS
Both epithelial barrier function and mucosal immunity depend on interactions between the microbiota and PRMs on epithelial and other mucosal cells. In particular, signaling through TLR2 has an essential role in maintaining the integrity of the epithelial barrier and controlling intestinal permeability. The finding that the spontaneous hyperactivation of systemic antibody responses against intestinal bacteria occurs in the absence of TLR signaling shows the vital role of innate immune signaling in corralling the microbiota in the mucosal compartment. The nature of adaptive immune responses at the mucosae also exhibits unique features including a preponderance of Th17 cells in the lamina propria and a preferential secretion of IgA. In both cases, the microbiota are required to drive these responses, although the specific PRMs responsible in most cases await determination. The finding that the induction of IgA responses requires the microbiota and TLR signaling, whereas Th17 cell induction is not TLR dependent,112 suggests that the relationships between innate signaling and adaptive immunity are diverse, indicating that other PRMs such as NLRs, RLRs, or C-type lectins are involved in promoting Th17 cell responses. A further example of this is the requirement for microbiota and TLR signaling for TNF-α/iNOS-producing DCs in the lamina propria of mice.67 NOD1 and NOD2 are important in the recognition of intracellular bacteria that have breached the epithelial wall and mutations result in the increased bacterial uptake into epithelial cells. The recent finding that the normal intestinal microbiota can alleviate the progression of autoimmune diabetes in a MyD88-independent manner135 suggests that the consequences of innate receptor signaling in the intestine may have systemic as well as local consequences. In addition, as UC is an important risk factor for the development of colorectal cancer, the consequences of aberrant innate signaling in the intestine are many. As a result, an increased understanding of the ligands and receptors responsible for promoting and inhibiting innate and adaptive immunity in the intestine has tremendous therapeutic implications.
ACKNOWLEDGMENTS
Ed Lavelle’s work on mucosal immunity and vaccines is funded by science foundation Ireland (08/RFP/BMT1363, 07/RFP/BICF537), Enterprise Ireland (IP 2007 0451) and the Meningitis Research Foundation (0610.0). Emma Creagh is supported by a Wellcome Trust Fellowship and the Broad Medical Research Program. Luke O’Neill and Caroline Murphy are funded by Science Foundation Ireland.
Footnotes
DISCLOSURE The authors declared no conflict of interest.
REFERENCES
- 1.Creagh EM, O’Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends. Immunol. 2006;27:352–357. doi: 10.1016/j.it.2006.06.003. [DOI] [PubMed] [Google Scholar]
- 2.Rakoff-Nahoum S, Pagina J, Eslami-Varzaneh F, Edberg S, Medzhitov R. Recognition of commensal microfl ora by toll-like receptors is required for intestinal homeostasis. Cell. 2004;118:229–241. doi: 10.1016/j.cell.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 3.Hooper LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. Molecular analysis of commensal host–microbial relationships in the intestine. Science. 2001;291:881–884. doi: 10.1126/science.291.5505.881. [DOI] [PubMed] [Google Scholar]
- 4.Mueller C, MacPherson AJ. Layers of mutualism with commensal bacteria protect us from intestinal infl ammation. Gut. 2006;55:276–284. doi: 10.1136/gut.2004.054098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Macpherson AJ, Slack E, Geuking MB, McCoy KD. The mucosal firewalls against commensal intestinal microbes. Semin. Immunopathol. 2009;31:145–149. doi: 10.1007/s00281-009-0174-3. [DOI] [PubMed] [Google Scholar]
- 6.Slack E, et al. Innate and adaptive immunity cooperate flexibly to maintain host–microbiota mutualism. Science. 2009;325 doi: 10.1126/science.1172747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Strober W, Fuss IJ, Blumberg RS. The immunology of mucosal models of inflammation. Annu. Rev. Immunol. 2002;20:495–549. doi: 10.1146/annurev.immunol.20.100301.064816. [DOI] [PubMed] [Google Scholar]
- 8.Matsushima N, et al. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics. 2007;8:124. doi: 10.1186/1471-2164-8-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr. Opin. Struct. Biol. 2001;11:725–732. doi: 10.1016/s0959-440x(01)00266-4. [DOI] [PubMed] [Google Scholar]
- 10.Kajava AV. Structural diversity of leucine-rich repeat proteins. J. Mol. Biol. 1998;277:519–527. doi: 10.1006/jmbi.1998.1643. [DOI] [PubMed] [Google Scholar]
- 11.Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu. Rev. Biochem. 2007;76:141–165. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 12.Sun Jin M, Lee J-O. Structures of the Toll-like receptor family and its ligand complexes. Immun. Rev. 2008;29:182–191. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 13.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 14.Kagan JC, Su T, Horng T, Chow A, Akira S, Medzhitov R. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferonbeta. Nat. Immunol. 2008;9:361–368. doi: 10.1038/ni1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fitzgerald KA, et al. Mal(MyD88 adapter like) is required for Toll-like receptor 4 signal transduction. Nature. 2001;413:78–83. doi: 10.1038/35092578. [DOI] [PubMed] [Google Scholar]
- 16.Kenny EF, Talbot S, Gong M, Golenbock DT, Bryant CE, O’Neill LAJ. MyD88 adaptor-like is not essential for TLR2 signaling and inhibits TLR3. J. Immunol. 2009;183:3642–3651. doi: 10.4049/jimmunol.0901140. [DOI] [PubMed] [Google Scholar]
- 17.Cario E, Podolsky DK. Differential alteration in intestinal epithelial cell expression of toll-like receptor 3 (TLR3) and TLR4 in inflammatory bowel disease. Infect. Immun. 2000;68:7010–7017. doi: 10.1128/iai.68.12.7010-7017.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu YJ. IPC: professional type 1 interferon-producing cells and plasmacytoid dendritic cell precursors. Annu. Rev. Immunol. 2005;23:275–306. doi: 10.1146/annurev.immunol.23.021704.115633. [DOI] [PubMed] [Google Scholar]
- 19.Wendland M, et al. CCR9 is a homing receptor for plasmacytoid dendritic cells to the small intestine. Proc. Natl. Acad. Sci. USA. 2007;104:6347–6352. doi: 10.1073/pnas.0609180104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hausmann M, et al. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 2002;122:1987–2000. doi: 10.1053/gast.2002.33662. [DOI] [PubMed] [Google Scholar]
- 21.Hart AL, et al. Characterisation of intestinal dendritic cells in inflammatory bowel disease. Gastroenterology. 2005;129:50–56. doi: 10.1053/j.gastro.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 22.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology. 2004;127:224–238. doi: 10.1053/j.gastro.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 23.Melmed G, et al. Human intestinal epithelial cells are broadly unresponsive to Toll-like receptor 2-dependent bacterial ligands: implications for host–microbial interactions in the gut. J. Immunol. 2003;170:1406–1415. doi: 10.4049/jimmunol.170.3.1406. [DOI] [PubMed] [Google Scholar]
- 24.Ey B, Eyking A, Gerken G, Podolsky DK, Cario E. TLR2 mediates gap junctional intercellular communication through connexin-43 in intestinal epithelial barrier injury. J. Biol. Chem. 2009;284:22332–22343. doi: 10.1074/jbc.M901619200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cario E, Gerken G, Podolsky DK. Toll-like receptor 2 controls mucosal inflammation by regulating epithelial barrier function. Gastroenterology. 2007;132:1359–1374. doi: 10.1053/j.gastro.2007.02.056. [DOI] [PubMed] [Google Scholar]
- 26.Schroder NW, Hermann C, Hamann L, Gobel UB, Hartung T, Schumann RR. High frequency of polymorphism Arg753Gln of the Toll-like receptor-2 gene detected by a novel allele-specific PCR. J. Mol. Med. 2003;81:368–372. doi: 10.1007/s00109-003-0443-x. [DOI] [PubMed] [Google Scholar]
- 27.Pierik M, et al. Toll-like receptor-1, -2, and -6 polymorphisms influence disease extension in inflammatory bowel diseases. Inflamm. Bowel Dis. 2006;12:1–8. doi: 10.1097/01.mib.0000195389.11645.ab. [DOI] [PubMed] [Google Scholar]
- 28.Le Mandat Schultz A, et al. Expression of TLR-2, TLR-4, NOD2 and pNF-kappaB in a neonatal rat model of necrotizing enterocolitis. PLoS One. 2007;2:e1102. doi: 10.1371/journal.pone.0001102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith PD, et al. Intestinal macrophages lack CD14 and CD89 and consequently are down-regulated for LPS- and IgA-mediated activities. J. Immunol. 2001;167:2651–2656. doi: 10.4049/jimmunol.167.5.2651. [DOI] [PubMed] [Google Scholar]
- 30.Hornef MW, Normark BH, Vandewalle A, Normark S. Intracellular recognition of lipopolysaccharide by toll-like receptor 4 in intestinal epithelial cells. J. Exp. Med. 2003;198:1225–1235. doi: 10.1084/jem.20022194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cerovic V, Jenkins CD, Barnes AG, Milling SW, MacPherson GG, Klavinskis LS. Hyporesponsiveness of intestinal dendritic cells to TLR stimulation is limited to TLR4. J. Immunol. 2009;182:2405–2415. doi: 10.4049/jimmunol.0802318. [DOI] [PubMed] [Google Scholar]
- 32.Uematsu S, et al. Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intestinal CD11c+ lamina propria cells. Nat. Immunol. 2006;7:868–874. doi: 10.1038/ni1362. [DOI] [PubMed] [Google Scholar]
- 33.Otte JM, Cario E, Podolsky DK. Mechanisms of cross hyporesponsiveness to Toll-like receptor bacterial ligands in intestinal epithelial cells. Gastroenterology. 2004;126:1054–1070. doi: 10.1053/j.gastro.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Ruemmele FM, et al. Lipopolysaccharide modulation of normal enterocyte turnover by Toll-like receptors is mediated endogenously by tumor necrosis factor (alpha) Gut. 2002;51:842–848. doi: 10.1136/gut.51.6.842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abreu MT, et al. TLR4 and MD-2 expression is regulated by immunemediated signals in human intestinal epithelial cells. J. Biol. Chem. 2002;277:20431–20437. doi: 10.1074/jbc.M110333200. [DOI] [PubMed] [Google Scholar]
- 36.Cetin S, et al. Endotoxin inhibits intestinal epithelial restitution through activation of Rho-GTPase and increased focal adhesions. J. Biol. Chem. 2004;128:24592–24600. doi: 10.1074/jbc.M313620200. [DOI] [PubMed] [Google Scholar]
- 37.Fort MM, et al. A synthetic TLR4 antagonist has anti-inflammatory effects in two murine models of inflammatory bowel disease. J. Immunol. 2005;174:6416–6423. doi: 10.4049/jimmunol.174.10.6416. [DOI] [PubMed] [Google Scholar]
- 38.Leaphart CL, et al. A critical role for TLR4 in the pathogenesis of necrotizing enterocolitis by modulating intestinal injury and repair. J. Immunol. 2007;179:4808–4820. doi: 10.4049/jimmunol.179.7.4808. [DOI] [PubMed] [Google Scholar]
- 39.Ungaro R, et al. A novel Toll-like receptor 4 antagonist antibody ameliorates inflammation but impairs mucosal healing in murine colitis. Am. J. Physiol. Gastrointest Liver Physiol. 2009;296:G1167–1179. doi: 10.1152/ajpgi.90496.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Man AL, et al. Macrophage migration inhibitory factor plays a role in the regulation of microfold (M) cell-mediated transport in the gut. J. Immunol. 2008;181:5673–5680. doi: 10.4049/jimmunol.181.8.5673. [DOI] [PubMed] [Google Scholar]
- 41.Iiyama R, et al. Normal development of the gut-associated lymphoid tissue except Peyer’s patch in MyD88-deficient mice. Scand. J. Immunol. 2003;58:620–627. doi: 10.1111/j.1365-3083.2003.01346.x. [DOI] [PubMed] [Google Scholar]
- 42.Meynell HM, Thomas NW, James PS, Holland J, Taussig MJ, Nicoletti C. Up-regulation of microsphere transport across the follicleassociated epithelium of Peyer’s patch by exposure to Streptococcus pneumoniae R36a. FASEB J. 1999;13:611–619. doi: 10.1096/fasebj.13.6.611. [DOI] [PubMed] [Google Scholar]
- 43.Cuvelier CA, Quatacker J, Mielants H, De Vos M, Veys E, Roels HJ. M-cells are damaged and increased in number in inflamed human ileal mucosa. Histopathology. 1994;24:417–426. doi: 10.1111/j.1365-2559.1994.tb00550.x. [DOI] [PubMed] [Google Scholar]
- 44.Tsuji M, et al. Requirement for lymphoid tissue-inducer cells in isolated follicle formation and T cell-independent immunoglobulin A generation in the gut. Immunity. 2008;29:261–271. doi: 10.1016/j.immuni.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 45.Bouskra D, et al. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature. 2008;456:507–510. doi: 10.1038/nature07450. [DOI] [PubMed] [Google Scholar]
- 46.Vijay-Kumar M, et al. Deletion of TLR5 results in spontaneous colitis in mice. J. Clin. Invest. 2007;117:3909–3921. doi: 10.1172/JCI33084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rumbo M, Nempont C, Kraehenbuhl J-P, Sirard J-C. Mucosal interplay among commensal and pathogenic bacteria: lessons from flagellin and Toll-like receptor 5. FEBS Lett. 2006;580:2976–2984. doi: 10.1016/j.febslet.2006.04.036. [DOI] [PubMed] [Google Scholar]
- 48.Zeng H, et al. Flagellin/TLR5 responses in epithelia reveal intertwined activation of inflammatory and apoptotic pathways. Am. J. Physiol. Gastrointest Liver Physiol. 2006;290:G96–G108. doi: 10.1152/ajpgi.00273.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinfl ammatory gene expression. J. Immunol. 2001;167:1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 50.Rhee SH, Im E, Riegler M, Kokkotou E, O’Brien M, Pothoulakis C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc. Natl. Acad. Sci. USA. 2005;102:13610–13615. doi: 10.1073/pnas.0502174102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fournier B, Williams IR, Gewirtz AT, Neish AS. Toll-like receptor 5-dependent regulation of inflammation in systemic Salmonella enterica Serovar typhimurium infection. Infect. Immun. 2009;77:4121–4129. doi: 10.1128/IAI.00656-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arques JL, et al. Salmonella induces flagellin- and MyD88-dependent migration of bacteria-capturing dendritic cells into the gut lumen. Gastroenterology. 2009;137:579–587. e571–572. doi: 10.1053/j.gastro.2009.04.010. 587. [DOI] [PubMed] [Google Scholar]
- 53.Uematsu S, et al. Regulation of humoral and cellular gut immunity by lamina propria dendritic cells expressing Toll-like receptor 5. Nat. Immunol. 2008;9:769–776. doi: 10.1038/ni.1622. [DOI] [PubMed] [Google Scholar]
- 54.Pedersen G, Andresen L, Matthiessen MW, Rask-Madsen J, Brynskov J. Expression of Toll-like receptor 9 and response to bacterial CpG oligodeoxynucleotides in human intestinal epithelium. Clin. Exp. Immunol. 2005;141:298–306. doi: 10.1111/j.1365-2249.2005.02848.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee J, et al. Maintenance of colonic homeostasis by distinctive apical TLR9 signalling in intestinal epithelial cells. Nat. Cell Biol. 2006;8:1327–1336. doi: 10.1038/ncb1500. [DOI] [PubMed] [Google Scholar]
- 56.Rumio C, et al. Degranulation of Paneth cells via toll-like receptor 9. Am. J. Pathol. 2004;165:373–381. doi: 10.1016/S0002-9440(10)63304-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rachmilewitz D, et al. Toll-like receptor 9 signaling mediates the antiinfl ammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–528. doi: 10.1053/j.gastro.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 58.Katakura K, Lee J, Rachmilewitz D, Li G, Eckmann L, Raz E. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J. Clin. Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Torok HP, Glas J, Tonenchi L, Bruennler G, Folwaczny M, Folwaczny C. Crohn’s disease is associated with a toll-like receptor-9 polymorphism. Gastroenterology. 2004;127:365–366. doi: 10.1053/j.gastro.2004.05.051. [DOI] [PubMed] [Google Scholar]
- 60.van Heel DA, et al. Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet. 2005;365:1794–1796. doi: 10.1016/S0140-6736(05)66582-8. [DOI] [PubMed] [Google Scholar]
- 61.Sakaguchi S, Yamaguchi T, Nomuka T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 62.Hall JA, et al. Commensal DNA limits regulatory T cell conversion and is a natural adjuvant of intestinal immune response. Immunity. 2006;29:637. doi: 10.1016/j.immuni.2008.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Macpherson AJ, Harris NL. Interactions between commensal intestinal bacteria and the immune system. Nat. Rev. Immunol. 2004;4:478–485. doi: 10.1038/nri1373. [DOI] [PubMed] [Google Scholar]
- 64.Fagarasan S, Muramatsu M, Suzuki K, Nagaoka H, Hiai H, Honjo T. Critical roles of activation-induced cytidine deaminase in the homeostasis of gut flora. Science. 2002;298:1424–1427. doi: 10.1126/science.1077336. [DOI] [PubMed] [Google Scholar]
- 65.Petersen DA, MacNulty NP, Gurge JL, Gordon JI. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–339. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 66.Cerutti A. The regulation of IgA class switching. Nat. Rev. Immunol. 2008;8:421–434. doi: 10.1038/nri2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tezuka H, et al. Regulation of IgA production by naturally occurring TNF/iNOS-producing dendritic cells. Nature. 2007;448:929–933. doi: 10.1038/nature06033. [DOI] [PubMed] [Google Scholar]
- 68.Rakoff-Nahoum S, Hao L, Medzhitov R. Role of Toll-like receptors in spontaneous commensal dependent colitis. Immunity. 2006;25:319–329. doi: 10.1016/j.immuni.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 69.Kobayashi M, et al. Toll-like receptor-dependent production of IL-12p40 causes chronic enterocolitis in myeloid cell-specifi c Stat3-deficient mice. J. Clin. Invest. 2003;111:1297–1308. doi: 10.1172/JCI17085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horino J, et al. Suppressor of cytokine signaling-1 ameliorates dextran sulfate sodium-induced colitis in mice. Int. Immunol. 2008;20:753–762. doi: 10.1093/intimm/dxn033. [DOI] [PubMed] [Google Scholar]
- 71.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- 72.Mansell A, et al. Suppressor of cytokine signalling 1 negatively regulates Toll-like receptor signalling by mediating Mal degradation. Nat. Immunol. 2006;7:148–155. doi: 10.1038/ni1299. [DOI] [PubMed] [Google Scholar]
- 73.Xiao H, et al. The Toll-interleukin-1 receptor member SIGIRR regulates colonic epithelial homeostasis, inflammation, and tumorigenesis. Immunity. 2007;26:461–475. doi: 10.1016/j.immuni.2007.02.012. [DOI] [PubMed] [Google Scholar]
- 74.Garlanda C, et al. Intestinal inflammation in mice deficient in Tir8, an inhibitory member of the IL-1 receptor family. Proc. Natl. Acad. Sci. USA. 2004;101:3522–3526. doi: 10.1073/pnas.0308680101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kobayashi K, Hernandez LD, Galan JE, Janeway CA., Jr IRAK-M is a negative regulator of Toll-like receptor signalling. Cell. 2002;110:191–202. doi: 10.1016/s0092-8674(02)00827-9. [DOI] [PubMed] [Google Scholar]
- 76.Boone DL, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat. Immunol. 2004;5:1052–1060. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 77.Turer EE, et al. Homeostatic MyD88-dependent signals cause lethal inflammation in the absence of A20. J. Exp. Med. 2008;205:451–464. doi: 10.1084/jem.20071108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shaw MH, Reimer T, Kim YG, Nunez G. NOD-like receptors (NLRs): bona fide intracellular microbial sensors. Curr. Opin. Immunol. 2008;20:377–382. doi: 10.1016/j.coi.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tschopp J, Martinon F, Burns K. NALPs: a novel protein family involved in inflammation. Nat. Rev. Mol. Cell Biol. 2003;4:95–104. doi: 10.1038/nrm1019. [DOI] [PubMed] [Google Scholar]
- 80.Inohara C, McDonald C, Nunez G. NOD-LRR proteins: role in host–microbial interactions and inflammatory disease. Annu. Rev. Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 81.Kroemer G, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Chamaillard M, et al. An essential role for NOD1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat. Immunol. 2003;4:702–707. doi: 10.1038/ni945. [DOI] [PubMed] [Google Scholar]
- 83.Girardin SE, et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 2003;278:8869–8872. doi: 10.1074/jbc.C200651200. [DOI] [PubMed] [Google Scholar]
- 84.Girardin SE, et al. Nod1 detects a unique muropeptide from Gramnegative bacterial peptidoglycan. Science. 2003;300:1584–1587. doi: 10.1126/science.1084677. [DOI] [PubMed] [Google Scholar]
- 85.Lee J, Tattoli I, Wojtal KA, Vavricka SR, Philpott DJ, Girardin SE. pH-dependent internalization of muramyl peptides from early endosomes enables Nod1 and Nod2 signaling. J. Biol. Chem. 2009;284:23818–23829. doi: 10.1074/jbc.M109.033670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Inohara N, et al. Nod1, an Apaf-1-like activator of caspase-9 and nuclear factor-kappaB. J. Biol. Chem. 1999;274:14560–14567. doi: 10.1074/jbc.274.21.14560. [DOI] [PubMed] [Google Scholar]
- 87.Gutierrez O, et al. Induction of Nod2 in myelomonocytic and intestinal epithelial cells via nuclear factor-kappa B activation. J. Biol. Chem. 2002;277:41701–41705. doi: 10.1074/jbc.M206473200. [DOI] [PubMed] [Google Scholar]
- 88.Rosenstiel P, et al. TNF-alpha and IFN-gamma regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–1009. doi: 10.1053/gast.2003.50157. [DOI] [PubMed] [Google Scholar]
- 89.Reardon C, Mak TW. cIAP proteins: keystones in NOD receptor signal transduction. Immunity. 2009;30:755–756. doi: 10.1016/j.immuni.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 90.Barreau F, et al. CARD15/NOD2 is required for Peyer’s patches homeostasis in mice. PLoS One. 2007;2:e523. doi: 10.1371/journal.pone.0000523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Viala J, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 92.Kobayashi KS, et al. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science. 2005;307:731–734. doi: 10.1126/science.1104911. [DOI] [PubMed] [Google Scholar]
- 93.Herskovits AA, Auerbuch V, Portnoy DA. Bacterial ligands generated in a phagosome are targets of the cytosolic innate immune system. PLoS Pathog. 2007;3:e51. doi: 10.1371/journal.ppat.0030051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wehkamp J, et al. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut. 2004;53:1658–1664. doi: 10.1136/gut.2003.032805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Inohara N, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003;278:5509–5512. doi: 10.1074/jbc.C200673200. [DOI] [PubMed] [Google Scholar]
- 96.Chamaillard M, et al. Gene–environment interaction modulated by allelic heterogeneity in inflammatory diseases. Proc. Natl. Acad. Sci. USA. 2003;100:3455–3460. doi: 10.1073/pnas.0530276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124:993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 98.Maeda S, et al. Nod2 mutation in Crohn’s disease potentiates NF-kappaB activity and IL-1beta processing. Science. 2005;307:734–738. doi: 10.1126/science.1103685. [DOI] [PubMed] [Google Scholar]
- 99.Wehkamp J, et al. Reduced Paneth cell alpha-defensins in ileal Crohn’s disease. Proc. Natl. Acad. Sci. USA. 2005;102:18129–18134. doi: 10.1073/pnas.0505256102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grimm MC, Pavli P. NOD2 mutations and Crohn’s disease: are Paneth cells and their antimicrobial peptides the link? Gut. 2004;53:1558–1560. doi: 10.1136/gut.2004.043307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Koslowski MJ, et al. Genetic variants of Wnt transcription factor TCF-4 (TCF7L2) putative promoter region are associated with small intestinal Crohn’s disease. PLoS One. 2009;4:e4496. doi: 10.1371/journal.pone.0004496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Okumura S, et al. Hyperexpression of NOD2 in intestinal mast cells of Crohn’s disease patients: preferential expression of inflammatory cell-recruiting molecules via NOD2 in mast cells. Clin. Immunol. 2009;130:175–185. doi: 10.1016/j.clim.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 103.McGovern DP, et al. Association between a complex insertion/deletion polymorphism in NOD1 (CARD4) and susceptibility to inflammatory bowel disease. Hum. Mol. Genet. 2005;14:1245–1250. doi: 10.1093/hmg/ddi135. [DOI] [PubMed] [Google Scholar]
- 104.Bryant C, Fitzgerald KA. Molecular mechanisms involved in inflammasome activation. Trends Cell Biol. 2009;19:455–464. doi: 10.1016/j.tcb.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 105.Ting JP, Willingham SB, Bergstralh DT. NLRs at the intersection of cell death and immunity. Nat. Rev. Immunol. 2008;8:372–379. doi: 10.1038/nri2296. [DOI] [PubMed] [Google Scholar]
- 106.Franchi L, et al. Cytosolic fl agellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006;7:576–582. doi: 10.1038/ni1346. [DOI] [PubMed] [Google Scholar]
- 107.Miao EA, et al. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006;7:569–575. doi: 10.1038/ni1344. [DOI] [PubMed] [Google Scholar]
- 108.Mariathasan S, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- 109.Martinon F, Petrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 110.Willingham SB, et al. Microbial pathogen-induced necrotic cell death mediated by the inflammasome components CIAS1/cryopyrin/NLRP3 and ASC. Cell Host Microbe. 2007;2:147–218. doi: 10.1016/j.chom.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharp FA, et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. USA. 2009;106:870–875. doi: 10.1073/pnas.0804897106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Atarashi K, et al. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008;455:808–812. doi: 10.1038/nature07240. [DOI] [PubMed] [Google Scholar]
- 113.Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- 114.Duncan JA, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. PNAS. 2007;104:8041–8046. doi: 10.1073/pnas.0611496104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mangan PR, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 116.Weaver CT, Hatton RD, Mangan PR, Harrington L. IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu. Rev. Immunol. 2007;25:821–852. doi: 10.1146/annurev.immunol.25.022106.141557. [DOI] [PubMed] [Google Scholar]
- 117.Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat. Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- 118.Ivanov II, et al. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 119.Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006;203:1685–1691. doi: 10.1084/jem.20060285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hayday A, Tigelaar R. Immunoregulation in the tissues by gammadelta T cells. Nat. Rev. Immunol. 2003;3:233–242. doi: 10.1038/nri1030. [DOI] [PubMed] [Google Scholar]
- 121.Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 122.Lyakh L, Trinchieri G, Provezza L, Carra G, Gerosa F. Regulation of interleukin-12/interleukin-23 production and the T-helper 17 response in humans. Immunol. Rev. 2008;226:112–131. doi: 10.1111/j.1600-065X.2008.00700.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hue S, et al. Interleukin-23 drives innate and T cell-mediated intestinalinfl ammation. J. Exp. Med. 2006;203:2473–2483. doi: 10.1084/jem.20061099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Izcue A, et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity. 2008;28:559–570. doi: 10.1016/j.immuni.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Duerr RH, et al. A genome-wide association study identifies IL23R as an infl ammatory bowel disease gene. Science. 2006;314:1461–1463. doi: 10.1126/science.1135245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kawai T, Akira S. Toll-like receptor and RIG-I like receptor signalling. Ann. N.Y. Acad. Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- 127.Seth RB, Sun L, Ea CK, Chen ZJ. Identifi cation and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 128.Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol. 2009;10:1065–1072. doi: 10.1038/ni.1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-dependent phosphorylation of the IRF-3 transcription factor regulates nuclear translocation, transactivation potential, and proteasome-mediated degradation. Mol. Cell Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lin R, Mamane Y, Hiscott J. Structural and functional analysis of interferon regulatory factor 3: localization of the transactivation and autoinhibitory domains. Mol. Cell Biol. 1999;19:2465–2474. doi: 10.1128/mcb.19.4.2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Qin BY, et al. Crystal structure of IRF-3 reveals mechanism of autoinhibition and virus-induced phosphoactivation. Nat. Struc. Biol. 2003;10:913–921. doi: 10.1038/nsb1002. [DOI] [PubMed] [Google Scholar]
- 132.Yoneyama M, Suhara W, Fukuhara Y, Sato M, Ozato K, Fujita T. Direct triggering of type I interferon system by virus infection: activation of a transcription factor complex containing IRF3 and CBP/p300. EMBO J. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang X, et al. Influenza A virus NS1 protein prevents activation of NF-kappaB and induction of alpha/beta interferon. J. Virol. 2000;74:11566–11573. doi: 10.1128/jvi.74.24.11566-11573.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wang Y, et al. Rig-I-(−/−) mice develop colitis associated with downregulation of Galphai2. Cell Res. 2007;17:858–868. doi: 10.1038/cr.2007.81. [DOI] [PubMed] [Google Scholar]
- 135.Wen L, et al. Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature. 2008;455:1109–1113. doi: 10.1038/nature07336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Rakoff-Nahoum S, Medzhitov R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science. 2007;317:124–127. doi: 10.1126/science.1140488. [DOI] [PubMed] [Google Scholar]
- 137.Beisswenger C, Coyne CB, Shchepetov M, Weiser JM. Role of p38 MAP kinase and transforming growth factor-signaling in transepithelial migration of invasive bacterial pathogens. J. Biol. Chem. 2007;282:28700–28707. doi: 10.1074/jbc.M703576200. [DOI] [PubMed] [Google Scholar]