

Abstract

The human mitotic kinesin Eg5 represents a novel mitotic spindle target for cancer chemotherapy. We previously identified S-trityl-l-cysteine (STLC) and related analogues as selective potent inhibitors of Eg5. We herein report on the development of a series of 4,4,4-triphenylbutan-1-amine inhibitors derived from the STLC scaffold. This new generation systematically improves on potency: the most potent C-trityl analogues exhibit Kiapp ≤ 10 nM and GI50 ≈ 50 nM, comparable to results from the phase II clinical benchmark ispinesib. Crystallographic studies reveal that they adopt the same overall binding configuration as S-trityl analogues at an allosteric site formed by loop L5 of Eg5. Evaluation of their druglike properties reveals favorable profiles for future development and, in the clinical candidate ispinesib, moderate hERG and CYP inhibition. One triphenylbutanamine analogue and ispinesib possess very good bioavailability (51% and 45%, respectively), with the former showing in vivo antitumor growth activity in nude mice xenograft studies.
Introduction
The mitotic kinesin Eg5 is a promising potential therapeutic target for small molecule inhibitors and has been validated in preclinical in vivo models of cancer to be essential for the progression of mitosis. Eg5 is instrumental in forming the bipolar spindle by separation of the duplicate spindle poles in the early prometaphase stage of mitosis.1,2 Several Eg5 and other mitotic kinesin inhibitors are at preclinical and clinical stages (reviewed in ref (3)), including ispinesib, one of the most advanced Eg5 inhibitors currently in multiple phase II clinical trials.4−6 Previously, we identified S-trityl-l-cysteine (STLC) as a potent and selective inhibitor of human Eg5 (Figure 1, 1).7,8 STLC and its derivatives induce mitotic arrest by activation of the mitotic checkpoint, which eventually leads to apoptosis in certain tumor cell lines by both caspase-dependent and -independent pathways.9,10 Furthermore, the p-methoxy analogue 5 has recently been shown to significantly prolong survival in in vivo experiments using bladder and prostate cancer xenografts in nude mice.11,12 As with most other allosteric Eg5 specific inhibitors, STLC binds in an allosteric pocket formed by helix α2/loop L5 and helix α3.13,14 Initial structure–activity relationship (SAR) studies performed prior to detailed structural analysis of Eg5–STLC complexes resulted in the development of analogues with GI50 values in the range of ∼200 nM (Figure 1, 2 and 3).15,16 Recent investigations have identified novel analogues with improved potency in cell-based assays.17,18
Figure 1.

STLC and representative examples of previously reported analogues.
We have determined previously the structure of Eg5 complexed with STLC (1) and identified the key binding interactions in the inhibitor-binding pocket (Figure 2A and 2B).14 The three phenyl rings of the trityl headgroup are situated in a predominantly hydrophobic core region of the allosteric inhibitor-binding site, in the three pockets denoted P1–P3 (Figure 2B), while the cysteine tail forms several important hydrogen bond interactions with Eg5, in agreement with those observed in another Eg5–STLC complex by Kim et al.19 Prior SAR studies had demonstrated that STLC analogues with a lipophilic substituent on one phenyl ring generally displayed increased potency,15,16,18 which we confirmed structurally by solving the crystal structure of Eg5 with an STLC analogue incorporating a p-chlorophenyl substituent in the hydrophobic P3 region (Figure 2B).17 Crystal structures of other ligands bound to Eg5 have revealed that small polar substituents like the m-hydroxy group in (S)-monastrol and its analogues interact with residues lining the P2 pocket, such as Glu118, Arg119, and Ala133.20,21 Crystallographic studies on STLC and its analogues have also established that the phenyl ring in P1 π-stacked with the Tyr 211 residue and exposed its leading edge to bulk solvent near Glu215 (Figure 2A). These data from both structural observations and previous SAR studies provided the foundation to investigate the optimal lipophilic and polar substituents on the trityl system to improve potency against Eg5.
Figure 2.

Eg5–inhibitor structures showing important hydrophobic, aromatic, and hydrogen bond (represented by broken lines) interactions between the protein and ligands. Structural water molecules are represented by red spheres. (A) STLC (1), colored green, in the Eg5 inhibitor-binding pocket. (B) Surface diagram showing an STLC analogue (gray) with a para-substituted chlorine on one phenyl ring, in the inhibitor-binding pocket. The chlorine (green) sits in the hydrophobic P3 pocket. (C) Stereoplot showing 25 (blue) in the inhibitor-binding pocket. (D) Stereoplot showing (S)-29 (pink) and (R)-29 (yellow) in the inhibitor-binding pocket.
In any hit-to-lead optimization process, improving potency against the target protein must be pursued in tandem with optimizing the physicochemical properties of the ligand, as physicochemical properties have profound influences on absorption, distribution, and elimination and ultimately determine drug efficacy.22 Our optimization strategy therefore needed to consider the potential metabolic liability of the thioether linkage to the trityl headgroup and the zwitterionic nature of the cysteine amino acid group in 1. A parallel approach was adopted for SAR investigations: we explored a range of substituents on the trityl system by thioetherification of tertiary alcohols to improve potency against Eg5 and in the case of polar substituents act as alternatives to the zwitterionic carboxylic acid in the cysteine tail. Second, isosteric replacements of the sulfur heteroatom (N, O, CH2) were investigated, with the ultimate aim of transferring the optimal trityl head from the STLC series to the sulfur-isosteric analogues.
We herein report on this new series of STLC analogues, the triphenylbutanamines. They display strong in vitro basal Eg5 inhibitory activity (≤10 nM) and potently inhibit a number of cancer tumor cell lines (≤100 nM) and improve on comparable STLC derivatives. The in vitro and in vivo pharmacokinetic properties of these analogues are promising and warrant further development, and initial in vivo studies with one compound in nude mice bearing a cell lung cancer explant xenograft LXFS 538 exhibited tumor stasis.
Results and Discussion
Synthesis
Novel compounds were synthesized as follows. O-Trityl analogue 6 was prepared by coupling of N-(2-hydroxyethyl)acetamide with trityl chloride followed by acetyl hydrazinolysis (Supporting Information, Scheme S7). N-Trityl analogues 7 and 8 were prepared via substitution of trityl chloride with FMOC-protected diamines (Scheme 1). Thioethers were prepared from tertiary alcohols in trifluoroacetic acid (Scheme 2)58 or as previously described with boron trifluoride and acetic acid.15 Intermediate trityl alcohols were furnished by organometallic mediated reduction of substituted benzophenones and methyl esters or alternatively lithiated aryl bromides, which allowed introduction of a wider range of substituents to the trityl headgroup (Scheme 2). Compounds 3–5 were obtained from the National Cancer Institute (NCI).
Scheme 1.

Reagents and conditions: (i) TMSCl, CH2Cl2/THF (5:1), reflux, 2 h; (ii) TrCl, NEt3, CH2Cl2, rt, 1 h; (iii) piperidine (20% in MeOH), rt, 2 h; (iv) TrCl, NEt3, DMF, rt, 3h.
Scheme 2. General Route for Synthesis of Thioethers.

Reagents and conditions: (i) n-BuLi, −78 °C, 1 h; (ii) benzophenone, THF, −78 °C 4–6 h, then rt overnight; (iii) cysteamine hydrochloride or l-cysteine, TFA, rt, 3 h.
A more elaborate methodology was required for creation of the C-trityl series, exemplified by compounds 9 and 10. To introduce the carbon scaffold, the lithium alkoxide salt of trityl alcohol was subjected to a mild allylation procedure employing iron trichloride and allyltrimethylsilane (Scheme 3).23 The alkene 55 was then oxidized by hydroboration–oxidation and subsequent Swern oxidation to afford the aldehyde 57, which was converted into the racemic protected α-aminonitrile intermediate 58 in a variation of the Strecker synthesis employing Montmorillonite KSF clay.24 After the nitrile was hydrolyzed, a mild ammonium formate based deprotection of the benzylamine 59 afforded C-trityl analogue 9 as a racemic mixture, in an overall yield of 45%.25 This route also allowed access to racemic 29 and 30. Triphenylbutan-1-amine analogues without the carboxylic acid, exemplified by 10, were synthesized by conversion of the intermediate alcohol 56 to the azide 60, with consequent Staudinger reduction to afford the target compound 10 in an overall yield of 49% (Scheme 4).
Scheme 3.

Reagents and conditions: (i) n-BuLi, CH2Cl2, rt, 30 min; (ii) allyltrimethylsilane, FeCl3, rt, 6 h; (iii) NaBH4, conc H2SO4 in Et2O, diglyme, rt, 3.5 h, then 75 °C, 1.5 h; (iv) 30% aq H2O2, 3 M aq NaOH, rt, 6.5 h; (v) Dess–Martin periodinane, CH2Cl2, rt, 3 h; (vi) benzylamine, TMSCN, Montmorillonite KSF clay, CH2Cl2, rt, 2 h; (vii) 6 M HCl in dioxane, reflux, 48 h; (viii) propylene oxide, EtOH, reflux, 30 min; (ix) HCOONH4, 10% Pd/C, MeOH, 80 °C, 1 h.
Scheme 4.

(i) MsCl, pyridine, rt, overnight; (ii) NaN3, DMF, microwave, 175 °C, 10 min; (iii) Ph3P, THF/H2O (10:1), 60 °C, overnight.
Determination of Stereochemistry
The racemic mixtures of the C-trityl amino acids rac-9, rac-29, and rac-32 were separated by semipreparative chiral HPLC, and their absolute stereochemistry was determined by application of a modification of Marfey’s method.26,27 This involved derivatization of the racemic mixture with the chiral agent Nα-(2,4-dinitro-5-fluorophenyl)-l-valinamide (FDVA), followed by analysis of the HPLC retention times of the resulting diastereomers (Figure 3B(i)). In these diastereomers, the dinitrobenzene moiety of FDVA is in a planar conformation with the diamino substituents adopting a cis- or trans-arrangement relative to this (Figure 3A(ii)).27 The diastereomer with the two more hydrophobic substituents in a cis-arrangement undergoes stronger interactions with the reverse stationary phase and is retained for longer than the corresponding diastereomer in the trans-configuration, which enables a nonempirical determination of the stereochemistry of amino acids and chiral amines.27,28 Applying this method to the resolved enantiomers of 32, the first diastereomer to elute was 32-1-FDVA. We therefore assigned 32-1 as the (S) configuration and 32-2 as the (R) enantiomer. As Marfey’s method is an indirect method of determining absolute stereochemistry, to ensure the diastereomers 32-1-FDVA and 32-2-FDVA eluted in the order that would be expected under the conditions employed, STLC 1 and STDC were evaluated by the same methodology. STLC-FDVA was eluted first, as would be expected from their relative stereochemistries.27 Further investigations with d- and l-cysteine and d- and l-phenylalanine also proved consistent with designated assignments (Figure 3B). Further assignments were based on the results of optical rotation and circular dichroism measurements when compared to the assigned enantiomers.
Figure 3.
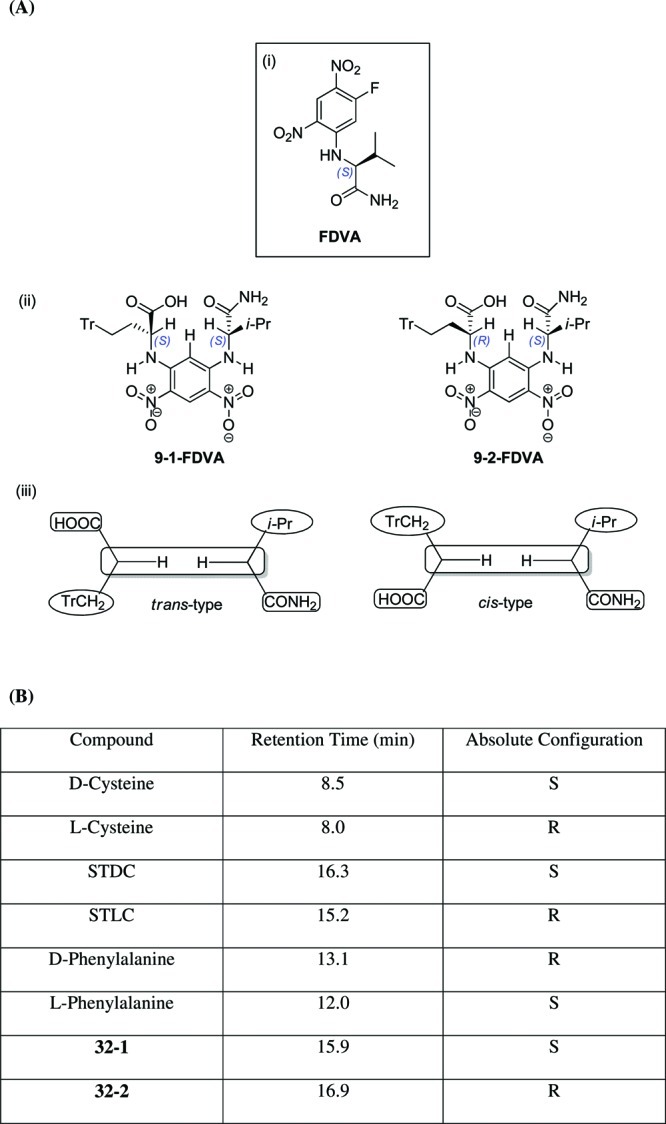
Stereochemical determination by Marfey’s method. (A) (i) Structure of Nα-(2,4-dinitro-5-fluorophenyl)-l-valinamide (FDVA); (ii) derivatised diastereoisomers 29-1-FDVA and 29-2-FDVA; (iii) schematic representations of proposed cis- and trans-like diastereomer conformations.27 (B) Retention times of 32-1-FDVA and 32-2-FDVA and corresponding standard amino acids. The absorption peak at ∼18.50 corresponds to elution of unreacted FDVA.
Optimizing the Amino Acid Tail
Replacements of the cysteine sulfur atom of STLC were investigated in order to establish its overall effect on potency and ADME characteristics (6–10, Table 1). A trityl group is often used as a protecting group in organic synthesis that is removed under acidic conditions; hence, it may be removed from STLC on encountering the acidic environment of the stomach if administered orally. Using LC–HRMS, we established that STLC was stable at pH 1.0 over 24 h (Supporting Information, Table S1 and Figure S1). On the other hand, corresponding N-trityl derivatives 7 and 8 not only displayed weak inhibition of the basal ATPase activity of Eg5 compared with STLC but also were completely hydrolyzed at pH 1.0 (t1/2 of ≤2.9 and 7.0 h, respectively). The oxygen analogue 6 was also a weak inhibitor of Eg5 and decomposed after 16 h at pH 1.0 (t1/2 = 2.5 h). We did not investigate further whether the lower activity of these O-trityl and N-trityl analogues was due to their instability in the assay medium or the reduced pKa of the primary amine and its consequent effect on the proportion of molecules ionized to facilitate interaction with the protein.
Table 1. Heteroatom Analogues of STLCa.

| compd | X | R | inhibition of basal ATPase activity, Kiapp (nM) | ligand efficiency | K562 cells, GI50 (nM) |
|---|---|---|---|---|---|
| 1 (STLC) | S | (R)-CO2H | 135.9 ± 20.5 | 0.36 | 1452 ± 76 |
| 2 | S | H | 245.5 ± 51.2 | 0.39 | 2286 ± 213 |
| 6 | O | H | 2157.6 ± 342.2 | 0.34 | nd |
| 7 | NH | (S)-CO2H | 3018.5 ± 398.5 | 0.29 | nd |
| 8 | NH | H | 2659.4 ± 295.6 | 0.33 | nd |
| rac-9 | CH2 | CO2H | 311.6 ± 53.9 | 0.34 | 865 ± 131 |
| (S)-9 | CH2 | (S)-CO2H | 416.5 ± 64.2 | 0.33 | 2065 ± 168 |
| (R)-9 | CH2 | (R)-CO2H | 173.5 ± 24.5 | 0.35 | 776 ± 26 |
| 10 | CH2 | H | 214.7 ± 30.1 | 0.40 | 577 ± 61 |
nd = not determined.
A thioether functionality is a site for metabolism via S-oxidation, and the enzyme phenylalanine monoxygenase has recently been identified as a possible source for the conversion of the mucoregulatory agents S-methyl-l-cysteine and S-carboxymethyl-l-cysteine to the corresponding inactiveS-oxides.29 An obvious isostere for sulfur that would not be susceptible to acid hydrolysis or S-oxidation is a methylene group. The carbon analogues 9 and 10 were prepared and shown to be stable for 5 days at pH 1.0. Furthermore, they had activity similar to that of STLC against Eg5, and when 9 was resolved, (R)-9 was the more potent enantiomer. These compounds also demonstrated a systematic improvement in the K562 human leukemia cell based assays compared to their sulfur counterparts 1 and 2, and (R)-9 reproduced its higher activity in the cellular assay compared with its enantiomer. Interestingly, in the Eg5 assay, the carbon analogue without the carboxylic acid in the tail (10) showed improved activity over the amphoteric form (9), a reversal of the trend observed with sulfur-based analogues 1 and 2.
One of the key interactions observed in the cocrystallized STLC–Eg5 structure was the hydrogen bond network between the primary ammonium group of the cysteine tail and the backbone carbonyl of Gly117 and the side chain of Glu116. To establish whether this was being reproduced in the C-trityl analogues 9 and 10, derivatives were prepared in which the length of the amine tail was varied incrementally (Table S2). A 20-fold and 8-fold decrease in Eg5 inhibitory activity was observed for the propyl-1-amine and pentyl-1-amine analogues, respectively, suggesting that the optimal network is achieved with the four-carbon butyl-1-amine chain (10, Table S2). Corresponding compounds were also prepared for the N-trityl analogue 8 to ensure that the decreased length of the primary amine tail was not the reason for the weak inhibitory activity observed. In this case, an ethylene unit proximal to the N-trityl unit again proved to be the optimal chain length (8, Table S2). These chain length manipulations concur with the earlier observations made for the alkyl chain in STLC.15
Having established that carbon was an appropriate isostere in the tail for sulfur and the optimum length was a butyl chain, we next evaluated triphenylbutan-1-amine analogues featuring functional group replacements and/or modifications to the alkyl chain, including those prepared as intermediates (55–60, Table S3). However, all proved to be inactive. Replacement of the primary amine with the secondary methylamine resulted in a marked dropoff in activity, reinforcing the importance of a three hydrogen bond network between the ammonium group and the glutamate and glycine residues in the pocket for efficient inhibition (S50, Table S3). The bulkier secondary benzylamine derivatives rac-58 and rac-59 displayed further reductions in activity. Replacement of the primary amine with a primary alcohol (56, Table S3) abolished activity, which again reinforces the need for three hydrogen bond donors and a role for an ionic interaction between ligand and protein. Additional N-trityl analogues incorporating cyclic piperazines in the amino acid tail region displayed no notable activity in comparison to either STLC 1 or 9 (data not shown).
Optimization of the Trityl Headgroup
The most potent STLC derivatives prepared previously incorporated a lipophilic para-substituent in one phenyl ring of the trityl moiety (Figure 1). We have recently solved the structure of an STLC derivative with a p-chloro on one phenyl ring, which, as expected, occupied the environment of the P3 pocket formed by Ile136, Pro137, Leu160, Leu214, and Phe239 (Figure 2B).18 A series of S-trityl thioethers were prepared to investigate this environment further. Compounds exhibiting Eg5 inhibition Kiapp ≤ 100 nM were also evaluated in growth inhibition assays using K562 cells.
While the o-chloro substituent of 11 significantly reduced activity against Eg5 compared with STLC 1, a m-chloro (13) produced an approximately 2-fold improvement in Kiapp for basal inhibition of Eg5 (Table 2). Further thioethers incorporating meta-substituents of varying electronic, hydrophobic, and steric character were synthesized to optimize this interaction, with alkyl groups particularly beneficial for Eg5 inhibitory activity. The stepwise improvement in activity from 16 (Me, Kiapp = 80 ± 24 nM), 19 (n-Pr, Kiapp = 26.9 ± 11 nM), 18 (i-Pr, Kiapp = 10.3 ± 3.4 nM) to 17 (Et, Kiapp = 5.9 ± 2.3 nM) illustrates that the interaction is very sensitive to steric bulk at this position and most likely reflects an interaction between the proximal lipophilic side chain of Leu214 and these alkyl substituents. Furthermore, electronic influences on the attached phenyl ring are also important: 20 with a m-CF3group, which can be considered as similar in size to a methyl substituent but electron-withdrawing, is 4-fold less active than 16.In our evaluation of all the crystal structures of STLC and its analogues with Eg5, we observed in the P3 pocket a C–H···π interaction between the phenyl group of STLC and the side chain of Leu214 (Figure 2A). This interaction is mediated via the π-electron clouds of the phenyl ring, so changes in its distribution will affect the strength of its interaction with nearby residues. Analysis of these results using a Craig plot substantiated the optimal meta-substituents as both hydrophobic and electron-donating (Figure 3).30
Table 2. STLC Analogues with Modified Trityl Ringsa.

| compd | X | R1 | R2 | inhibition of basal ATPase activity, Kiapp (nM) | ligand efficiency | K562 cells, GI50 (nM) |
|---|---|---|---|---|---|---|
| 11 | S | 2-Cl | (R)-CO2H | 1783.9 ± 384.0 | 0.29 | nd |
| 12 | S | 3-F | H | 377.4 ± 38.4 | 0.36 | nd |
| 13 | S | 3-Cl | (R)-CO2H | 89.9 ± 18.5 | 0.36 | 2404 ± 222 |
| 14 | S | 3-Cl | H | 297.8 ± 60.0 | 0.37 | 1045 ± 42 |
| 15 | S | 3-Br | H | 293.9 ± 61.8 | 0.37 | nd |
| 16 | S | 3-Me | H | 80.0 ± 23.9 | 0.40 | 698 ± 115 |
| 17 | S | 3-Et | H | 5.9 ± 2.3 | 0.45 | 680 ± 84 |
| 18 | S | 3-i-Pr | H | 10.3 ± 3.4 | 0.42 | 581 ± 68 |
| 19 | S | 3-n-Pr | H | 29.6 ± 11.0 | 0.39 | 1760 ± 124 |
| 20 | S | 3-CF3 | H | 352.7 ± 27.9 | 0.33 | nd |
| 21 | S | 3-OMe | H | 149.8 ± 18.5 | 0.37 | nd |
| 22 | S | 3-SMe | H | 520.4 ± 87.6 | 0.34 | 1474 ± 330 |
| 23 | S | 3-OCF3 | H | 27.8 ± 5.0 | 0.37 | 1518 ± 164 |
| 24 | S | 3-COMe | (R)-CO2H | 519.8 ± 102.0 | 0.30 | 964 ± 82 |
| 25 | S | 3-COMe | H | 185.8 ± 30.4 | 0.35 | 706 ± 47 |
| 26 | S | 4-COMe | (R)-CO2H | 271.7 ± 36.6 | 0.31 | 4266 ± 274 |
| 27 | S | 4-COMe | H | 51.0 ± 21.6 | 0.38 | 705 ± 77 |
| 28 | C | 3-Cl | H | 120.6 ± 20.7 | 0.35 | 596 ± 68 |
| rac-29 | C | 3-Me | CO2H | 12.2 ± 3.8 | 0.40 | 73 ± 3 |
| (S)-29 | C | 3-Me | (S)-CO2H | 11.1 ± 3.9 | 0.40 | 128 ± 15 |
| (R)-29 | C | 3-Me | (R)-CO2H | 6.4 ± 3.9 | 0.41 | 91 ± 9 |
| 30 | C | 3-Me | H | 8.8 ± 1.8 | 0.46 | 200 ± 16 |
| 31 | C | 4-Cl | H | 16.2 ± 3.1 | 0.44 | 337 ± 28 |
| rac-32 | C | 4-Me | CO2H | 7.5 ± 1.7 | 0.41 | 96 ± 5 |
| (S)-32 | C | 4-Me | (S)-CO2H | 16.7 ± 3.0 | 0.39 | 149 ± 6 |
| (R)-32 | C | 4-Me | (R)-CO2H | 5.4 ± 1.7 | 0.42 | 82 ± 4 |
| 33 | C | 4-Me | H | 16.4 ± 1.9 | 0.44 | 219 ± 21 |
nd = not determined.
In the cellular assay, submicromolar activity was evident for alkyl (16–18) and acetate substituents (24, 25, 27), which demonstrated an approximate 2-fold improvement in this assay versus the unsubstituted S-trityl benchmarks (1 and 2). Paradoxically, 19 with an n-Pr substituent exhibited a dramatic loss in cellular activity, probably as a result of limited aqueous solubility (turbidimetric solubility of 3.75 μM). Introduction of a carboxylic acid into the tail of 27 to produce 26 resulted in a 4-fold reduction in Eg5 activity, comparable to that seen with 25 and 24, but produced a much more pronounced reduction in activity in the K562 cell line, which was a reversal of the pattern seen with 1 and 2. Insufficient related compounds were prepared in this series to determine if this trend was consistent. From the comparable S-trityl inhibitors prepared, two pairs with modified trityl moieties had better activity in the Eg5 basal assay when prepared without the carboxyl acid (24 and 25; 26 and 27), while all modified S-trityl analogues with the carboxylic acid moiety exhibited lower activity in the cellular assay than their corresponding counterparts without the carboxylate (13 and 14; 24 and 25; 26 and 27). Clearly, further investigations are needed before definitive conclusions can be made regarding the SAR role of the carboxylic acid with respect to Eg5 inhibitory and cellular activity.
The positioning of the P1 ring extending toward the solvent front and the nearby Glu215 residue afforded an additional optimization opportunity to introduce more polar substituents (Figure 2A). A series of analogues that incorporated polar H-bond acceptor/donors on one phenyl ring was synthesized to target bulk solvent interactions and the proximal acidic residue. It was envisaged that these substituents could attenuate the highly lipophilic nature of the headgroup and produce compounds with physicochemical properties (e.g., log D) compatible with predicted favorable ADME profiles.22,31S-Trityl thioethers were again prepared to investigate these potential interactions (Table 3), and only one compound, the m-hydroxy STLC analogue 35, demonstrated any significant improvement in basal Eg5 inhibitory activity over 1. This is likely due to formation of hydrogen bonds with the main chain nitrogen of Leu132 and the side chain of Arg119 in the P2 pocket, as observed in the crystal structures for dihydropyrimidine–Eg5 ternary complexes.20,21 The direct analogue 36, which differed only by the absence of the carboxylic acid in the tail, while 4-fold less potent in the basal assay, was 5-fold more potent than 35 in the cellular assay. This difference can be attributed to the difference in cell permeability resulting from the presence of the carboxylic acid. Evaluation of the passive in vitro cellular permeability by parallel artificial membrane permeability assays (PAMPA) demonstrated 36 to have higher permeability (Papp = (16.7 ± 2.6) × 10–6 cm s–1), with no permeability recorded for 35. All other compounds in this series displayed only moderate inhibition of the basal activity of Eg5 but generally similar cellular activity to 2, demonstrating a wide tolerability of polar substrates. A modest improvement in K562 cellular activity was observed in the meta-primary and secondary amides 42 and 43, although their Eg5 inhibitory activity was reduced 2-fold and 8-fold, respectively. Likewise, the p-CH2OH containing compound 47 had improved cellular activity but reduced Eg5 inhibitory activity. This may suggest that their cellular activity is being expressed through an alternative mechanism to Eg5 inhibition. The meta-primary amide 41, which has the carboxylic acid group in the tail, has similar Eg5 inhibitory activity, compared to 42, but significantly reduced cellular activity (32-fold less than 42 and 18-fold less than STLC), probably due to a very low log D (log D7.4 = 1.13 for 41; log D7.4 = 1.41 for 42). Measurement of the cell permeability by PAMPA assays found 42 to possess high permeability of (Papp = (42.8 ± 15.0) × 10–6 cm s–1), while 41 had no membrane diffusion detected when evaluated by this method. Thus, while the meta-primary amide and secondary amides of 42 and 43 in the trityl headgroup may act as a suitable alternatives to the zwitterionic carboxylate in the cysteine tail for enhancing the physicochemical balance of STLC analogues, the dual presence of the trityl amide modification and carboxylate appears to be incompatible with effective cell penetration. To conclude, overall improvements to both basal and cellular activity were associated with hydrophobic rather than hydrophilic para- or meta-substituents.
Table 3. STLC Analogues with Modified Trityl Ringsa.

| compd | R1 | R2 | inhibition of basal ATPase activity, Kiapp (nM) | ligand efficiency | K562 cells, GI50 (nM) |
|---|---|---|---|---|---|
| 34 | 2-OH | CO2H | 1978.5 ± 587.3 | 0.29 | nd |
| 35 | 3-OH | CO2H | 48.8 ± 22.0 | 0.37 | 2559 ± 302 |
| 36 | 3-OH | H | 200.3 ± 51.9 | 0.38 | 555 ± 121 |
| 37 | 3-CN | H | 450.6 ± 197.4 | 0.35 | 2128 ± 108 |
| 38 | 3-CH2NH2 | H | 838.2 ± 164.1 | 0.33 | 2018 ± 244 |
| 39 | 3-CH2NHCOMe | H | 829.2 ± 100.5 | 0.30 | 2133 ± 135 |
| 40 | 3-CO2H | H | 990.6 ± 156.9 | 0.31 | 2138 ± 241 |
| 41 | 3-CONH2 | CO2H | 329.9 ± 49.2 | 0.30 | 16749 ± 6112 |
| 42 | 3-CONH2 | H | 419.7 ± 38.8 | 0.33 | 802 ± 51 |
| 43 | 3-CONHMe | H | 887.8 ± 74.9 | 0.31 | 982 ± 72 |
| 44 | 3-CONMe2 | H | 6055.6 ± 1123.0 | 0.25 | 2831 ± 171 |
| 45 | 3-SO2Me | H | 2089.6 ± 246.6 | 0.29 | 2559 ± 180 |
| 46 | 4-CN | H | 432.4 ± 91.2 | 0.35 | 2178 ± 149 |
| 47 | 4-CH2OH | H | 311.2 ± 31.8 | 0.35 | 783 ± 50 |
| 48 | 4-CH2NH2 | H | 2942.4 ± 782.8 | 0.30 | 2594 ± 191 |
| 49 | 4-CH2NHCOMe | H | 1721.3 ± 294.5 | 0.28 | 2904 ± 150 |
| 50 | 4-CONH2 | H | 3228.6 ± 447.4 | 0.29 | 4335 ± 341 |
| 51 | 4-CONHMe | H | 2030.8 ± 671.6 | 0.29 | 3954 ± 293 |
| 52 | 4-CONMe2 | H | 1526.7 ± 359.7 | 0.28 | 2911 ± 271 |
| 53 | 4-SO2Me | H | 1212.1 ± 179.9 | 0.30 | 2735 ± 482 |
nd = not determined.
Combining the Optimized Head and Tail Moieties into the Carbon Scaffold
After establishment of the SAR of the trityl headgroup in thioethers and the optimal characteristics of the tail group with isosteric butan-1-amine, the appropriate para- or meta-substituted trityl groups were next translated into the C-trityl series (28–33, Table 2). We found a corresponding systematic increase in activity against the target Eg5 protein when evaluated in both basal in vitro and cellular growth inhibition assays; an approximate 3-fold increase was evident for comparable analogues 14 and 28 and 16 and 30 in the K562 assay. The most potent analogues prepared were racemic mixtures of 29 and 32, which had the 2-aminobutanoic acid tail and the C-trityl scaffold with a m- or p-methyl substituent on one phenyl ring, respectively (EC50 of 73 ± 3 and 95 ± 5 nM). The Kiapp and EC50 of the resolved enantiomers were also determined (Table 2). The more active (R)-enantiomers (R)-29 and (R)-32 were low nanomolar inhibitors of Eg5 and have estimates of Kiapp values of 6.4 ± 3.9 and 5.4 ± 1.7 nM, respectively, which are close to the reported Kiapp of 2 nM for ispinesib.4 The corresponding butan-1-amine analogues (30 and 33) possess similarly low Kiapp estimates (8.8 ± 1.8 and 16.4 ± 1.9 nM, respectively), indicating that the presence of the carboxylic acid is not essential for inhibition. On the other hand the carboxylic acid containing (R)-29 and (R)-32 analogues are approximately 2-fold more active in cell-based growth inhibition assays. This reverses the trend observed with the sulfur analogues, where removal of the carboxylic acid from the tail of compounds with substituted trityl head groups in general improved cellular activity.
The ligand efficiencies, which depend on protein targets, were calculated for all the compounds based on their Kiapp values (Tables 1–3) to enable useful comparison between compounds with a range of molecular weights and activities.32,33 The compounds without the carboxylic acid groups generally showed better ligand efficiencies than their counterparts with the carboxylic acid groups: compounds 29 and 32 had ligand efficiencies of 0.4 and 0.41, whereas 30 and 33 showed higher ligand efficiency of 0.46 and 0.44, respectively. Ispinesib has a calculated ligand efficiency of 0.32 based on the reported Kiapp (1.7 ± 0.1 nM),4 indicating that our smaller triphenylbutanamine scaffold possesses greater enhancement potential.
Next, we examined the most potent analogues together with selected control compounds, for the inhibition of the MT-stimulated Eg5 ATPase activity (Table 5). This assay, with a salt concentration of 150 mM, performed in the presence of MTs, is closer to physiological conditions and allows Kiapp to be determined in the presence of ∼5 nM Eg5 instead of ∼80 nM employed for the basal assay, resulting in more reliable values, in particular for the more potent analogues. The estimates for Kiapp for both the inhibition of the basal and MT-stimulated ATPase activities are in good agreement. The most potent compounds are ispinesib with Kiapp of 2.5 ± 1.2 nM followed by (R)-32 with 3.4 ± 0.9 nM, which is further confirmed in cell-based assays.
Table 5. Testing of a Series of Representative STLC Analogues for the Inhibition of the MT-Stimulated ATPase Activity and in Human Leukemia (K562), Lung (NCI-H1299), Pancreas (BxPC3), and Colon (HCT116) Tumor Cell Lines and a Normal Breast Cell Line (hTERT-HME1).
| GI50 (nM) |
||||||
|---|---|---|---|---|---|---|
| compd | inhibition MT-stimulated ATPase activity, Kiapp (nM) | HCT116 | K562 | hTERT-HME1 | NCI-H1299 | BxPC-3 |
| 1 (STLC) | 143.8 ± 21.6 | 553 ± 57 | 1452 ± 76 | 361 ± 75 | 1549 ± 111 | 1563 ± 155 |
| 2 | 152.3 ± 15.0 | 1213 ± 40 | 2286 ± 213 | 1607 ± 199 | 2301 ± 511 | 2463 ± 908 |
| 3 (NSC123139) | 13.6 ± 2.4 | 130 ± 11 | 242 ± 22 | 57 ± 15 | 242 ± 28 | 284 ± 32 |
| 4 (NSC123529) | 4.2 ± 2.3 | 153 ± 10 | 247 ± 38 | 119 ± 3 | 227 ± 28 | 245 ± 43 |
| 5 (NSC123528) | 9.1 ± 2.2 | 130 ± 7 | 240 ± 17 | 81 ± 12 | 294 ± 24 | 291 ± 54 |
| rac-9 | 235.4 ± 47.5 | 828 ± 29 | 865 ± 131 | 494 ± 46 | 2825 ± 219 | 1489 ± 111 |
| (S)-9 | 384.4 ± 61.3 | 1603 ± 127 | 2065 ± 168 | 721 ± 99 | 4018 ± 303 | 4864 ± 545 |
| (R)-9 | 211.3 ± 24.6 | 472 ± 20 | 776 ± 26 | 321 ± 28 | 1324 ± 55 | 1119 ± 138 |
| 10 | 203.0 ± 44.5 | 213 ± 18 | 577 ± 61 | 572 ± 92 | 964 ± 214 | 1225 ± 296 |
| rac-29 | 5.1 ± 3.3 | 99 ± 5 | 91 ± 9 | 38 ± 7 | 137 ± 8 | 144 ± 6 |
| (R)-29 | 10.2 ± 2.0 | 51 ± 2 | 98 ± 12 | 26 ± 3 | 132 ± 7 | 75 ± 13 |
| (S)-29 | 12.8 ± 1.6 | 95 ± 7 | 128 ± 15 | 42 ± 5 | 281 ± 26 | 180 ± 35 |
| 30 | 10.8 ± 1.8 | 95 ± 5 | 200 ± 16 | 150 ± 32 | 360 ± 73 | 1211 ± 265 |
| 31 | 10.7 ± 3.6 | 230 ± 27 | 337 ± 28 | 308 ± 54 | 828 ± 37 | 2312 ± 418 |
| (R)-32 | 3.4 ± 1.6 | 40 ± 2 | 82 ± 4 | 27 ± 3 | 111 ± 6 | 85 ± 12 |
| (S)-32 | 19.3 ± 6.4 | 115 ± 6 | 149 ± 6 | 49 ± 4 | 305 ± 30 | 268 ± 27 |
| rac-32 | 9.8 ± 1.3 | 84 ± 6 | 98 ± 5 | 44 ± 4 | 204 ± 19 | 151 ± 29 |
| 33 | 9.7 ± 1.8 | 154 ± 7 | 233 ± 21 | 601 ± 107 | 376 ± 37 | 1603 ± 168 |
| 54 (ispinesib) | 2.5 ± 1.2 | 25 ± 3 | 71 ± 8 | 32 ± 8 | 82 ± 10 | 80 ± 15 |
Finally, we investigated the cross-reactivity of rac-29 against a kinesin panel of the five human kinesins Kif5a, Kif7, MPP1, MKLP-2, and CENP-E by investigating their inhibition of the basal and MT-stimulated ATPase activities. We did not observe any significant inhibition for any of the kinesins tested (data not shown), indicating that the selectivity previously observed for STLC is maintained in this more potent analogue.8
Crystal Structures of Active Compounds
The SAR reported here confirms that a lipophilic substituent on one phenyl ring generally improves potency, and we have demonstrated that the sulfur in the tail group could be replaced by methylene to yield compounds with systematically improved activity both against Eg5 and in the K562 cell line. Here, we report the crystal structures of Eg5 complexed with S-trityl analogue 25 and triphenylbutanamine analogue 29 to a resolution of 2.75 Å. Data collection and refinement statistics are presented in Table 4. Eg5–25 and Eg5–29 complexes crystallized in space group P212121 and I213 with 7 and 1 molecule per asymmetric unit, respectively. In the Eg5–25 complex, 6 out of 7 of the Eg5 molecules were in the final inhibitor-bound state, whereby loop L5 has swung downward to close the inhibitor-binding pocket, the switch II cluster has moved upward, and the neck-linker has adopted a docked conformation. The seventh molecule (chain G) was trapped in an intermediate state similar to that in other Eg5–inhibitor structures, whereby local changes at the inhibitor-binding pocket have not propagated to complete structural changes at the switch II cluster and the neck-linker remains undocked.14,18 In the Eg5–29 complex, Eg5 was in the final inhibitor-bound state.
Table 4. Data Collection and Refinement Statistics for Eg5–25 and Eg5–29 Complexes.
| Eg5–25b | Eg5–29a,b | |
|---|---|---|
| unit cell dimensions | ||
| a (Å) | 145.72 | 158.61 |
| b (Å) | 156.20 | 158.61 |
| c (Å) | 170.00 | 158.61 |
| γ (deg) | 90 | 90 |
| space group | P212121 | I213 |
| beamline/detector | ID23-1/ADSC Q315R | I23-2/MAR 225 |
| molecules per asymmetric unit | 7 | 1 |
| resolution range (Å) | 30.0–2.75 | 30–2.75 |
| no. of unique reflections | 98930 (14132) | 17322 (2496) |
| completeness (%) | 97.7 (96.6) | 99.8 (99.8) |
| multiplicity | 5.4 (5.5) | 5.8 (5.4) |
| Rsym (%) | 9.4 (57.7) | 8.3 (60.9) |
| I/σ(I) | 13.0 (2.7) | 12.2 (3.2) |
| Wilson B (Å2)/DPIc (Å) | 70.75/0.33 | 77.98/0.27 |
| Refinement Statistics | ||
| Rwork/Rfree (%) | 22.06/28.04 | 19.76/24.30 |
| average B factors | ||
| overall | 64.67 | 66.54 |
| main chain/side chain | 63.94/65.39 | 65.34/67.72 |
| no. of. ADP/inhibitor/water | 7/7/302 | 1/1/41 |
| rmsdd in bond length (Å) | 0.013 | 0.012 |
| rmsdd in bond angle (deg) | 1.70 | 1.75 |
The racemic mixtures were used for crystallization.
Values in parentheses pertain to the highest resolution shell.
DPI: diffraction-component precision index.55
rmsd is the root-mean-square deviation from ideal geometry.
Examination of both complexes revealed that the three phenyl rings of 25 (Figure 2C) and 29 (Figure 2D) were buried in the same pockets as STLC, with the same hydrophobic and aromatic interactions described previously.14 As we used the racemic mixture of 29 to crystallize the Eg5–inhibitor complex, we observed electron density for both enantiomers in the structure. Several hydrogen bond interactions between Eg5 and the cysteine moiety of (S)-29 were apparent: the amino group with the main chain carbonyl oxygen of Gly117 and the side chain oxygen (OE1) of Glu116, and an oxygen (OXT) of the carboxylic group of the cysteine moiety with one of the side chain amino groups (NH1) of Arg221 (Figure 2D). These interactions were not observed for the (R)-enantiomer. Instead, the amino group of the cysteine moiety interacts with the protein via hydrogen bonding with a structural water molecule, which does not produce any significant change in affinity. Although the sulfur in STLC has been substituted by a carbon in 29, the interactions between Eg5 and both inhibitors are virtually identical, with the exception of the shorter C–C–C bonds (3.9 Å) compared to the C–S–C bonds (4.5 Å).
In the complex with 25 where the carboxylic acid is absent, only hydrogen bond interactions were observed between the amino group and the main chain carbonyl oxygen of Gly117 and the side chain oxygen (OE1) of Glu116 (Figure 2C). The loss of the carboxylate group not only removed a hydrogen bond interaction but also appeared to reduce restraints on the amine tail and increased its conformational freedom, as evident from one molecule (chain D) where the amino group pointed away from the side chain oxygen (OE1) of Glu116 and toward the amino group of Arg221 instead. This compound has equivalent potency with STLC, which suggests that the loss of the carboxylate group and the greater flexibility of tail group may be compensated by an improved interaction with the trityl headgroup bearing a meta-substituent. Paradoxically, 24 (the direct analogue of 25 with a carboxylate in the tail) has an approximately 3-fold reduction in activity against Eg5 in the basal in vitro assay, which suggests that the substituent in the trityl group may influence the relative position of the tail so that the carboxylate has a detrimental effect on binding.
Figure 4.

Craig plot for aromatic substituents of σ vs π. π is the substituent hydrophobicity constant and σ the Hammett substitution constant, a measure of the electronic influence of the substituent. Values were obtained from model systems from refs (30), (56), and (57).
With well-defined electron density maps, we were able to determine unequivocally the location of the meta-substituted acetate and methyl groups of 25 and 29, respectively. Both substituents sit in the hydrophobic P3 pocket formed by Ile136, Pro137, Leu160, Leu214, Phe239, and the salt bridge between Glu116 and Arg221. The carbonyl oxygen of the acetate group of 25 appeared to from a hydrogen bond with one of the side chain amino groups (NH1) of Arg221 and the main chain nitrogen of Ala218. The methyl of the acetate group was buried in a largely hydrophobic pocket formed by Phe239, Leu160, Ile136, and Leu214.
Overall, the crystal structures of Eg5–25 and Eg5–29 provide insight into the arrangement of the substituents within the binding site and help explain some of the structure–activity relationships of the ligands. For example, replacement of the m-acetyl with a m-ethyl (17) or m-isopropyl (18) group significantly improved activity by approximately 20-fold, which could be attributed to the replacement of a polar carbonyl group in an essentially hydrophobic pocket with the more lipophilic alkyl group and facilitating a favorable interaction with the side chain of Leu214. Furthermore, the electronic properties of the phenyl ring will be altered by an alkyl (+I) compared with an acetyl (−I), which would impact on the strength of the π–CH interaction with Leu214. The generally lower activities of the m-hydrophilic group series (Table 3) compared with 25 provide further evidence that this hydrophobic pocket has a low tolerance for polar groups, despite the proximity of the Glu116–Arg221 salt bridge. Shifting the m-acetyl to the p-position (compound 27) produced a 4-fold increase in activity, which could be due to an improved alignment of the carbonyl with the side chain of Arg221.
Evaluation of Inhibitors across Multiple Cell Lines
Of the 47 compounds tested in proliferation assays using human K562 leukemia cells (Tables 1–3), the most potent were subsequently investigated in four additional human cell lines (Table 5). These were derived from colon, lung, and pancreatic cancer, as well as immortalized cells derived from normal breast tissue. From our previous study, we included three potent S-trityl analogues (3–5) and STLC 1 as controls and to monitor progress and the clinical phase II candidate ispinesib to benchmark the new analogues.4,5,15 (R)-32 improves at least 2-fold on our previously reported most potent STLC analogues (3–5) in the selected cell lines and has comparable activity with ispinesib across all cell lines. (R)-29, which differs from (R)-32 only by the methyl group being in the para-position, shows only slightly lower activity in the lung cell line. The most potent analogue lacking the carboxylic acid group is 30. Although significant differences in activity were not observed between S-trityl-l-cysteine and S-trityl-d-cysteine in previous studies, detectable differences were evident among the enantiomers of the 2-aminobutanoic acid series.8,18 When the sulfur of STLC was replaced with carbon to produce (S)-9, there was lower activity across all cell lines, while the corresponding carbon analogue of STDC [(R)-9] had comparable activity. It is only when substituents were introduced into the headgroup of the tritylbutanamines that significant improvements in activity became apparent.
ADME Profiling of the Triphenylbutanamines and in Vivo Xenograft Studies
Our two most potent compounds with and without the carboxylic acid (29 and 30, respectively) together with ispinesib to provide a benchmark were assessed in a series of in vitro and in vivo ADME assays (Table 6; for 29, the separated enantiomers were used for profiling). Ispinesib had the lowest turbidimetric solubility (65 μM, partially soluble), whereas both 30 and rac-29 were soluble (>100 μM). Despite the nonoptimal log P of 30 and ispinesib, the log D at pH 7.4 was <3 and favorable for all compounds.22,31 During the time course of the experiments, all inhibitors either proved stable or exhibited low clearance in human microsomal and hepatocyte stability assays. In mouse microsomal assays, ispinesib and both enantiomers of 29 were stable whereas 30 showed high microsomal clearance, indicating species-dependent differences between mouse and human for this compound. Consequently, the high clearance of 30 excluded it from in vivo mouse xenograft studies. A further example of the need to balance efficacy with structural modifications to improve potency is demonstrated by hERG inhibition. Both ispinesib and 30 were moderately potent hERG inhibitors with IC50 of 4.7 ± 1.8 and 6.5 ± 1.6 μM, respectively. Among the most successful reported approaches for diminishing binding to the hERG channel are modulation of lipophilicity and structural modifications that disrupt the π-stacking and hydrophobic interactions between the drug candidate and the channel cavity.34 It is therefore not unexpected that installation of a polar, carboxylic acid functionality into 30 to produce 29 results in a dramatic reduction in hERG binding. Plasma protein binding influences the distribution and elimination of compounds: the fraction unbound of all compounds tested was >90% with (S)-29 being slightly less bound than the other compounds and is acceptable for this program. We also tested the compounds for inhibition of the main CYP isoforms. (S)-29 and (R)-29 did not show any inhibition up to 25 μM, but ispinesib moderately to significantly inhibited isoform CYP3A4 in vitro with an IC50 of 4.05 ± 0.44 μM, in agreement with two recent studies.35,36 The four isoforms CYP2C9, CYP2C19, CYP2D6, and CYP3A4 were inhibited by 30 with moderate to significant IC50 between 3.17 ± 0.45 and 8.46 ± 0.84 μM.
Table 6. ADME Profiling of Triphenylbutanamines and Ispinesib.
| assay/compd | 54 (ispinesib) | 30 | (S)-29 | (R)-29 |
|---|---|---|---|---|
| MW (Da) | 517.06 | 315.45 | 359.46 | |
| turbidimetric solubility pH 2.0, 6.0, 7.4 (μM) | 65, 65, 65 | >100, >100, >100 | >100, >100, >100a | |
| log P | 4.75 ± 0.04 | 4.69 ± 0.05 | 2.45 ± 0.07a | |
| pKa | 9.34 ± 0.08 | 10.09 ± 0.02 | pKa 1: 9.07 ± 0.03a | |
| pKa 2: 2.55 ± 0.07a | ||||
| log D7.4 | 2.66 ± 0.24 | 2.33 ± 0.16 | 2.45 ± 0.19a | |
| microsomal stability [Clint [(μL/min)/mg protein] | ||||
| human | stable | stable | stable | stable |
| mice | stable | 45.0 ± 3.09 | stable | stable |
| human hepatocytes [(μL/min)/million cells] | 0.75 ± 2.35 | 3.93 ± 2.48 | stable | stable |
| hERG (μM) | 4.81 ± 1.36 | 6.48 ± 1.63 | >25 | >25 |
| human plasma protein binding (fu100%) (% recovery) | 0.0835 ± 0.0023 | 0.0697 ± 0.0178 | 0.191 ± 0.0028 | 0.168 ± 0.0012 |
| 50.6 | 69.1 | 52.3 | 50.1 | |
| CYP450 inhibition (μM) | ||||
| 1A2 | >25 | >25 | >25 | >25 |
| 2C9 | >25 | 4.3 ± 0.5 | >25 | >25 |
| 2C19 | 24.3 ± 6.5 | 3.2 ± 0.5 | >25 | >25 |
| 2D6 | >25 | 8.5 ± 0.8 | >25 | >25 |
| 3A4 | 4.1 ± 0.4 | 5.5 ± 0.8 | >25 | >25 |
| bioavailability (%) | 45 | 4 | 51a | |
The racemic mixture was used.
We subsequently determined the bioavailability for 30, rac-29, and ispinesib (Table 6). Compound 30 had particularly low bioavailability of 4% and low po levels, which confirms the in vitro high microsomal clearance values seen in mice. Ispinesib shows moderate clearance with good po levels and has a moderate bioavailability of 45%. Finally, rac-29 showed low clearance, good po levels, and a bioavailability of 51%. On the basis of rac-29 having the better ADME profile compared with 30, particularly with respect to bioavailability, we tested it in vivo using lung cancer patient explants (LXFS 538) passaged as subcutaneous xenografts in nude mice.
In this model, by transplantation of a tumor from a patient to a mouse, many of the characteristics of the parental patient tumors including histology and sensitivity to anticancer drugs are retained. Moreover, earlier studies have shown that such explants correctly replicate the response of the donor patient to standard anticancer drugs in >90% of the cases.37
Rac-29 displayed good antitumor activity in LXFS 538: a minimum T/C of 26.3% was recorded, corresponding to transient partial tumor remission (i.e., individual relative tumor volumes of <100%) in four out of five tumors around day 10 and an obvious reduction of growth rates compared to the vehicle control group in the latter part of the experiment (Figure 5). This resulted in an increase of tumor volume doubling times from 8.8 days in the control group to 28.7 days in the rac-29-treated group. The results became statistically significant on day 28.
Figure 5.

Anticancer efficacy of rac-29 in a subcutaneous xenograft tumor model. The rac-29 treatment group (◆) received 20 mg/kg on days 0, 2, and 4 and 15 mg/kg rac-29 on days 17, 19, 24 (indicated by arrows), whereas the control group (□) received only vehicle on the same days. The data are plotted as the mean of the relative tumor volume ± standard deviation. The difference between treated group and vehicle is statistically significant (p = 0.016).
Conclusion
Triphenylbutan-1-amines represent a potent class of Eg5 inhibitors, which demonstrate good in vivo antitumor activity against lung cancer xenografts in mouse models. The SAR modifications of meta or para lipophilic trityl substituents, isosteric replacement of the sulfur with methylene, and inversion of the amino acid stereocenter with respect to STLC have produced analogues that systematically improve on the comparable S-trityl thioethanamines. With very potent nanomolar inhibitory activity exhibited against the target kinesin Eg5, favorable ligand efficiencies, and similar GI50 values over five tumor cell lines compared to the phase II candidate of equivalent potency, they represent excellent compounds for continued development and optimization. The major effect of the α-carboxylic acid is in determining the cellular permeability, and its presence in the amine tail reduces log P, enhances bioavailability, and attenuates interactions with hERG and several CYP isoforms. The continued development and further studies on this scaffold will be reported in due course.
Experimental Section
(A) Chemistry
All reagents and solvents were of commercial quality and used without further purification. STLC 1 was purchased from Nova Biochem and used without further purification. Ispinesib was a gift from Sanofi-Aventis. Compounds 3–5 and STDC (NSC123139, NSC123528, NSC123529, and NSC124767) were obtained from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov), National Cancer Institute. Anhydrous reactions were carried out in oven-dried glassware under a nitrogen atmosphere unless otherwise noted. Microwave reactions were performed using a Biotage Initiator-8 microwave synthesizer (operating at 2.45 GHz). Thin-layer chromatography (TLC) was carried out on aluminum-backed SiO2 plates (silica gel 60, F254), and spots were visualized using ultraviolet light (254 nm) or by staining with phosphomolybdic acid. Flash column chromatography was performed on silica gel [60 Å, 35–70 μm or SNAP KP-Sil, 60 Å, 40–63 μm cartridges] manually or using a Biotage SP4 automated chromatography system (detection wavelength, 254 nm; monitoring, 280 nm). 1H and 13C NMR spectra were recorded on a JEOL ECX-400 (400 MHz), Avance DPX400 (400 MHz), or Avance DPX500 (500 MHz) spectrometer. 19F NMR spectra were recorded on an Avance AV400 (400 MHz) instrument equipped with a multinuclear probe. 1H chemical shifts (δ) are reported in ppm relative to the residual signal of the deuterated solvent (7.26 in CDCl3, 3.31 in CD3OD, and 2.50 in DMSO-d6). Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), p (pentet), m (multiplet), and br (broad). 13C chemical shifts (δ) are reported in ppm relative to the carbon resonance of the deuterated solvent (77.16 in CDCl3, 49.00 in CD3OD, and 39.52 in DMSO-d6). 19F spectra are referenced relative to CFCl3. Mass spectra were recorded on a Thermo Electron LTQ ORBITRAP mass spectrometer using electrospray ionization. Data from gas chromatography–mass spectrometry (GC–MS) were recorded on a Thermo Scientific Focus GC with DSQ2 single quadrupole mass spectrometer. Melting points were determined using a Stuart Scientific SMP1 melting point apparatus and are uncorrected. Elemental analysis data were recorded on a Perkin-Elmer 2400 series 2 CHN analyzer. All tested compounds were in general ≥95% pure. HPLC analysis was carried out using a Gilson HPLC system and DIONEX P 680 HPLC system. New compounds were named according to IUPAC nomenclature using ACD ChemSketch 12.01 (Windows, Advanced Chemistry Development, Toronto, Canada). Detailed synthetic procedures and characterization for all other compounds are available in the Supporting Information.
1,1′,1″-But-1-ene-4,4,4-triyltribenzene (55)
The title compound was prepared using an adaptation of the method reported by Kabalka et al.23 A solution of triphenylmethanol (7.81 g, 30.0 mmol) in anhydrous CH2Cl2 (150 mL) was treated with n-butyllithium (1.6 M in hexane, 20.0 mL, 32.0 mmol) at 0 °C and the mixture then warmed to room temperature. After the mixture was stirred for 30 min, allyltrimethylsilane (5.75 mL, 36.0 mmol) and iron trichloride (5.19 g, 32.0 mmol) were added. The mixture was allowed to stir for a further 6 h and then quenched with water (40 mL). The reaction mixture was extracted with EtOAc (100 mL), dried (MgSO4), and concentrated in vacuo. The crude product was purified by flash chromatography [SiO2, hexane] to give 55 as a yellow syrup (7.9 g, 93%). 1H NMR (400 MHz, CDCl3) δ = 3.46 (d, 2H, CH2), 4.96–5.06 (m, 2H, CH2), 5.64–5.68 (m, 1H, CH), 7.10–7.27 (m, 15H, Ph). 13C NMR (100 MHz, CDCl3) δ = 45.6, 56.4, 117.4, 126.1, 127.9, 129.5, 147.4, 136.1. HRMS (ESI+) calcd for C22H20 (M + H)+, 285.163 78; found, 285.163 60. Anal. Calcd for C22H20: C, 92.91; H, 7.09. Found: C, 92.98; H, 7.27.
4,4,4-Triphenyl-1-butanol (56)
The title compound was prepared using an adaptation of the method reported by Starnes.38 A solution of concentrated sulfuric acid (15.3 μL, 0.27 mmol) in anhydrous diethyl ether (0.37 mL) was added dropwise (at room temperature) to a solution of sodium borohydride (20.6 mg, 0.54 mmol) and 1,1′,1″-but-1-ene-4,4,4-triyltribenzene 55 (405 mg, 1.42 mmol) in anhydrous diglyme (3 mL). The mixture was stirred at room temperature for 3.5 h and then heated at 75 °C for a further 1.5 h. The mixture was cooled to 0 °C and treated successively with water (39 μL), aqueous NaOH (3M, 0.18 mL), and 30% aqueous hydrogen peroxide (0.18 mL). The mixture was stirred for 20 min at 0 °C and then 6.5 h at room temperature. The mixture was extracted with Et2O (50 mL), washed with water (25 mL), dried (MgSO4), and concentrated in vacuo. The crude product was purified by flash chromatography [SiO2, 10–40% EtOAc in hexane] to afford the alcohol 56 as a white solid (305 mg, 71%). Mp 118 °C (lit. 120–122 °C).391H NMR (400 MHz, DMSO-d6) δ = 1.07–1.11 (m, 2H, CH2), 2.54–2.58 (m, 2H, CH2), 3.37–3.39 (m, 2H, CH2), 4.46 (t, 1H, OH), 7.22–7.28 (m, 15H, Ph). 13C NMR (100 MHz, DMSO-d6) δ = 29.0, 36.0, 55.9, 60.9, 125.6, 127.8, 128.7, 147.2. HRMS (ESI+) calcd for C22H22O (M + NH4)+. 320.200 89; found, 320.200 58. Anal. Calcd for C22H22O: C, 87.38; H, 7.33. Found: C, 85.27; H, 7.54.
4,4,4-Triphenylbutanal (57)
The title compound was prepared using an adaptation of the procedure reported by Rodriguez et al.40 Dess–Martin periodinane (356 mg, 0.84 mmol) was added to a stirred solution of 4,4,4-triphenyl-1-butanol 55 (212 mg, 0.7 mmol) in CH2Cl2 (8 mL). The mixture was stirred at room temperature for 3 h and then quenched with saturated aqueous Na2S2O3 solution (5 mL) followed by saturated aqueous NaHCO3 solution (5 mL). The mixture was extracted with CH2Cl2 (50 mL), dried (MgSO4), and concentrated in vacuo. The crude product was purified by flash chromatography [SiO2, 2–15% EtOAc in hexane] to give 57 as a white solid (187 mg, 89%). Mp 108 °C. 1H NMR (400 MHz, DMSO-d6) δ = 2.14–2.18 (t, 2H, CH2), 2.85–2.89 (t, 2H, CH2), 7.23–7.29 (m, 15H, Ph), 9.47 (s, 1H, CHO). 13C NMR (100 MHz, DMSO-d6) δ = 31.4, 38.9, 55.6, 125.7, 127.8, 128.7, 146.5, 202.3. HRMS (ESI+) calcd for C22H20O (M + NH4)+, 318.185 24; found, 318.184 81. Anal. Calcd for C22H20O: C, 87.96; H, 6.71. Found: C, 87.99; H, 6.76.
2-(Benzylamino)-5,5,5-triphenylpentanenitrile (rac-58)
The title compound was prepared using an adaptation of the method reported by Yadav et al.24 Montmorillonite KSF clay (1.2 g) was added to a solution of 4,4,4-triphenylbutanal 57 (300 mg, 1.0 mmol), benzylamine (129 mg, 132 μL, 1.2 mmol), and trimethylsilyl cyanide (144 mg, 192 μL, 1.4 mmol) in CH2Cl2 (12 mL). The mixture was stirred at room temperature for 2 h, filtered and the clay rinsed with CH2Cl2 (15 mL). The combined organic layers were dried (MgSO4), concentrated in vacuo and the crude product was purified by flash chromatography [SiO2, 0–15% EtOAc in hexane] to give rac-58 as a white solid (388 mg, 93%). 1H NMR (400 MHz, DMSO-d6) δ = 1.35–1.41 (m, 2H, CH2), 2.67–2.78 (m, 2H, CH2), 3.19–3.23 (m, 1H, NH), 3.59–3.64 (m, 2H, PhCH2), 3.79 (dd, 1H, CH), 7.23–7.30 (m, 20H, Ph). 13C NMR (100 MHz, DMSO-d6) δ = 29.2, 35.4, 49.5, 50.7, 55.8, 120.7, 125.9, 127.6, 128.6, 128.8, 146.7. HRMS (ESI+) calcd for C30H28N2 (M + H)+, 417.232 53; found, 417.232 17. Anal. Calcd for C30H28N2·2H2O: C, 79.61; H, 7.13; N, 6.19. Found: C, 79.03; H, 6.79; N, 5.50.
2-(Benzylamino)-5,5,5-triphenylpentanoic Acid (rac-59)
The title compound was prepared using an adaptation of the procedures reported by Bigge et al. and Warmuth et al.41,42 A solution of 2-(benzylamino)-5,5,5-triphenylpentanenitrile rac-58 (130 mg, 0.31 mmol) in HCl (6 M in dioxane, 8 mL) was heated at reflux for 2 days and then concentrated in vacuo. The residue was redissolved in EtOH (3 mL), propylene oxide (1 mL) added, and the mixture heated at reflux for 30 min. The volatiles were removed in vacuo and the crude product was purified by flash chromatography [SiO2, 0–18% MeOH in CH2Cl2 with 0.5% NH4OH] to give the protected amino acid rac-59 as a white solid (118 mg, 87%). 1H NMR (400 MHz, CD3OD) δ = 1.57–1.64 (m, 2H, CH2), 2.70–2.84 (m, 2H, CH2), 3.30 (m, 2H, PhCH2), 3.39–3.43 (m, 1H, NH), 3.89–4.04 (dd, 1H, CH), 7.23–7.37 (m, 20H, Ph). 13C NMR (100 MHz, CD3OD) δ = 26.6, 35.4, 50.0, 56.1, 62.0, 125.7, 127.6, 128.8, 129.0, 147.0. HRMS (ESI+) calcd for C30H29NO2 (M + H)+: 436.227 11; found, 436.227 20. Anal. Calcd for C30H29NO2: C, 82.73; H, 6.71; N, 3.22. Found: C, 82.82; H, 6.93; N, 2.52.
2-Amino-5,5,5-triphenylpentanoic Acid (rac-9)
The title compound was prepared using an adaptation of the method reported by Siya Ram et al.25 A mixture of 2-(benzylamino)-5,5,5-triphenylpentanoic acid rac-58 (189 mg, 0.43 mmol), 10% Pd/C (94.5 mg), and HCOONH4 (137 mg, 2.17 mmol) in anhydrous MeOH (15 mL) was heated at 80 °C for 1 h. The mixture was cooled and then filtered through Celite, which was then washed with MeOH (10 mL). The filtrate was concentrated in vacuo and the crude product purified by flash chromatography [SiO2, 5–25% MeOH in CH2Cl2 with 0.5% NH4OH] to give rac-9 as a white solid (133 mg, 89%). Mp 176 °C. 1H NMR (500 MHz, DMSO-d6) δ = 1.26–1.45 (m, 2H, CH2), 2.60–2.77 (m, 2H, CH2), 3.16 (m, 1H, CH), 7.15–7.28 (m, 15H, Ph). 13C NMR (125 MHz, DMSO-d6) δ = 27.7, 36.1, 55.1, 56.4, 126.2, 128.3, 129.4, 147.5, 170.5. HRMS (ESI+) calcd for C23H23NO2 (M + H)+, 346.180 16; found, 346.179 87. Anal. Calcd for C23H23NO2: C, 79.97; H, 6.71; N, 4.05. Found: C, 79.17; H, 7.09; N, 4.64.
1,1′,1″-(4-Azidobutane-1,1,1-triyl)tribenzene (60)
Methanesulfonyl chloride (85 μL, 1.1 mmol) was added to a solution of 56 (166 mg, 0.55 mmol) in anhydrous pyridine (5 mL) at 0 °C (ice–water). The mixture was stirred for 12 h at room temperature, and the volatiles were removed in vacuo. The residue was extracted with CH2Cl2 (30 mL), dried (MgSO4), and concentrated in vacuo. The crude product was purified by flash chromatography [SiO2, 2–30% EtOAc in hexane] to give 4,4,4-triphenylbutylmethanesulfonate as a white solid (199 mg, 95%). Sodium azide (130 mg, 2.0 mmol) was added to a solution of 4,4,4-triphenylbutyl methanesulfonate (190 mg, 0.5 mmol) in anhydrous DMF (2 mL) and the mixture irradiated with microwave radiation at 175 °C. The mixture was cooled. The solid residue was filtered off, and the filtrate was concentrated in vacuo. The crude product was purified by flash chromatography [SiO2, 2–30% EtOAc in hexane] to give the azide 60 as a white solid (150 mg, 92%). Mp 114 °C. 1H NMR (400 MHz, CDCl3) δ = 1.37 (m, 2H, CH2), 2.64 (m, 2H, CH2), 3.24 (t, 2H, CH2), 7.10–7.27 (m, 15H, Ph). 13C NMR (100 MHz, CDCl3) δ = 25.6, 37.4, 52.1, 56.4, 126.1, 128.0, 129.2, 147.1. Anal. Calcd for C22H21N3: C, 80.70; H, 6.46; N, 12.83. Found: C, 79.96; H, 6.58; N, 11.78.
4,4,4-Triphenylbutan-1-amine (10)
The title compound was prepared using an adaptation of the procedure reported by Dockendorff et al.43 Triphenylphosphine (603 mg, 2.3 mmol) was added to a solution of 1,1′,1″-(4-azidobutane-1,1,1-triyl)tribenzene 60 (150 mg, 0.46 mmol) in THF/H2O (5:0.5 mL). The reaction mixture was stirred at 60 °C for 12 h and then concentrated in vacuo. The crude product was purified by flash chromatography [SiO2, 3–15% MeOH in CH2Cl2 with 0.5% NH4OH] to give the title compound 10 as a white solid (117 mg, 85%). Mp 72 °C. 1H NMR (400 MHz, DMSO-d6) δ = 1.02 (m, 2H, CH2), 2.49–2.57 (m, 4H, CH2 and CH2), 7.22–7.40 (m, 15H, Ph). 13C NMR (100 MHz, DMSO-d6) δ = 29.9, 37.5, 42.4, 56.6, 126.3, 128.4, 129.4, 147.9. HRMS (ESI+) calcd for C22H23N (M + H)+, 302.190 33; found, 302.189 91. Anal. Calcd for C22H23N·0.5H2O: C, 85.12; H, 7.79; N, 4.51. Found: C, 85.23; H, 7.69; N, 4.46.
Enantiomeric Separation by Semipreparative Chiral Chromatography
Separation was performed on a Dionex P680 HPLC system using a 10 mm × 250 mm ChiralPack IC column containing cellulose tris(3,5-dichlorophenylcarbamate) immobilized on 5 μm silica gel as the chiral stationary phase. Mobile phase was n-heptane/ethanol/trifluoroacetic acid/triethylamine [95:5:0.1:0.1% v/v]. The flow rate was 4 mL/min, and detection wavelength (UV) was 254 nm. The separated enantiomers were impure with TFA and NEt3. Purification by flash chromatography [SiO2, 0–24% MeOH in CH2Cl2 with 1% NH4OH] afforded the pure enantiomers as white solids (∼5 mg each). Retention times for compound 9: 30 and 36 min. For 29: 32 and 40 min. For 32: 33 and 40 min.
Application of Marfey’s Method To Determine Absolute Stereochemistry
The following general procedure was used for derivatization of 32-1 and 32-2.44N-α-(2,4- Dinitro-5-fluorophenyl)-l-valinamide (1% w/v in acetone, 100 μL, 3.6 μmol) was added to a small vial containing a solution of the amino acid (2.5 μmol) in methanol (50 μL), followed by NaHCO3 (1 M in H2O, 20 μL, 20 μmol), and the mixture was heated at 40 °C for 1 h with stirring. After cooling to room temperature, the mixture was quenched with aqueous HCl (1 M, 20 μL), diluted to 1 mL with MeOH, and then measured by LC–MS. HPLC conditions: Dionex Ultimate 3000 system, acetonitrile (0.1% formic acid) and water (0.1% formic acid), 15–100% acetonitrile in 35 min. Flow rate: 0.3 mL/min. Wavelength: 254 nm.
(B) Biology. Inhibition of the Basal and MT-Stimulated Eg5 ATPase Activities
For ATPase assays and structure determination, Eg51–368 was cloned, expressed, and purified as previously described.14 The inhibition of the Eg5 ATPase activity was determined as previously described.18 In short, ATPase rates were measured using the pyruvate kinase/lactate dehydrogenase-linked assay. The measurements were performed at 25 °C in 96-well μclear plates (340 nm) using a 96-well Tecan Sunrise photometer. The salt concentration was 150 mM NaCl for both the inhibition of the basal and the MT-stimulated ATPase activity with an Eg5 concentration of ∼80 and ∼5 nM, respectively. It is noteworthy that for this Eg5 construct the IC50 or Kiapp values can depend on the ionic strength of the buffer.45 All data were measured at least in triplicate. To obtain estimates for the apparent Ki, data were fitted to the Morrison equation:46
whereby vi/v0 is the fractional activity, vmax is the uninhibited protein activity, vmin is the remaining activity at the highest inhibitor concentration used, [E] and [I] represent the enzyme and inhibitor concentrations used in the assays, and Kiapp is the apparent Ki determined from the data.
Calculation of Ligand Efficiencies
The equation for calculating ligand efficiencies is LE = −ΔG/HAC ≈ –RT ln(Kiapp)/HAC, where ΔG is the change in Gibbs free energy, T is the absolute temperature, R represents the gas constant, and HAC is the heavy atom count for non-hydrogen atoms.
Tissue Culture
HCT116 (ATCC CCL-247) cells were cultured in DMEM (Invitrogen, Paisley, U.K.), supplemented with 10% fetal bovine serum (PAA, Pasching, Austria). K562 (ATCC CCL-243), LNCaP (ATCC CRL-1740), and NCI-H1299 (CRL-5803) cells were cultured in RPMI (Invitrogen, Paisley, U.K.), supplemented with 10% fetal bovine serum (PAA, Pasching, Austria). BxPC-3 (ATCC CRL-1687) cells were cultured in RPMI (Invitrogen, Paisley, U.K.), supplemented with 1% nonessential amino acids (Invitrogen, Paisley, U.K.), 1% sodium pyruvate (Invitrogen, Paisley, U.K.), 1% glutamine (Invitrogen, Paisley, U.K.), and 10% fetal bovine serum (PAA, Pasching, Austria). hTERT-HME1 cells (Clontech, Basingstoke, U.K.) were cultured in mammary epithelial cell growth medium (PromoCell, Heidelberg, Germany). All cells were maintained at 37 °C, 95% humidity, and 5% carbon dioxide in a humidified incubator. They were used for experiments for 6–8 weeks before they were replaced with fresh stocks that were stored in liquid nitrogen.
Proliferation Assays
Cells were seeded in triplicate in 96-well assay plates at 1.250 cells (BxPC-3, HCT116), 2.500 cells (hTERT-HME1, NCI-H1299), or 5.000 cells (K562) per well in 100 μL of the respective growth medium. Medium blanks and cell blanks for every cell line were also prepared. On the next day, inhibitors were added with a starting concentration of 100 μM in a 3-fold serial dilution series. At 72 h after inhibitor addition, 10% Alamar blue (Invitrogen, Paisley, U.K.) was added, and depending on the cell line, 2–12 h later the absorbance was measured at 570 and 600 nm. All values were corrected for the absorbance of the medium blank, and the corrected cell blanks were set to 100%. Calculations for determining the relative proliferation were performed using equations described in the manufacturer’s manual. Finally, the GI50 values were determined using a sigmoidal dose–response fitting (variable slope) with GraphPad Prism 5.03 for Windows (GraphPad Software, San Diego, CA, U.S.).
Tumor Xenografts
The animal experiments were performed at Oncotest GmbH with female NMRI nu/nu mice (Charles River, Sulzfeld, Germany). Tumor fragments were obtained from xenografts in serial passage in nude mice. After removal from donor mice, tumors were cut into fragments (4–5 mm diameter) and placed in PBS until subcutaneous implantation. Recipient mice were anesthetized by inhalation of isoflurane. A small incision was made, and one tumor fragment per animal was transplanted with tweezers. The approximate age at implantation was 5–7 weeks. At 10–12 weeks, mice were randomized to the various groups and dosing started when the required number of mice carried a tumor of 50–250 mm3 volume, preferably 80–200 mm3. Vehicle details and drug formulation are described in the Supporting Information.
Vehicle control mice (group 1) were treated with 10 mL/kg rac-29 vehicle on days 0, 2, and 4 and with 7.5 mL/kg on days 17, 19, 24, 31, and 34. The rac-29 treatment group (group 2) received 20 mg/kg rac-29 on days 0, 2, and 4 and 15 mg/kg rac-29 on days 17, 19, 24, 31, and 34. All treatments were given intraperitoneally. The experiment was terminated on day 34, and tumor samples were collected.
Mortality checks were conducted at least daily during routine monitoring. Body weight was used as a means of determining toxicity. Mice were weighed twice a week. The tumor volume was determined by two-dimensional measurement with a caliper on the day of randomization (day 0) and then twice weekly (i.e. on the same days on which mice were weighed). Tumor volumes were calculated according to the formula (ab2) × 0.5 where a represents the largest and b the perpendicular tumor diameter.
Tumor inhibition for a particular day (T/C in %) was calculated from the ratio of the median RTV values of test versus control groups multiplied by 100%. For the evaluation of the statistical significance of tumor inhibition, the Mann–Whitney U-test was performed. Individual RTVs were compared on days on which the minimum T/C was achieved, as long as sufficient animals were left for statistical analysis or otherwise on days as indicated. By convention, p ≤ 0.05 indicates significance of tumor inhibition. Statistical calculations were performed using GraphPad Prism 5.01 (GraphPad Software, San Diego, CA, U.S.).
Crystallization of the Eg5–25 and Eg5–29 Complexes
Eg51–368 was cloned, expressed, and purified as previously described.14 Purified Eg5 (20 mg/mL), in complex with 1 mM Mg2+ATP, was incubated with 2 mM 25 or rac-29 (in DMSO) for 2 h on ice. Crystals of Eg5 with 25 appeared after 4 days in hanging drops by mixing 1 μL of protein–inhibitor complex with 1 μL of reservoir solution containing 22% polyethylene glycol-3350, 0.25 M ammonium sulfate, 0.1 M potassium sodium tartrate tetrahydrate, and 0.1 M MES, pH 5.8, in VDX plates (Hampton Research) at 4 °C. A thick needle-shaped crystal with dimensions of approximately 0.1 mm × 0.01 mm × 0.01 mm was immersed in cryoprotectant solution (27.6% polyethylene glycol-3350, 0.36 M of ammonium sulfate, 0.12 M MES, pH 5.5, 0.06 M potassium chloride, and 20% erythritol) and flash frozen in liquid nitrogen. Crystals of Eg5 with rac-29 appeared after 1 week in hanging drops by mixing 1 μL of protein–inhibitor complex with 0.2 μL of 1 M potassium sodium tartrate tetrahydrate and 1 μL of reservoir solution containing 23% polyethylene glycol-3350, 0.25 M ammonium sulfate, and 0.1 M MES, pH 5.5, in VDX plates (Hampton Research) at 4 °C. Dehydrating solution (33% polyethylene glycol-3350, 0.25 M ammonium sulfate, 0.1 M potassium sodium tartrate tetrahydrate, 0.1 M MES, pH 5.5, and 10% glycerol) was added slowly to the crystal droplet until the total volume of the drop was 8 times the original. The drop was then equilibrated against air in 4 °C for 30 min. A cubic crystal with dimensions of approximately 0.1 mm on each side was then flash frozen in liquid nitrogen.
Data Collection and Processing
Diffraction data for the Eg5–inhibitor complexes Eg5–25 and Eg5–29 were recorded at the European Synchrotron Radiation Facility (ESRF) beamlines ID23-1 and ID23-2, respectively. Data were processed using iMosflm47 and scaled using Scala48 from the CCP4 suite of programs.49 The structures were solved by molecular replacement (Phaser)50 using one molecule of Eg5 motor domain from structures with PDB codes 1X88(20) and 3KEN,19 respectively. Refinement was carried out with Phenix51 and Refmac5, respectively. The calculation of Rfree used 5% of data. Electron density and difference density maps, all σA-weighted, were inspected, and the model was improved using Coot.52 Model geometry was analyzed using Molprobity.53 For the Eg5–25 structure, 95.7% (2106) of the residues are in the preferred regions, 3.54% (78) are in the allowed regions, and 0.77% (17) are outliers as shown by the Ramachandran plot. For the Eg5–29 complex, 95.2% (316) of the residues are in the preferred regions, 3.61% (12) are in the allowed regions, and 1.20% (4) are outliers. Figures are prepared using PyMOL.54
Acknowledgments
We are grateful to Ruangelie Edrada-Ebel at Strathclyde University, U.K., for performing HR-MS experiments, Denise Gilmour for elemental analysis, Craig Irving for NMR support, and Marta Kozlowska and Sandeep Talapatra from The Beatson Institute for Cancer Research for providing purified human kinesins to analyse the specificity of rac-29. We thank Dr. Alexander Popov and Dr. Gordon Leonard of ESRF and of EMBL-Grenoble for assistance and support in using beamlines ID23-1 and ID23-2. We are grateful to the NCI/NIH for providing some of the STLC analogues investigated in this work. We thank Cancer Research UK for financial support.
Glossary
Abbreviations Used
- CD
circular dichroism
- DMF
dimethylformamide
- DMSO
dimethylsulfoxide
- EC50
half-maximal effective concentration
- FDVA
Nα-(2,4-dinitro-5-fluorophenyl)-l-valinamide
- FMOC
fluorenylmethyloxycarbony
- GI50
the concentration required to achieve 50% growth inhibition
- h
hour
- hERG
human ether-ago-go-related gene
- HPLC
high performance liquid chromatography
- IC50
median inhibitory concentration
- Kiapp
apparent Ki
- KSP
kinesin spindle protein
- MT
microtubule
- SAR
structure–activity relationship
- STDC
S-trityl-d-cysteine
- STLC
S-trityl-l-cysteine
- MDR
multidrug resistance
- NCI
National Cancer Institute
- nd
not determined
- ni
no inhibition
- PAMPA
parallel artificial membrane permeability assay
- PBS
phosphate buffered saline
- Pgp
P-glycoprotein
- rt
room temperature
- T/C
relative test tumor versus control value
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
Supporting Information Available
Additional experimental procedures and compound characterization data, ADME profiling and tumor experiment methods, supplementary results on acid stability of compounds, and validation of Marfey’s method. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
† Coordinate and structure factor files for the Eg5−25 and Eg5−29 complexes (PDB codes 4A51 and 4A50) were deposited at the PDB.
Author Contributions
∥ These authors contributed equally.
Author Present Address
⊥ Institute of Biophysics, Chinese Academy of Sciences, 15 Datun Road, Chaoyang District, Beijing, 100101, China.
Author Present Address
# Institute of Molecular and Cell Biology, 61 Biopolis Drive, Proteos, 138673, Singapore.
Supplementary Material
References
- Blangy A. L.; Heidi A.; d’Herin P.; Harper M.; Kress M.; Niggt E. A. Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 1995, 83, 1159–1169. [DOI] [PubMed] [Google Scholar]
- Sawin K. E.; LeGuellec K.; Philippe M.; Mitchison T. J. Mitotic spindle organization by a plus-end-directed microtubule motor. Nature 1992, 359, 540–543. [DOI] [PubMed] [Google Scholar]
- Huszar D.; Theoclitou M. E.; Skolnik J.; Herbst R. Kinesin motor proteins as targets for cancer therapy. Cancer Metastasis Rev 2009, 28, 197–208. [DOI] [PubMed] [Google Scholar]
- Lad L.; Luo L.; Carson J. D.; Wood K. W.; Hartman J. J.; Copeland R. A.; Sakowicz R. Mechanism of inhibition of human KSP by ispinesib. Biochemistry 2008, 47, 3576–3585. [DOI] [PubMed] [Google Scholar]
- Purcell J. W.; Davis J.; Reddy M.; Martin S.; Samayoa K.; Vo H.; Thomsen K.; Bean P.; Kuo W. L.; Ziyad S.; Billig J.; Feiler H. S.; Gray J. W.; Wood K. W.; Cases S. Activity of the kinesin spindle protein inhibitor ispinesib (SB-715992) in models of breast cancer. Clin. Cancer Res. 2010, 16, 566–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakowicz R.; Finer J. T.; Beraud C.; Crompton A.; Lewis E.; Fritsch A.; Lee Y.; Mak J.; Moody R.; Turincio R.; Chabala J. C.; Gonzales P.; Roth S.; Weitman S.; Wood K. W. Antitumor activity of a kinesin inhibitor. Cancer Res. 2004, 64, 3276–3280. [DOI] [PubMed] [Google Scholar]
- DeBonis S.; Skoufias D. A.; Lebeau L.; Lopez R.; Robin G.; Margolis R. L.; Wade R. H.; Kozielski F. In vitro screening for inhibitors of the human mitotic kinesin Eg5 with antimitotic and antitumor activities. Mol. Cancer Ther. 2004, 3, 1079–1090. [PubMed] [Google Scholar]
- Skoufias D. A.; DeBonis S.; Saoudi Y.; Lebeau L.; Crevel I.; Cross R.; Wade R. H.; Hackney D.; Kozielski F. S-Trityl-l-cysteine is a reversible, tight binding inhibitor of the human kinesin Eg5 that specifically blocks mitotic progression. J. Biol. Chem. 2006, 281, 17559–17569. [DOI] [PubMed] [Google Scholar]
- Shimizu M.; Ishii H.; Ogo N.; Unno Y.; Matsuno K.; Sawada J.-i.; Akiyama Y.; Asai A. S-Trityl-l-cysteine derivative induces caspase-independent cell death in K562 human chronic myeloid leukemia cell line. Cancer Lett. 2010, 298, 99–106. [DOI] [PubMed] [Google Scholar]
- Kozielski F.; Skoufias D. A.; Indorato R. L.; Saoudi Y.; Jungblut P. R.; Hustoft H. K.; Strozynski M.; Thiede B. Proteome analysis of apoptosis signaling by S-trityl-l-cysteine, a potent reversible inhibitor of human mitotic kinesin Eg5. Proteomics 2008, 8, 289–300. [DOI] [PubMed] [Google Scholar]
- Ding S.; Nishizawa K.; Kobayashi T.; Oishi S.; Lv J.; Fujii N.; Ogawa O.; Nishiyama H. A potent chemotherapeutic strategy for bladder cancer: (S)-methoxy-trityl-l-cystein, a novel Eg5 inhibitor. J. Urol. 2010, 184, 1175–1181. [DOI] [PubMed] [Google Scholar]
- Xing N.-D.; Ding S.-T.; Saito R.; Nishizawa K.; Kobayashi T.; Inoue T.; Oishi S.; Fujii N.; Lv J.-J.; Ogawa O.; Nishiyama H. A potent chemotherapeutic strategy in prostate cancer: S-(methoxytrityl)-l-cysteine, a novel Eg5 inhibitor. Asian J. Androl. 2011, 13, 236–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brier S.; Lemaire D.; Debonis S.; Forest E.; Kozielski F. Identification of the protein binding region of S-trityl-l-cysteine, a new potent inhibitor of the mitotic kinesin Eg5. Biochemistry 2004, 43, 13072–13082. [DOI] [PubMed] [Google Scholar]
- Kaan H. Y. K.; Ulaganathan V.; Hackney D. D.; Kozielski F. An allosteric transition trapped in an intermediate state of a new kinesin–inhibitor complex. Biochem. J. 2009, 425, 55–60. [DOI] [PubMed] [Google Scholar]
- DeBonis S.; Skoufias D. A.; Indorato R.-L.; Liger F.; Marquet B.; Laggner C.; Joseph B.; Kozielski F. Structure–activity relationship of S-trityl-l-cysteine analogues as inhibitors of the human mitotic kinesin Eg5. J. Med. Chem. 2008, 51, 1115–1125. [DOI] [PubMed] [Google Scholar]
- Ogo N.; Oishi S.; Matsuno K.; Sawada J.-i.; Fujii N.; Asai A. Synthesis and biological evaluation of l-cysteine derivatives as mitotic kinesin Eg5 inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3921–3924. [DOI] [PubMed] [Google Scholar]
- Wiltshire C.; Singh B. L.; Stockley J.; Fleming J.; Doyle B.; Barnetson R.; Robson C. N.; Kozielski F.; Leung H. Y. Docetaxel-resistant prostate cancer cells remain sensitive to S-trityl-l-cysteine-mediated Eg5 inhibition. Mol. Cancer Ther. 2010, 9, 1730–1739. [DOI] [PubMed] [Google Scholar]
- Kaan H. Y. K.; Weiss J.; Menger D.; Ulaganathan V.; Tkocz K.; Laggner C.; Popowycz F.; Joseph B. t.; Kozielski F. Structure–activity relationship and multidrug resistance study of new S-trityl-l-cysteine derivatives as inhibitors of Eg5. J. Med. Chem. 2011, 54, 1576–1586. [DOI] [PubMed] [Google Scholar]
- Kim E. D.; Buckley R.; Learman S.; Richard J.; Parke C.; Worthylake D. K.; Wojcik E. J.; Walker R. A.; Kim S. Allosteric drug discrimination is coupled to mechanochemical changes in the kinesin-5 motor core. J. Biol. Chem. 2010, 285, 18650–18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y.; Sardana V.; Xu B.; Homnick C.; Halczenko W.; Buser C. A.; Schaber M.; Hartman G. D.; Huber H. E.; Kuo L. C. Inhibition of a mitotic motor protein: where, how, and conformational consequences. J. Mol. Biol. 2004, 335, 547–554. [DOI] [PubMed] [Google Scholar]
- Kaan H. Y. K.; Ulaganathan V.; Rath O.; Prokopcová H.; Dallinger D.; Kappe C. O.; Kozielski F. Structural basis for inhibition of Eg5 by dihydropyrimidines: stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J. Med. Chem. 2010, 53, 5676–5683. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P.; Hersey A.; Montanari D.; Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discovery 2011, 10, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabalka G. W.; Yao M.-L.; Borella S.; Goins L. K. Iron trichloride mediated allylation of lithium alkoxides through an unusual carbon–oxygen bond cleavage. Organometallics 2007, 26, 4112–4114. [Google Scholar]
- Yadav J. S.; Reddy B. V. S.; Eeshwaraiah B.; Srinivas M. Montmorillonite KSF clay catalyzed one-pot synthesis of [alpha]-aminonitriles. Tetrahedron 2004, 60, 1767–1771. [Google Scholar]
- Ram S.; Spicer L. D. Debenzylation of N-benzylamino derivatives by catalytic transfer hydrtyation with ammonium formate. Synth. Commun. 1987, 17, 415–418. [Google Scholar]
- Marfey P. Determination of d-amino acids. II. Use of a bifunctional reagent, 1,5-difluoro-2,4-dinitrobenzene. Carlsberg Res. Commun. 1984, 49, 591–596. [Google Scholar]
- Fujii K.; Ikai Y.; Mayumi T.; Oka H.; Suzuki M.; Harada K.-i. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: elucidation of limitations of Marfey’s method and of its separation mechanism. Anal. Chem. 1997, 69, 3346–3352. [Google Scholar]
- Fujii K.; Shimoya T.; Ikai Y.; Oka H.; Harada K.-i. Further application of advanced Marfey’s method for determination of absolute configuration of primary amino compound. Tetrahedron Lett. 1998, 39, 2579–2582. [Google Scholar]
- Boonyapiwat B.; Panaretou B.; Forbes B.; Mitchell S. C.; Steventon G. B. Human phenylalanine monooxygenase and thioether metabolism. J. Pharm. Pharmacol. 2009, 61, 63–67. [DOI] [PubMed] [Google Scholar]
- Craig P. N. Interdependence between physical parameters and selection of substituent groups for correlation studies. J. Med. Chem. 1971, 14, 680–684. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Tounge B. A.; Bembenek S. D. Ligand binding efficiency: trends, physical basis, and implications. J. Med. Chem. 2008, 51, 2432–2438. [DOI] [PubMed] [Google Scholar]
- Reynolds C. H.; Bembenek S. D.; Tounge B. A. The role of molecular size in ligand efficiency. Bioorg. Med. Chem. Lett. 2007, 17, 4258–4261. [DOI] [PubMed] [Google Scholar]
- Jamieson C.; Moir E. M.; Rankovic Z.; Wishart G. Medicinal chemistry of hERG optimizations: highlights and hang-ups. J. Med. Chem. 2006, 49, 5029–5046. [DOI] [PubMed] [Google Scholar]
- Tang P.; Siu L.; Chen E.; Hotte S.; Chia S.; Schwarz J.; Pond G.; Johnson C.; Colevas A.; Synold T.; Vasist L.; Winquist E. Phase II study of ispinesib in recurrent or metastatic squamous cell carcinoma of the head and neck. Invest. New Drugs 2008, 26, 257–264. [DOI] [PubMed] [Google Scholar]
- Souid A.-K.; Dubowy R. L.; Ingle A. M.; Conlan M. G.; Sun J.; Blaney S. M.; Adamson P. C. A pediatric phase I trial and pharmacokinetic study of ispinesib: a children’s oncology group phase I consortium study. Pediatr. Blood Cancer 2010, 55, 1323–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiebig H. H.; Dengler W. A.; Roth T.. Human Tumor Xenografts. Predictivity, Characterization and Discovery of New New Anticancer Agents. In Relevance of Tumor Models for Anticancer Drug Development; Fiebig H. H., Burger A. M., Eds.; Karger: Basel, Switzerland, 1999; Vol. 54, pp 29–50. [Google Scholar]
- Starnes W. H. Lead tetraacetate oxidation of 4,4,4-triphenyl-1-butanol, 3,3,3-triphenyl-1-propanol, and 4,4,4-triphenylbutyric acid. J. Org. Chem. 1968, 33, 2767–2774. [Google Scholar]
- Harrison C. R.; Hodge P.; Hunt B. J.; Khoshdel E.; Richardson G. Preparation of alkyl chlorides, acid chlorides, and amides using polymer-supported phosphines and carbon tetrachloride: mechanism of these reactions. J. Org. Chem. 1983, 48, 3721–3728. [Google Scholar]
- Rodriguez R.; Chapelon A.-S.; Ollivier C.; Santelli M. Stereoselective synthesis of CD-ring precursors of vitamin D derivatives. Tetrahedron 2009, 65, 7001–7015. [Google Scholar]
- Warmuth R.; Munsch T. E.; Stalker R. A.; Li B.; Beatty A. Enantioselective synthesis of benzocyclic α,α-dialkyl-amino acids: new insight into the solvent dependent stereoselectivity of the TMSCN addition to phenylglycinol derived imines. Tetrahedron 2001, 57, 6383–6397. [Google Scholar]
- Bigge C. F.; Drummond J. T.; Johnson G.; Malone T.; Probert A. W.; Marcoux F. W.; Coughenour L. L.; Brahce L. J. Exploration of phenyl-spaced 2-amino-(5-9)-phosphonoalkanoic acids as competitive N-methyl-d-aspartic acid antagonists. J. Med. Chem. 1989, 32, 1580–1590. [DOI] [PubMed] [Google Scholar]
- Dockendorff C.; Jin S.; Olsen M.; Lautens M.; Coupal M.; Hodzic L.; Spear N.; Payza K.; Walpole C.; Tomaszewski M. J. Discovery of μ-opioid selective ligands derived from 1-aminotetralin scaffolds made via metal-catalyzed ring-opening reactions. Bioorg. Med. Chem. Lett. 2009, 19, 1228–1232. [DOI] [PubMed] [Google Scholar]
- Bhushan R.; Brückner H. Marfey’s reagent for chiral amino acid analysis: a review. Amino Acids 2004, 27, 231–247. [DOI] [PubMed] [Google Scholar]
- Luo L.; Carson J. D.; Dhanak D.; Jackson J. R.; Huang P. S.; Lee Y.; Sakowicz R.; Copeland R. A. Mechanism of inhibition of human KSP by monastrol: insights from kinetic analysis and the effect of ionic strength on KSP inhibition. Biochemistry 2004, 43, 15258–15266. [DOI] [PubMed] [Google Scholar]
- Morrison J. F. Kinetics of the reversible inhibition of enzyme-catalysed reactions by tight-binding inhibitors. Biochim. Biophys. Acta, Enzymol. 1969, 185, 269–286. [DOI] [PubMed] [Google Scholar]
- Leslie A. G. W.Jt. CCP4 ESF-EAMCB Newsl. Protein Crystallogr. 1992, No. 26. [Google Scholar]
- Evans P. Scaling and assessment of data quality. Acta Crystallogr., Sect. D 2006, 62, 72–82. [DOI] [PubMed] [Google Scholar]
- The CCP4 suite: programs for protein crystallography. Acta Crystallogr., Sect. D 1994, 50, 760–763 [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B.; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System, version 1.3r1; Schrodinger, LLC: New York, NY, 2009. [Google Scholar]
- Cruickshank D. Remarks about protein structure precision. Acta Crystallogr., Sect. D 1999, 55, 583–601. [DOI] [PubMed] [Google Scholar]
- Jaffe H. H. A rexamination of the Hammett equation. Chem. Rev. 1953, 53, 191–261. [Google Scholar]
- Fujita T.; Iwasa J.; Hansch C. A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar]
- Maltese M. Reductive demercuration in deprotection of trityl thioethers, trityl amines, and trityl ethers. J. Org. Chem. 2001, 66, 7615–7625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.