

Abstract
We demonstrate the applicability of Sharpless “click” chemistry, specifically Huisgen 1,3-dipolar cycloadditions, as a general methodology for functionalizing surfaces coated with self-assembled monolayers. Ferrocene immobilization was used as our model, and the resulting monolayers were analyzed using traditional surface analytical techniques. Our preliminary results indicate that this reaction proceeds to completion at room temperature in aqueous solvent. The triazole group is a thermally and hydrolytically stable, conjugated linkage. The reactants, acetylenes and azides, are independently stable; they do not react with common organic reagents or with themselves. Thus the potential for this reaction to immobilize a wide range of functionally complex substances on metal surfaces is significant. To our knowledge this is the first report of the use of “click” chemistry to modify a well-defined electrode surface.
Self-assembled monolayers (SAMs) on gold surfaces offer a well-characterized platform on which electron-transfer processes can be studied.1 Surface modification with electroactive monolayers is typically accomplished by synthesizing a molecule with a pendant alkanethiol group followed by coadsorption of the molecule with a more plentiful coadsorbate onto gold to form the SAM. To achieve a specific surface composition, the ratio of derivatized to diluent thiols must be empirically optimized.2 Such preassembly has significant disadvantages as the tethered group increases in size and functional complexity. For instance, altering the length or functionality of the thiol may not be a trivial synthetic modification. Furthermore, there is no guarantee that the molecule will pack to form a well-behaved, electroactive SAM. Chemical modification of a functionalized monolayer offers an attractive alternative to preassembly,3 but the yields are often low and the conditions do not allow for a broad range of applicability. Ideally, one would like an extremely high yielding coupling reaction that uses chemistry and functional groups that are compatible with a wide range of molecules and solvents.
In our search for versatile chemistry to modify surfaces, we were attracted to the recently reported Sharpless “click” chemistry, especially the formation of triazoles through the classic Huisgen 1,3-dipolar cycloaddition.4 Functionalization of electrodes through cycloadditions has already been powerfully demonstrated with Diels–Alder chemistry.5 However, this method is limited by the reactivity of the reagents and the potential reversibility of the Diels–Alder reaction at elevated temperatures.6 Huisgen 1,3-dipolar cycloadditions offer a more general and robust method of modification. Azides and acetylenes are convenient to introduce, do not react among themselves, and show extraordinary tolerance of other functionality. Triazole formation is irreversible and usually quantitative. In addition, this reaction benefits from an extremely mild and regioselective copper(I) catalyst system that is surprisingly indifferent to solvent and pH.7 Although this chemistry has been used to functionalize polymers,8 to our knowledge this is the first report of the use of “click” chemistry to modify a well-defined electrode surface.
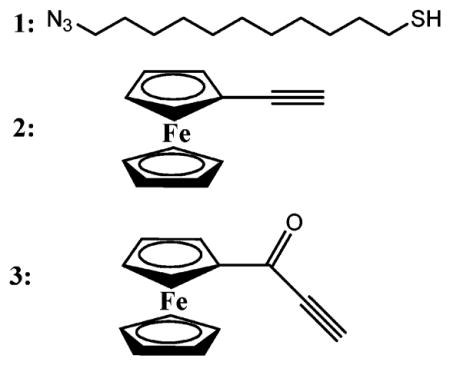
To demonstrate the utility of “click” chemistry, SAMs incorporating various mole fractions of azidoundecanethiol, N3(CH2)11SH (1),9 and a diluent alkanethiol (decanethiol) were formed by placing freshly prepared (111)-oriented gold substrates in dilute ethanolic solutions of the two thiols.10 Once formed, the surfaces were rinsed with ethanol, dried under nitrogen, and immersed at room temperature in 1 mM solutions of either ethynyl ferrocene, FcCCH (2), or propynone ferrocene, FcCOCCH (3) (Figure 1).11 For catalyzed reactions, 1 mol % copper(II) sulfate pentahydrate and 15 mol % sodium ascorbate were added. Exposure to light was kept to a minimum in order to prevent photo-oxidation of the SAM.12 No attempt was made to exclude oxygen.
Figure 1.

Depiction of electrode surface before and after “click” chemistry between azidoundecanethiol, 1, and ferrocenepropynone, 3.
The monolayers before and after reaction with 2 or 3 were characterized with ellipsometry, grazing angle infrared spectroscopy, and electrochemistry using previously described techniques.13 Ellipsometry was used to determine film thicknesses before and after reaction. Derivatization of SAMs formed from thiol solutions containing 30 mol % 1 (χsoln = 0.3) resulted in an average increase in thickness of 2 and 3 Å for 2 and 3,respectively.14 Figure 2 depicts typical cyclic voltammograms before and after the reaction of mixed SAMs (χsoln = 0.3) in 1 mM solutions of 2 or 3 with a solvent comprised of 2:1 by volume of water and ethanol.15 The dependence of the peak current on the scan rate was linear, demonstrating surface immobilization of the redox species.16 In both cases, the positive and negative current peaks occur at nearly the same potential. The standard potential of the SAMs terminated with 2 and 3 were, respectively, 336 and 465 mV versus a Ag/AgCl/1 M KCl reference electrode. Azide-free SAMs were treated with 2 or 3 under identical conditions, but only background charging current, without current peaks, was observed.
Figure 2.

Cyclic voltammograms of mixed 1 monolayers before (dashed) and after derivatization by 2 (dotted) and 3 (solid). Scans done in 1 M HClO4 with a scanning rate of 0.3 V/s. Potentials are measured relative a Ag/AgCl/1 M KCl reference electrode.
The width of the current peaks depends strongly on the distribution of individual ferrocenes.2 The entropically determined, ideal full width at half-maximum (fwhm) is 90 mV for identical, noninteracting ferrocenes.17 Monolayers functionalized with 2 had a fwhm of 114 mV, whereas surfaces functionalized with 3 had a fwhm of 96 mV. We conclude that the ferrocenes are not significantly aggregated and are subject to almost identical redox conditions. We believe the differences between 2 and 3 arise due to the greater solubility of 3 in water and the expected increased distance of 3 from the monolayer due to the carbonyl group. Both of these factors are expected to allow greater screening of the ferrocenes one from the other and prevent monolayer disorder from affecting the ferrocene redox potential, the result being more nearly ideal redox behavior.18
Grazing-angle infrared spectroscopy of mixed monolayers containing 1 revealed the typical methylene and methyl modes as well as a strong absorption at 2101 cm−1. We assign this feature to the asymmetric stretching mode of the organic azide.19 The methylene stretch was located at 2920 cm−1, which indicates dense packing of the monolayer.20 The azide stretch serves as a convenient analytical probe because it can be monitored as a function of reaction time in order to estimate the degree of completion.21 Infrared spectra were taken before and after catalyzed reaction of mixed SAMs in 1 mM solutions of 3 with a solvent mixture of 2:1 water and ethanol (Figure 3a). After 1 h of reaction, the integrated intensity of the azide was diminished. After 3 h of reaction, the intensity diminished to the noise level (Figure 3b). To test whether side reactions were responsible for azide consumption, an identically prepared monolayer was soaked in a 2:1 water and ethanol solution containing the catalyst but without 3. Six hours of immersion resulted in an insignificant change in intensity (Figure 3c).
Figure 3.

Grazing angle infrared spectra of χsoln = 0.3 azidoundecanthiol, 1, mixed monolayers. Monolayers were immersed in aqueous ethanol with catalyst and 1 mM 3 after; 0 h of reaction (A) and 3 h of reaction (B). Monolayers were also immersed for 6 h in a control solution of aqueous ethanol containing the catalyst but without 3 (C).
Ferrocene immobilization correlates with azide disappearance. Cyclic voltammograms of mixed monolayers were taken after 0, 1, and 3 h of reaction (Figure 4). Electrochemically determined ferrocene coverages were 3.46 × 1013 molecules/cm2 after 1 h and 7.78 × 1013 molecules/cm2 after 3 h.22 There was negligible change in the current peak after 3 h, providing further evidence that the reaction had reached completion. Well packed n-alkanethiol SAMs on (111)-oriented gold surfaces have coverages of 4.67 × 1014 molecules/cm2.23 Assuming that the organic azide reacts quantitatively to form the triazole, the total amount of immobilized ferrocene also gives the coverage of 1 in the initial mixed SAM. For a mixed monolayer formed from a deposition solution of χsoln = 0.3, the surface ratio of 1 to diluent was thus 0.17. This suggests that 1 is more soluble in ethanol than the diluent, perhaps due to the polarity of the azido group. Quantifying the surface ratio of 1 eliminates the need for repeated empirical optimization to achieve a desired coverage for different redox molecules. Coverage is dependent on the surface azide and not on the molecule being immobilized, highlighting a distinct advantage of derivatization.
Figure 4.

Cyclic voltammograms of χsol = 0.3 azidoundecanthiol (1) mixed monolayers taken after immersion in aqueous ethanol with catalyst and FcCOCCH (3) for 0 h (solid), 1 h (dashed), and 3 h (dotted). Scans were done in 1 M HClO4 with a scanning rate of 0.3 V/s. Potentials are measured relative to a Ag/AgCl/1 M KCl reference electrode
This click reaction was tolerant of various solvent conditions, working in pure water, aqueous ethanol, and aqueous tert-butyl alcohol. Catalyzed reaction times varied depending on the ferrocene. Thorough reaction of the surface azide in 1 mM solutions of 2 in 2:1 water and ethanol required between 14 and 16 h, whereas for solutions of 3 only 2–3 h was needed under the same conditions. Without catalyst, the cycloaddition proceeds spontaneously for 3, though the time required for completion increases to 9–12 h and with lesser expected regioselectivity. Mixed monolayers immersed for 24 h in a 1 mM solution of 2, without copper catalyst, showed no evidence of reaction. Electron-rich azides are known to react more rapidly with electron poor acetylenes. Thus the greater reactivity of 3 compared to 2 is consistent with the presence of the electron-withdrawing carbonyl.24
In summary, “click” chemistry applied to SAMs offers a mild and convenient method to modify well-defined electrode surfaces. This surface chemistry is extremely specific and high yielding. The thermally and hydrolytically stable triazole offers a robust, conjugated link for coupling a wide array of molecules to SAMs on electrodes. Currently, we are pursuing the applicability of this reaction for metalloporphyrin functionalization of electrodes.
Supplementary Material
Acknowledgments
This material is based upon work supported by the National Science Foundation under Grant No. CHE0131206 and a Stanford Graduate Fellowship. We acknowledge insightful discussions with Todd Eberspacher, Christopher Sunderland, and Richard Decreau.
Footnotes
Supporting Information Available: Detailed experimental procedures and a discussion of monolayer charging currents. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For some recent examples: Morita T, Kimura S. J Am Chem Soc. 2003;125:8732–8733. doi: 10.1021/ja034872n.Xiao Y, Patolsky F, Katz E, Hainfeld J, Willner I. Science. 2003;299:1877–1881. doi: 10.1126/science.1080664.Yamada H, Imahori H, Nishimura Y, Yamazaki I, Ahn TK, Kim SK, Kim D, Fukuzumi S. J Am Chem Soc. 2003;125:9129–9139. doi: 10.1021/ja034913f.Long Y, Li C, Sunderland TC, Chahma M, Lee JS, Kraatz H. J Am Chem Soc. 2003;125:8724–8725. doi: 10.1021/ja034684x.
- 2.Chidsey CED, Bertozzi CR, Putvinski TM, Mujsce AM. J Am Chem Soc. 1990;112:4301–4306. [Google Scholar]
- 3.(a) Yan L, Huck WT, Zhao X, Whitesides GM. Langmuir. 1999;15:1208–1214. [Google Scholar]; (b) Frey BL, Corn RM. Anal Chem. 1996;68:3187–3193. [Google Scholar]
- 4.Kolb HC, Finn MG, Sharpless KB. Angew Chem. 2001;113:1198–1220. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 5.Yousaf MN, Mrksich M. J Am Chem Soc. 1999;121:4286–4287. [Google Scholar]
- 6.As specific examples, the β carbon of benzoquinone reacts readily with nucleophiles and cyclopentadiene is prone to spontaneous dimerization at room temperature. The Diels–Alder adduct between cyclopentadiene and benzoquinone has been observed to slowly revert at room temperature. See: Kemp DS, Vellaccio F. Organic Chemistry. Worth Publishers Inc; New York: 1980. p. 758.p. 1108.Pool BR, White JM. Org Lett. 2000;22:3505–3507. doi: 10.1021/ol006553w.
- 7.Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem. 2002;114:2708–2711. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; Angew Chem, Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 8.Fazio F, Bryan MC, Blixt O, Paulson JC, Wong C. J Am Chem Soc. 2002;124:14397–14402. doi: 10.1021/ja020887u. [DOI] [PubMed] [Google Scholar]
- 9.Synthesis adapted from: Shon Y, Kelly KF, Halas NJ, Lee TR. Langmuir. 1999;15:5329–5332.See Supporting Information for full experimental details.
- 10.Porter MD, Bright TB, Allara DL, Chidsey CED. J Am Chem Soc. 1987;109:3559–3568. [Google Scholar]
- 11.Ferroceneacetylenes were synthesized following previously published literature procedures. Rosenblum M, Brawn N, Papenmeier J, Applebaum M. J Organomet Chem. 1966;6:173–180.Barriga S, Marcos CF, Riant O, Torroba T. Tetrahedron. 2002;58:9785–9792.
- 12.Tarlov M, Newman J. Langmuir. 1992;8:1398–1405. [Google Scholar]
- 13.(a) Chidsey CED, Loicacono D. Langmuir. 1990;6:682–691. [Google Scholar]; (b) Offord DA, Sachs SB, Ennis MS, Eberspacher TA, Griffin JH, Chidsey CED, Collman JP. J Am Chem Soc. 1998;120:4478–4487. [Google Scholar]
- 14.Δδ values were −0.3 and −0.4 for 2 and 3, respectively.
- 15.Where χ is equal to the mole fraction of azidoundecanethiol compared to total thiol in the deposition solution.
- 16.Bard AJ, Faulkner LR. Electrochemical Methods: Fundamentals and Applications. Wiley; New York: 2001. [Google Scholar]
- 17.Chidsey CED. Science. 1991;251:919–922. doi: 10.1126/science.251.4996.919. [DOI] [PubMed] [Google Scholar]
- 18.(a) Rowe GK, Creager SE. Langmuir. 1991;7:2307–2312. [Google Scholar]; (b) Calvente JJ, Andreau R, Molero M, Perez G, Dominguez M. J Phys Chem B. 2001;105:9557–9568. [Google Scholar]
- 19.The location of the asymmetric vibration of the azido group is located between 2170 and 2080 for nearly all organic azides. Ramachandra CN, Chao TS, Hoffman CWW. Anal Chem. 1957;29:916–918.
- 20.Snyder RG, Strauss HL, Elliger CA. J Phys Chem. 1982;86:5145–5150. [Google Scholar]
- 21.Wollman E, Kang D, Frisbie CD, Lorkovic IM, Wrighton MS. J Am Chem Soc. 1994;16:4395–4404. [Google Scholar]
- 22.An analysis of the electrochemical data reveals that the pseudo-first-order rate constant of product accumulation appears to increase such that the reaction is complete at 3 h even though the degree of completion at 1 h predicts that a longer time would be required. This suggests an increase in catalyst activity as the reaction proceeds.
- 23.Strong L, Whitesides GM. Langmuir. 1988;4:546–558. [Google Scholar]
- 24.Lwowski W. In: 1,3 Dipolar Cycloaddition Chemistry. Chapter 5 Padwa A, editor. Vol. 1. Wiley; New York: 1984. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.