

Summary
Background
Inflammatory infiltrates, airway hyper-responsiveness, goblet cell hyperplasia and subepithelial thickening are characteristic of chronic asthma. Current animal models of allergen-induced airway inflammation generally concentrate on the acute inflammation following allergen exposure and fail to mimic all of these features.
Objective
The aim of this study was to use a murine model of prolonged allergen-induced airway inflammation in order to characterize the cells and molecules involved in the ensuing airway remodelling. Moreover, we investigated whether remodelling persists in the absence of continued allergen challenge.
Methods
Acute pulmonary eosinophilia and airways hyper-reactivity were induced after six serial allergen challenges in sensitized mice (acute phase). Mice were subsequently challenged three times a week with ovalbumin (OVA) (chronic phase) up to day 55. To investigate the persistence of pathology, one group of mice were left for another 4 weeks without further allergen challenge (day 80).
Results
The extended OVA challenge protocol caused significant airway remodelling, which was absent in the acute phase. Specifically, remodelling was characterized by deposition of collagen as well as airway smooth muscle and goblet cell hyperplasia. Importantly, these airway changes, together with tissue eosinophilia were sustained in the absence of further allergen challenge. Examination of cytokines revealed a dramatic up-regulation of IL-4 and tumour growth factor-β1 during the chronic phase. Interestingly, while IL-4 levels were significantly increased during the chronic phase, levels of IL-13 fell. Levels of the Th1-associated cytokine IFN-γ also increased during the chronic phase.
Conclusion
In conclusion, we have demonstrated that prolonged allergen challenge results in persistent airway wall remodelling.
Keywords: airway remodelling, allergic airway inflammation, asthma, eosinophils, Th2 cytokines
Introduction
Asthma is a chronic disease characterized by airway hyper-responsiveness (AHR), airway inflammation and reversible airway obstruction [1]. In addition to airway inflammation, structural changes are also evident within the airways [1]. This phenomenon is termed ‘airway remodelling’ and is thought to occur as a result of an imbalance in regeneration and repair mechanisms resulting in the abnormal regulation of extra-cellular matrix components [2]. Remodelling in the lung has been classified as an increase in airway wall thickness, due to many factors [3, 4]. Subepithelial fibrosis is a distinctive feature of airway remodelling and contributes to thickened airway walls due to the deposition of collagen types I, III and V along with other extracellular matrix proteins such as, tenascin and laminin. An increase in myocyte muscle mass is also characteristic of airway remodelling in asthma and may be due to myofibroblast proliferation that may also lead to further collagen production [5]. Excessive mucus secretion from hyperplastic goblet cells is a feature of the asthmatic airway and may lead to the occlusion of the airways [6]. The combination of these processes leads to airway narrowing and therefore, reduced lung function [3, 4]. Many mediators have been implicated in the fibrotic response. Lung specific over-expression of Th2 cell derived cytokines such as IL-4, IL-5, IL-9 or IL-13 have shown that this group of cytokines is capable of inducing subepithelial fibrosis in addition to their well-documented roles in airway inflammation and AHR [7–10]. TGF-β is a potent pro-fibrotic cytokine and is thought to play a crucial role in the progression of fibrotic events. TGF-β is released by many different cell types and has been shown to be a regulator of fibroblast/myofibroblast function with the production of many extracellular matrix proteins including collagen I [5]. Levels of TGF-β are elevated in asthmatic patients and expression has been shown to correlate with basement membrane thickness, fibroblast number and/or disease severity [11–13].
Animal models of the acute allergic response to inhaled antigens have been widely studied in order to try to elucidate the mechanisms leading to the development of inflammation and AHR during asthma. However, the majority of these models involve relatively short-term exposure to aerosolized antigen and many do not show the chronic inflammatory and epithelial changes or the mucosal inflammation characteristic of human asthma. Moreover, the inflammation and hyper-reactivity that are observed generally resolve after the cessation of allergen challenge. Repeated exposure of mice to ovalbumin (OVA) has been shown to increase the thickness of the epithelial layer of the trachea as well as deposition of collagenous material in the submucosal layer of the trachea [14]. However, remodelling was not determined in the lower airways nor was the matrix deposition associated with fibrotic mediators during this model. Recently, it has been demonstrated that mice exposed to prolonged allergen challenge and subsequently left for up to 8 weeks developed significant airway remodelling in association with an increase in lung resistance [15]. However, again the association between airway remodelling and possible causative mediators was not fully evaluated. The hypothesis behind the study was that prolonged allergen challenge would lead to irreversible structural changes of the airways characteristic of airway remodelling observed in chronic asthmatics. We hypothesize that prolonged allergen challenge would result in an increase in profibrotic factors, such as TGF-β, and lead to the characteristic features of airway remodelling such as increased matrix deposition and smooth muscle cell proliferation. Moreover, we hypothesized that this prolonged allergen challenge would lead to persistent changes to airway structure even if allergen challenge was stopped. To this end we have characterized a model of prolonged allergen-induced airway inflammation in order to investigate the development and persistence of remodelling immediately following chronic allergen exposure as well as after a period of non-exposure. We have determined the extent of matrix dysregulation and the contribution of inflammatory cells and pro-fibrotic mediators, as well as the occurrence of changes in airway function. This study reports an increase in allergen driven remodelling characterized by deposition of collagen in the airways and airway smooth muscle (ASM) cell proliferation, together with an increase in IL-4 and TGF-β1 expression. Moreover, airway remodelling and eosinophilic inflammation but not AHR, were shown to be persistent, even in the absence of further allergen challenge.
Methods
Allergen-induced airway inflammation
Acute OVA-induced airway inflammation was induced in female BALB/c mice as previously described [16]. Briefly, mice were sensitized using OVA (Sigma, Poole, UK) at a concentration of 0.01 mg/mouse in 0.2mL alum (Au-Gel-S, Serva Electrophoresis, Heidelberg, Germany) intraperitoneal on days 0 and 12. Control mice received the same volume of phosphate-buffered saline (PBS) in alum. All groups of mice were then challenged daily with 5% OVA (aerosolized for 20 min) via the airways between days 18 and 23. Mice were killed by exsanguination under terminal anaesthesia at 24 h after the last serial OVA administration on day 24. We have previously shown that this protocol results in increased AHR, inflammation of the airways and Th2 cytokine production, and is termed the acute period for the purposes of this study. Prolonged inflammation was induced by subsequent exposure of mice to aerosolized OVA (5%) three times a week for 20 min until groups of mice were killed on days 35 and 55 (chronic phase). In a separate group of mice, OVA challenging was ceased on day 55 and mice were killed 1 month later on day 80. Control mice were treated in parallel to test groups and were killed on days 24, 35, 55 and 80. There was no significant differences between these control mice killed at each time point, therefore groups were pooled and included on graphs as alum controls.
Measurement of AHR
Airway responsiveness was measured indirectly by whole body plethysmography to calculate enhanced pause (Penh: Buxco Technologies, Petersfield, UK). Response to inhaled methacholine (Sigma) at concentrations of 3–100 mg/mL were measured for 1 min, as described previously [17]. Results are shown for Penh after acute (day 24) and prolonged allergen challenge on days 35, 55 and 80 in mice sensitized to PBS/alum (alum controls) or OVA/alum.
Bronchoalveolar lavage
Bronchoalveolar lavage (BAL) was performed as described [16]. Briefly, the airways of the mice were lavaged three times with 0.4 mL of PBS via a tracheal cannula. BAL fluid was centrifuged (700 g, 5 min, 4 °C) and cells were recovered. BAL cell supernatants were removed and analysed for T cell derived cytokines by ELISA.
Histology
Paraffin-embedded sections (4 μm) were stained with haematoxylin/eosin (H&E) to evaluate general morphology, and goblet cells were visualized on periodic acid-Schiff (PAS)-stained lung sections. Matrix deposition was assessed on Martius scarlet blue (MSB)-stained sections. All slides were examined by two reviewers in a random blinded fashion. Image analysis was performed on MSB-stained lung sections using Scion Image Analysis software package (Scion Corporation, Frederick, MD, USA) adapted from the literature [18]. Digital photographs of four bronchioles per tissue section were taken at × 40 magnification and these images were converted into monochrome. Ten measurements of 20 μm lines from each of the four bronchioles were drawn at a right angle from the basement membrane into the submucosa and the mean density of staining intensity along the 20 μm was calculated and expressed as pixels per μm2.
Quantification of airway eosinophils and smooth muscle cells
Lung tissue eosinophils were counted in H&E-stained lung tissue sections at × 20 magnification. Five hundred inflammatory leukocytes were counted in random regions along major airways (30 μm length) and the percentage of eosinophils calculated from total cells counted.
ASM cells were counted in proliferating cell nuclear antigen (PCNA) immunostained lung tissue sections at × 40 magnification. Dark elongated smooth muscle cell nuclei and round nuclei in the muscle bundles were counted in three random measurements (30 μm length) per bronchiole in three to four bronchioles per mouse [19]. Cells positive for PCNA stained dark brown. Total cell counts and those positive for PCNA were counted, meaned and divided by the number of measurements taken. All slides were examined by both investigators, in a random blinded fashion at the end of the study.
Immunohistochemistry
Paraffin sections (4–5 μm) from alumand OVA-treated mice were stained with rabbit anti-mouse TGF-β1 (Santa Cruz, Heidelberg, Germany) using an avidin/biotin staining method. All incubations were carried out under humidified conditions and slides were washed twice between steps for 5 min each in 0.1 M PBS/0.1% Tween. Briefly, endogenous peroxidase was blocked by incubation for 20 min in methanol containing 3% hydrogen peroxide. Non-specific staining due to cross-reaction with endogenous avidin or biotin was then blocked by incubation with avidin solution followed by biotin solution, both for 15 min. Thereafter, sections were overlaid with 20% donkey serum in PBS for 15 min, and then incubated with antibodies specific for TGF-β1. Bound antibody was visualized by incubation with biotinylated rabbit immunoglobulin for TGF-β1 (diluted in 1% normal mouse serum in PBS), followed by steptavidin complex for 30 min each. Finally, slides were incubated with a peroxidase substrate, counterstained with haematoxylin, dehydrated, mounted and studied by light microscopy. Lung tissue sections were stained with PCNA according to manufacturer’s instructions (DAKO, Ely, UK).
Collagen analysis
Collagen content was measured in lung tissue homogenates by a biochemical assay according to the manufacturer’s instructions (Sircol collagen assay, Biocolor, Belfast, UK). Lung tissue (100 mg) was homogenized in 2 mL HBSS, centrifuged (800 g, 10 min) and the supernatant collected and analysed.
Cytokine analysis
Cytokines were analysed in BAL fluid and lung tissue homogenates. Paired antibodies for murine IL-4 and IFN-γ (PharMingen, Oxford, UK), IL-5 (Endogen-Pierce, Tattenhall, UK), TGF-β1 and eotaxin/CCL11 (R&D Systems, Abingdon, UK) were used in standardized sandwich ELISAs according to the manufacturer’s protocol. Kits to measure IL-13 were purchased from R&D Systems.
Statistical analysis
Results were expressed as mean ± SEM with a group size of 4–17 from four different experiments. Data was analysed by anova (Kruskal–Wallis) or Mann–Whitney U-test where appropriate and statistical significance was accepted when P<0.05.
Results
We have developed a model of prolonged allergen challenge that shares many characteristics similar to the airway re-modelling observed in human chronic asthma. Mice were subjected to a period of acute allergen exposure, which induced significant Th2-driven eosinophilic inflammation in conjunction with AHR (acute phase). Thereafter, in order to mimic repeated allergen exposure, mice were subjected to OVA challenge three times a week until they were killed on days 35 and 55 (chronic phase). Airway remodelling was also assessed in mice 1 month after the final day 55 OVA challenge in order to investigate persistence of airway remodelling (day 80).
Airway inflammation is sustained after prolonged OVA challenge
Acute allergen challenge resulted in large inflammatory infiltrates in perivascular and peribronchiolar regions, consisting mainly of eosinophils as previously described ([16]; Fig. 1a, arrows depict eosinophils). This inflammatory infiltrate was sustained during prolonged OVA challenge. Interestingly, the composition of the infiltrate became comparatively more mononuclear in nature, although eosinophils still represented a significant proportion of the infiltrate. On day 35, 22.7 ± 1.8% (n = 6) of the peribronchiolar infiltrates were eosinophils and by day 55, 15.6 ± 1.4% (n = 6) of cells in the infiltrates were eosinophils in comparison with alum controls (0 ± 0%, n = 6; Fig. 1a photomicrographs). Moreover, in the absence of further allergen challenge, eosinophil infiltration was still evident (7.31 ± 0.53%, n = 6; Fig. 1a, day 80).
Fig. 1.

Effect of prolonged ovalbumin (OVA) challenge on lung tissue pathology. Representative photomicrographs of (a) haematoxylin/eosin (H&E)- (arrows depict eosinophils), (b) periodic acid-Schiff (PAS)- and (c) Martius scarlet blue (MSB)-stained lung sections from alum control mice, OVA challenged mice at day 24 (acute phase) and day 55 (chronic phase), and mice that were no longer exposed to OVA from day 55 and killed at day 80 are shown (original magnification × 40). Data is representative of n = 9–17/group/time point.
Goblet cell hyperplasia occurs after acute and prolonged OVA challenge
Excessive mucus secretion from hyperplastic goblet cells is a characteristic feature of the asthmatic airway. In order to determine the extent of mucus cell metaplasia following prolonged allergen challenge, paraffin-embedded sections of lung were stained with PAS. An increase in PAS-positive cells was observed in the bronchial epithelium during acute inflammation in comparison with alum controls and was sustained throughout prolonged OVA challenge (Fig. 1b, pink staining). When allergen challenge was ceased, some PAS-positive cells were seen in the bronchiolar epithelium, albeit at a significantly reduced rate compared with day 55, and numbers were not significantly different from alum controls (Fig. 1b, day 80).
Prolonged OVA challenged results in increased matrix deposition, which is persistent in the absence of further allergen exposure
A prominent feature of airway remodelling observed in the airways of patients with asthma is an increase in matrix deposition in the subepithelial region of the bronchioles. In order to determine if prolonged OVA challenge affected matrix regulation, paraffin-embedded sections of lung were stained with MSB to show collagen deposition (Fig. 1c, blue represents collagen). Alum control mice showed little collagen staining around the airways and the vasculature. Acute OVA challenge was associated with an increase in fine collagen fibrils within some inflammatory peri-bronchiolar and perivascular infiltrates. In contrast, during prolonged OVA challenge an increase in matrix was observed in the subepithelial layer of the airways and also in peri-vascular regions as shown by a dramatic increase in both the extent of collagen fibril deposition and intensity of staining. Dense collagen fibrils were seen in the subepithelial and submucosal areas, and in between cells within inflammatory infiltrates. Importantly, this level of collagen deposition did not resolve when allergen challenge was ceased, indicating persistence of airway remodelling (Fig. 1c, day 80). In order to quantitate this increase in matrix deposition, image analysis was performed on MSB-stained lung sections. Image analysis showed a significant increase in the density of the submucosal region during the acute phase in comparison with alum controls (Fig. 2a). A further increase in density was observed on days 35 and 55 during the chronic phase. Density was maintained at day 80, in the absence of allergen challenge indicating the persistence of pathological changes.
Fig. 2.

Effect of prolonged ovalbumin (OVA) challenge on matrix deposition. (a) Image analysis of Martius scarlet blue-stained lung sections from mice treated with alum or OVA during acute and prolonged allergen challenge. Random measurements from the basement membrane into the submucosa (10 measurements of 20 μm in length) were taken and the mean density calculated from four bronchioles per mouse, n = 4 mice per group. (b) Levels of total collagen were measured in lung homogenate samples taken throughout the model. Data is expressed as mean ± SEM, n = 4/group; *P<0.05 in comparison with alum control mice.
Levels of matrix within the lung tissue were also measured quantitatively using a biochemical assay of total collagen levels. No significant increase in collagen levels was observed at day 24 in comparison with alum control mice. Levels of collagen were increased by 3-fold during the chronic phase in comparison with alum control and acute challenged mice on days 35 and 55 (Fig. 2b). In addition, even in the absence of allergen challenge, increased levels of total collagen were maintained at day 80 in comparison with alum control mice (Fig. 2b).
An increase in ASM cell proliferation occurs during prolonged allergen challenge
An increase in ASM mass is characteristic of human chronic asthma [20]. To determine the extent of ASM cell proliferation during prolonged allergen challenge, slides were stained to evaluate PCNA expression and total and PCNA-positive ASM cells were counted during acute and chronic allergen challenge. PCNA is a cell surface antigen that is expressed by proliferating cells in S-phase of the cell cycle. An increase in proliferating cells, as shown by positive PCNA staining in the smooth muscle layer was observed during the chronic phase in comparison with the acute phase (Fig. 3a, arrows depict positive cells). ASM counting showed that acute allergen challenge increased the number of smooth muscle cells, which was maintained on day 35 during prolonged allergen challenge in comparison with alum control mice (Fig. 3b). On day 55 during prolonged allergen challenge, a dramatic increase in the number of ASM cells was observed in comparison with alum and day 24 mice. In the absence of further allergen challenge (day 80), this increase in smooth muscle cell number was maintained. By counting the number of ASM cells positive for PCNA along 30 μm of the basement membrane, we observed that there was no increase in ASM proliferation after acute allergen challenge (alum: 0.61 cells ± 0.1 vs. day 24: 0.64 cells ± 0.2). However, during prolonged allergen challenge, numbers of proliferating cells increased 4-fold above day 24 on days 35 and 55 (2.53 cells ± 0.33 and 2.64 cells ± 1.2). Interestingly, in the absence of further allergen challenge on day 80, the number of proliferating cells was further increased 2.5-fold in comparison with day 55 (7.0 cells ± 0.69).
Fig. 3.
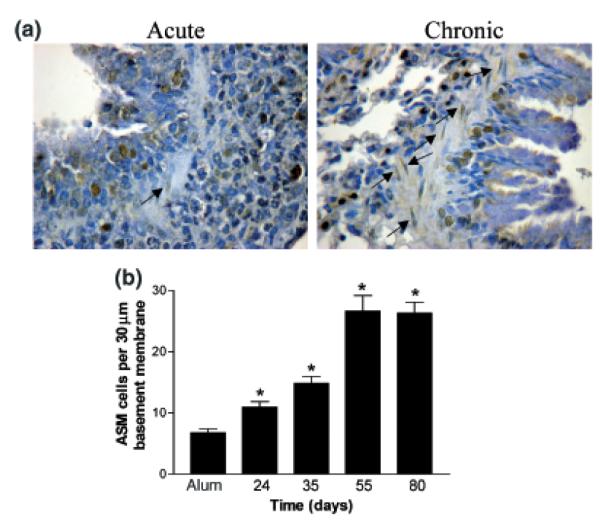
Effect of prolonged ovalbumin (OVA) challenge on airway smooth muscle cell proliferation. (a) Representative photomicrographs of paraffin-embedded lung sections from day 24 acute OVA challenged mice and mice from day 55 during prolonged allergen challenge, stained with antibodies against proliferating cell nuclear antigen (PCNA) (original magnification × 40. PCNA-positive cells stain brown). Data is representative of n = 4/ group/time point. Ig control sections were negative. (b) Total and proliferating airway smooth muscle cell counts from lung tissue sections taken from alumand OVA-treated mice throughout the model. Data is expressed as mean ± SEM, n = 4/group; *P<0.05 in comparison with alum control mice.
IL-4 is the dominant Th2 type cytokine in the lung during prolonged allergen challenge
Increased levels of Th2 cytokines are indicative of the allergic response. Moreover, Th2 cytokines, such as IL-13, have been implicated in the development of fibrosis in a variety of transgenic murine models. We used ELISA to measure concentrations of Th2-associated cytokines IL-4, IL-5 and IL-13 and the Th1-associated cytokine IFN-γ within BAL (Fig. 4, left panel) and lung tissue (Fig. 4, right panel) compartments after prolonged challenge with antigen. The response to acute OVA challenge in the BAL and lung tissue was associated with significant increases in IL-4, IL-5 and IL-13 in comparison with alum controls (Figs 4a–f). Following prolonged OVA challenge, BAL and lung tissue levels of IL-4 were significantly raised in comparison with alum controls (Figs 4a and b). Levels of IL-5 peaked on day 35 in both BAL and lung tissue during prolonged allergen challenge (Figs 4c and d). In contrast, BAL and lung tissue levels of IL-13 were shown to decrease during prolonged OVA challenge (Figs 4e and f). Interestingly, BAL and lung tissue levels of IFN-γ were also increased during the chronic phase, achieving significance on day 55 in the lung tissue in comparison with alum controls (Figs 4g and h). In the absence of further allergen challenge after day 55, cytokines were no longer detectable in the BAL. In contrast, IL-4 remained significantly elevated in the lung tissue in comparison with alum controls, whereas IL-5, IL-13 and IFN-γ returned to baseline (Fig. 4, day 80).
Fig. 4.

Effect of prolonged ovalbumin (OVA) challenge on cytokine levels. IL-4 (a, b), IL-5 (c, d), IL-13 (e, f) and IFN-γ (g, h) levels were measured in bronchoalveolar lavage (left panel) and lung tissue homogenates (right panel) by ELISA in samples from alum controls, acute and chronic phase killed 24 h after the final OVA challenge and mice from day 80. Data are expressed as mean ± SEM; n = 4–17/group and *P<0.05 in comparison with alum control mice. ND, non-detectable.
Active TGF-β1 is increased during prolonged allergen challenge
TGF-β is a fibrotic cytokine previously shown to be up-regulated in multiple fibrotic conditions including severe asthma [21]. TGF-β is potentially expressed by a variety of different cell types, therefore, cellular localization of total TGF-β1 was determined by IHC (Fig. 5ai–iv). TGF-β1 was found to be constitutively expressed in lung tissue before allergen challenge (Fig. 5ai). Low levels of expression was seen in some bronchiolar and alveolar epithelial cells. Expression of TGF-β1 changed significantly after acute OVA challenge (Fig. 5aii). Less staining was observed within the airway epithelium in comparison with alum controls. Positive staining was shown on the surface of mononuclear cells within the inflammatory infiltrate, but secreted TGF-β1 was also observed within the submucosa. Following chronic allergen exposure, TGF-β1 staining was more dense within the inflammatory infiltrate. Intense staining was seen between cells in the infiltrate in comparison with acute allergen challenge, as well as in the smooth muscle layer (Fig. 5aiii). In the absence of further allergen exposure after day 55, expression of TGF-β1 remained elevated above acute allergen challenge lungs (day 80). Staining was less cell bound than day 55, with more staining being observed between the cells in the submucosa (Fig. 5aiv). Levels of active TGF-β1 in lung homogenates were measured during prolonged allergen challenge in comparison with alum controls and acute phase mice using a specific ELISA (Fig. 5b). There was no difference in levels of active TGF-β1 following acute allergen challenge in comparison with alum control mice (Fig. 5b). A significant increase in levels of active TGF-β1 was observed on day 35 in comparison with alum-treated mice. This increase in levels of active TGF-β1 was maintained up to day 55 during prolonged allergen challenge (Fig. 5b). If allergen challenging was ceased at day 55 and mice were killed at day 80, levels of active TGF-β1 returned to levels comparable with those observed during the acute phase.
Fig. 5.

Effect of prolonged ovalbumin (OVA) challenge on levels of TGF-β. (a) Representative photomicrographs of paraffin-embedded lung sections from alum controls (i), OVA challenged mice at day 24 (ii), day 55 (iii) and day 80 (iv), stained with antibody against TGF-β1. Data is representative of n = 4/group/ time point. Ig controls were negative (original magnification × 40). (b) Levels of active TGF-β1 were measured in lung homogenates by ELISA taken from acute and chronic OVA challenged mice. Data is expressed as mean ± SEM, n = 5–10/group; *P<0.05 in comparison with alum control mice. (c) Levels of eotaxin were measured in lung tissue homogenates by ELISA from mice during prolonged allergen challenge. Data is expressed as mean ± SEM, n = 4/group; *P<0.05 in comparison with alum control mice.
Levels of eotaxin are increased during acute and prolonged allergen challenge
Levels of eotaxin were also measured in the lung tissue during prolonged allergen challenge (Fig. 5c). Levels of eotaxin were increased after acute allergen challenge and levels peaked on day 35 during prolonged allergen challenge (Fig. 5c). Levels remained elevated on day 55 in comparison with alum control mice. In the absence of further allergen challenge, levels of eotaxin in the lung tissue also remained elevated in comparison with alum controls (Fig. 5c).
Airway hyper-reactivity is sustained during prolonged OVA but decreases to baseline in the absence of further allergen exposure
Figure 6 shows airway hyper-reactivity to increasing concentrations of methacholine in OVA-treated mice and alum control mice. Acute allergen challenge (day 24) resulted in a dose-dependent increase in AHR in comparison with alum control mice (Fig. 6). This increase in AHR was shown to be sustained on day 35 during prolonged allergen challenge. On day 55, although AHR remains elevated in comparison with alum controls, the magnitude of the shift in the dose–response curve is decreased in comparison with earlier time points. In the absence of continued allergen exposure, by day 80 hyper-reactivity to cholinergic stimulation was not significantly different from alum control mice.
Fig. 6.

Effect of prolonged allergen challenge on airway hyper-responsiveness. Airway hyper-responsiveness was measured 24 h after each final OVA challenge on days 24 (acute phase), 35 and 55, and in the absence of further allergen exposure on day 80 using a Buxco system where mice were exposed to increasing concentrations of methacholine (3–100 mg/mL). Values are expressed as mean ± SEM, n = 8–12/group/time point. *P<0.05 in comparison with alum control mice.
Discussion
Chronic allergen-induced airway inflammation in atopic asthmatics is characterized by eosinophilia, AHR as well as irreversible changes to the airway structure, termed airway remodelling [1, 22]. However, animal models of airway inflammation are generally acute and induce eosinophilia and AHR only (reviewed by Lloyd et al. [23]). We set out to establish a model of prolonged allergen-induced airway inflammation to try and mimic the tissue remodelling observed in asthmatic patients. We found that extending the number and period of allergen challenges induced airway remodelling as well as airway inflammation and AHR in mice. This airway remodelling was characterized by deposition of collagen around the airways and vasculature in the lungs, and ASM cell and goblet cell hyperplasia. Deposition of collagen occurred alongside an increase in the levels of TGF-β1 and IL-4. Importantly, this airway remodelling was shown to persist even in the absence of further allergen challenge.
Histological assessment of MSB-stained lung sections, image analysis and biochemical measurements of collagen revealed a modest rise in matrix deposition during acute inflammation but levels increased significantly during the chronic phase. Moreover, prolonged allergen challenge was shown to induce ASM cell proliferation, which was further increased in the absence of further allergen exposure. These increases in proliferating smooth muscle cells coincides with the increase in matrix deposition. Collagen fibril deposition was evident in the subepithelial and smooth muscle layer, with fibrils observed between individual cells. It has been suggested that allergen challenge results in phenotypic changes in smooth muscle cells that are then able to secrete a range of factors capable of influencing secretion of extracellular matrix, such as TGF-β [19]. Matrix deposition may also result from the recruitment and activation of fibroblasts. The particular contribution of smooth muscle cells and fibroblasts to the changes in matrix regulation after chronic allergen challenge warrant further investigation.
Together these data indicate that our model of prolonged allergen challenge of sensitized mice induces matrix changes similar to those observed in asthmatic patients [22]. Repeated exposure of mice to OVA has been shown to increase the thickness of the epithelial layer of the trachea as well as deposition of collagenous material in the submucosal layer of the trachea [14]. However, remodelling was not determined in the lower airways during this model. Other groups have also indicated that extracellular matrix deposition occurs after prolonged OVA challenge [24–26]. However, our study demonstrates along with multiple markers of tissue fibrosis, that prolonged allergen leads to persistent airway remodelling, being maintained even after the cessation of allergen challenge.
Initiation and progression of fibrosis is thought to depend on the expression of mediators that influence local cell function. In asthma, exposure of patients to allergen results in chronic inflammation, with recruitment of inflammatory cells and subsequent secretion of Th2-associated cytokines, such as, IL-4, IL-5, IL-9 and IL-13, as well as growth factors such as, TGF-β from infiltrating leukocytes and lung resident cells (reviewed by Renauld [27]). TGF-β is a cytokine implicated in the development and maintenance of the fibrotic response. It has been shown that in vitro stimulation of bronchial fibroblasts from asthmatics with TGF-β resulted in their transformation into myofibroblasts, with the expression of α smooth muscle actin and pro-collagen I [28]. Moreover, up-regulation of TGF-β expression has been documented in the airways of asthmatic patients [11, 12]. In our mouse model, deposition of collagen and ASM proliferation occurred in conjunction with an increase in lung tissue TGF-β1 protein levels during the chronic phase. In addition, we determined that mononuclear cells, likely macrophages, were the main source of TGF-β1 during prolonged OVA challenge. Interestingly, others have suggested that eosinophils are potential sources of TGF-β [11, 12, 18]. However, we could not find TGF-β protein associated with eosinophils in lungs during our model. Previous data from clinical studies in asthmatic patients report a compartmentalization of TGF-β expression occurring after allergen challenge [29]. Non-asthmatic patients showed epithelial staining for TGF-β1 whereas asthmatic patients had no epithelial TGF-β1 staining but positive staining in the inflammatory infiltrates in the submucosal region. We documented a similar phenomenon in our mouse model, showing a switch from epithelial TGF-β1 staining in unchallenged mice to staining of submucosal and inflammatory infiltrate after acute and prolonged allergen challenge. It is possible that under normal conditions TGF-β1 produced by the bronchial epithelial cells acts in a regulatory capacity, functioning to limit inappropriate immune responses to external allergens, and promoting pulmonary homeostasis. However, after allergen challenge TGF-β1 is produced predominantly by inflammatory cells as well as resident lung tissue cells beneath the basement membrane that allows for interaction with fibroblasts in the connective tissue, influencing the development of fibrotic reactions.
Lung specific over-expression of Th2 cell derived cytokines such as IL-4, IL-5, IL-9, IL-11 or IL-13 have shown that this group of cytokines is capable of inducing subepithelial fibrosis in addition to their well-documented roles in airway inflammation and AHR [7–10, 30]. In our study, levels of IL-4 were found to dominate in the BAL and lung during the chronic inflammatory phase. An increase in IL-4 levels was also observed during bleomycin-induced pulmonary fibrosis in mice [31], and IL-4 deficient mice had significantly less fibrosis than wild-type controls in this model [32]. IL-4 has been described by several investigators to be a fibrogenic cytokine, and has been shown to regulate collagen biosynthesis by lung fibroblasts in vitro [33]. Levels of IL-5 in our model were also increased during chronic allergen exposure with levels peaking at day 35. Anti-IL-5 treatment has been shown to decrease reticulin deposition during a murine model of allergen-induced airway inflammation [34]. Moreover, recent clinical data using a humanized antibody to IL-5 has shown an anti-fibrotic role for IL-5 [18]. Treatment with anti-IL-5 decreased expression of tenascin and pro-collagen III in the lung, together with a decrease in airway eosinophils. Interestingly, although levels of IL-13 were increased in the acute phase of our model, levels were comparatively reduced during the chronic phase. However, this does not rule out IL-13 as a contributor to this pathology since lung tissue levels are still elevated in comparison with alum control mice. A role for IL-13 in the development of airway remodelling came from experiments where lung specific overexpression of IL-13 in mice induced subepithelial fibrosis [10]. However, the pathophysiology of this particular IL-13 transgenic mouse is more akin to that of emphysema/chronic obstructive pulmonary disease rather than the airway remodelling observed in chronic asthmatics. It has been shown that IL-4 and IL-13 act in conjunction to ensure the rapid onset of a Th2-like response (reviewed by McKenzie [35]). In our model, IL-13 may act in synergy with IL-4 and with CC-chemokines such as eotaxin and MCP-1 to contribute to the fibrotic response [10]. Stimulation of human fibroblasts with IL-4 or IL-13 resulted in proliferation and an increase in CCL11/eotaxin and CCL2/MCP-1 release [28, 36]. Moreover, it has been shown in vitro that IL-13 acting in concert with TGF-β can increase the release of eotaxin from human fibroblasts [37]. Indeed, we have shown that levels of lung CCL11/eotaxin were also elevated during prolonged allergen challenge. IL-13 has been shown to activate TGF-β1 production in vivo [38]. It has also been demonstrated that IL-13 and IL-4 can stimulate production of TGF-β2 from human bronchial epithelial cells in vitro [28, 39]. Thus, in our model it is conceivable that IL-13 and IL-4 produced during the acute phase may stimulate the production of TGF-β in vivo, which may contribute to the collagen deposition observed during prolonged allergen challenge. In our study, we hypothesize that levels of IL-4 combined with IL-5 and IL-13 within the lung tissue promote fibrosis and result in airway remodelling. In addition, there may be contribution from other profibrotic cytokines such as platelet derived growth factor (PDGF), connective tissue growth factor (CTGF), epidermal growth factor (EGF) as well as IL-11, all of which have been observed in biopsies of patients with remodelled airways [40–42]. Interestingly, levels of the Th1-associated cytokine IFN-γ were also shown to increase at day 55 in the chronic phase in comparison with control mice. An increase in levels of IFN-γ in BAL fluid and serum from asthmatic patients have also been documented [43, 44]. Moreover, IFN-γ has been shown to increase expression of markers of activation on eosinophils, for example, CD69 suggesting a role for IFN-γ in eosinophil activation [45].
AHR is a hallmark feature of human asthma and we were interested to see how the structural changes reported in the prolonged allergen challenge model impacted on lung function. AHR was increased after allergen challenge during both the acute allergen challenge phase of our model as well as during chronic allergen challenge in comparison with alum controls. However, the magnitude of the response on day 55 was less than that on day 24, perhaps due to the decline in levels of IL-13, a cytokine shown to be essential for AHR in mice [46, 47]. In the absence of continued allergen exposure after day 55, there was no significant difference between alum controls and day 80 mice. A similar improvement in AHR and a decline in eosinophilia was also observed in asthmatic patients when allergen exposure to house dust mites was eliminated via high altitude allergen avoidance [48]. In our model, we also saw a decline in the percentage of eosinophils in airway infiltrates after allergen challenge was stopped. Interestingly, eosinophils did not disappear completely, and were still significantly raised above baseline, probably due to elevated levels of eotaxin still present, although in vitro evidence suggests that TGF-β and IL-13 cooperate to induce eotaxin secretion from pulmonary fibroblasts, providing a possible mechanism for maintained eosinophil recruitment [37]. Overall our data indicate that eosinophils may contribute to the development and maintenance of remodelling, through release of cytokines and fibrogenic mediators, but not AHR. It is possible that there may be a threshold of eosinophils necessary to induce AHR, or the decline in AHR may be due to the decrease in levels of cytokines such as IL-13 or IL-5 that are thought to contribute to AHR in sensitized mice [46, 47, 49]. Although AHR was not maintained in the absence of allergen challenge, airway remodelling was shown to be persistent, with no change in symptoms after the cessation of allergen challenge.
In summary, the study presented here has shown that prolonged allergen challenge of sensitized mice results in persistent remodelling of the airways. Collagen deposition, ASM proliferation, fibrotic mediator secretion were observed together with the features of acute inflammation, namely eosinophilia and Th2 cytokine production. Moreover, the structural airway changes are sustained even in the absence of further allergen challenge. The protocol described represents an improved model for the study of airway inflammation in mice since it faithfully reproduces many of the characteristic features of the chronic human disease and may be beneficial for testing novel therapies for the treatment of chronic asthma.
Acknowledgements
The authors would like to thank Lorraine Lawrence for technical assistance with histology. This work was supported by The Wellcome Trust (ref #057704).
References
- 1.Bousquet J, Jeffery PK, Busse WW, Johnson M, Vignola AM. Asthma. From bronchoconstriction to airways inflammation and remodeling. Am J Respir Crit Care Med. 2000;161:1720–45. doi: 10.1164/ajrccm.161.5.9903102. [DOI] [PubMed] [Google Scholar]
- 2.Elias JA, Zhu Z, Chupp G, Homer RJ. Airway remodeling in asthma. J Clin Invest. 1999;104:1001–6. doi: 10.1172/JCI8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busse W, Elias J, Sheppard D, Banks-Schlegel S. Airway remodeling and repair. Am J Respir Crit Care Med. 1999;160:1035–42. doi: 10.1164/ajrccm.160.3.9902064. [DOI] [PubMed] [Google Scholar]
- 4.Jeffery PK, Wardlaw AJ, Nelson FC, Collins JV, Kay AB. Bronchial biopsies in asthma. An ultrastructural, quantitative study and correlation with hyperreactivity. Am Rev Respir Dis. 1989;140:1745–53. doi: 10.1164/ajrccm/140.6.1745. [DOI] [PubMed] [Google Scholar]
- 5.Brewster CE, Howarth PH, Djukanovic R, Wilson J, Holgate ST, Roche WR. Myofibroblasts and subepithelial fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1990;3:507–11. doi: 10.1165/ajrcmb/3.5.507. [DOI] [PubMed] [Google Scholar]
- 6.Wanner A. The role of mucus in chronic obstructive pulmonary disease. Chest. 1990;97:11S–5S. doi: 10.1378/chest.97.2_supplement.11s. [DOI] [PubMed] [Google Scholar]
- 7.Rankin JA, Picarella DE, Geba GP, et al. Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc Natl Acad Sci USA. 1996;93:7821–5. doi: 10.1073/pnas.93.15.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JJ, McGarry MP, Farmer SC, et al. Interleukin-5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med. 1997;185:2143–56. doi: 10.1084/jem.185.12.2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Temann UA, Geba GP, Rankin JA, Flavell RA. Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J Exp Med. 1998;188:1307–20. doi: 10.1084/jem.188.7.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu Z, Homer RJ, Wang Z, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Minshall EM, Leung DY, Martin RJ, et al. Eosinophil-associated TGF-beta1 mRNA expression and airways fibrosis in bronchial asthma. Am J Respir Cell Mol Biol. 1997;17:326–33. doi: 10.1165/ajrcmb.17.3.2733. [DOI] [PubMed] [Google Scholar]
- 12.Ohno I, Nitta Y, Yamauchi K, et al. Transforming growth factor beta 1 (TGF beta 1) gene expression by eosinophils in asthmatic airway inflammation. Am J Respir Cell Mol Biol. 1996;15:404–9. doi: 10.1165/ajrcmb.15.3.8810646. [DOI] [PubMed] [Google Scholar]
- 13.Vignola AM, Chanez P, Chiappara G, et al. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am J Respir Crit Care Med. 1997;156:591–9. doi: 10.1164/ajrccm.156.2.9609066. [DOI] [PubMed] [Google Scholar]
- 14.Temelkovski J, Hogan SP, Shepherd DP, Foster PS, Kumar RK. An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax. 1998;53:849–56. doi: 10.1136/thx.53.10.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leigh R, Ellis R, Wattie J, et al. Dysfunction and remodeling of the mouse airway persist after resolution of acute allergen-induced airway inflammation. Am J Respir Cell Mol Biol. 2002;27:526–35. doi: 10.1165/rcmb.2002-0048OC. [DOI] [PubMed] [Google Scholar]
- 16.Lloyd CM, Gonzalo JA, Nguyen T, et al. Resolution of bronchial hyperresponsiveness and pulmonary inflammation is associated with IL-3 and tissue leukocyte apoptosis. J Immunol. 2001 doi: 10.4049/jimmunol.166.3.2033. [DOI] [PubMed] [Google Scholar]
- 17.Hamelmann E, Schwarze J, Takeda K, et al. Noninvasive measurement of airway responsiveness in allergic mice using barometric plethysmography [see comments] Am J Respir Crit Care Med. 1997;156:766–75. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 18.Flood-Page P, Menzies-Gow A, Phipps S, et al. Anti-IL-5 treatment reduces deposition of ECM proteins in the bronchial subepithelial basement membrane of mild atopic asthmatics. J Clin Invest. 2003;112:1029–36. doi: 10.1172/JCI17974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moir LM, Leung SY, Eynott PR, et al. Repeated allergen inhalation induces phenotypic modulation of smooth muscle in bronchioles of sensitized rats. Am J Physiol Lung Cell Mol Physiol. 2003;284:L148–59. doi: 10.1152/ajplung.00105.2002. [DOI] [PubMed] [Google Scholar]
- 20.Dunhill MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of the bronchi in normal subjects, in status asthmaticus, chronic bronchitis, and in emphysema. Thorax. 1969;24:176–9. doi: 10.1136/thx.24.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Redington AE, Madden J, Frew AJ, et al. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am J Respir Crit Care Med. 1997;156:642–7. doi: 10.1164/ajrccm.156.2.9605065. [DOI] [PubMed] [Google Scholar]
- 22.Roche WR, Beasley R, Williams JH, Holgate ST. Subepithelial fibrosis in the bronchi of asthmatics. Lancet. 1989;i:520–4. doi: 10.1016/s0140-6736(89)90067-6. [DOI] [PubMed] [Google Scholar]
- 23.Lloyd CM, Gonzalo JA, Coyle AJ, Gutierrez-Ramos JC. Mouse models of allergic airway disease. Adv Immunol. 2001;77:263–95. doi: 10.1016/s0065-2776(01)77019-8. [DOI] [PubMed] [Google Scholar]
- 24.Blyth DI, Pedrick MS, Savage TJ, Hessel EM, Fattah D. Lung inflammation and epithelial changes in a murine model of atopic asthma. Am J Respir Cell Mol Biol. 1996;14:425–38. doi: 10.1165/ajrcmb.14.5.8624247. [DOI] [PubMed] [Google Scholar]
- 25.Henderson WRJ, Tang LO, Chu SJ, et al. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002;165:108–16. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka H, Masuda T, Tokuoka S, et al. The effect of allergen-induced airway inflammation on airway remodeling in a murine model of allergic asthma. Inflamm Res. 2001;50:616–24. doi: 10.1007/PL00000243. [DOI] [PubMed] [Google Scholar]
- 27.Renauld JC. New insights into the role of cytokines in asthma. J Clin Pathol. 2001;54:577–89. doi: 10.1136/jcp.54.8.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Richter A, Puddicombe SM, Lordan JL, et al. The contribution of interleukin (il)-4 and il-13 to the epithelial-mesenchymal trophic unit in asthma. Am J Respir Cell Mol Biol. 2001;25:385–91. doi: 10.1165/ajrcmb.25.3.4437. [DOI] [PubMed] [Google Scholar]
- 29.Magnan A, Retornaz F, Tsicopoulos A, et al. Altered compart-mentalization of transforming growth factor-beta in asthmatic airways. Clin Exp Allergy. 1997;27:389–95. [PubMed] [Google Scholar]
- 30.Tang W, Geba GP, Zheng T, et al. Targeted expression of IL-11 in the murine airway causes lymphocytic inflammation, bronchial remodeling, and airways obstruction. J Clin Invest. 1996;98:2845–53. doi: 10.1172/JCI119113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gharaee-Kermani M, Nozaki Y, Hatano K, Phan SH. Lung interleukin-4 gene expression in a murine model of bleomycin-induced pulmonary fibrosis. Cytokine. 2001;15:138–47. doi: 10.1006/cyto.2001.0903. [DOI] [PubMed] [Google Scholar]
- 32.Huaux F, Liu T, McGarry B, Ullenbruch M, Phan SH. Dual roles of IL-4 in lung injury and fibrosis. J Immunol. 2003;170:2083–92. doi: 10.4049/jimmunol.170.4.2083. [DOI] [PubMed] [Google Scholar]
- 33.Sempowski GD, Derdak S, Phipps RP. Interleukin-4 and interferon-gamma discordantly regulate collagen biosynthesis by functionally distinct lung fibroblast subsets. J Cell Physiol. 1996;167:290–6. doi: 10.1002/(SICI)1097-4652(199605)167:2<290::AID-JCP13>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 34.Blyth DI, Wharton TF, Pedrick MS, Savage TJ, Sanjar S. Airway subepithelial fibrosis in a murine model of atopic asthma: suppression by dexamethasone or anti-interleukin-5 antibody. Am J Respir Cell Mol Biol. 2000;23:241–6. doi: 10.1165/ajrcmb.23.2.3999. [DOI] [PubMed] [Google Scholar]
- 35.McKenzie AN. Regulation of T helper type 2 cell immunity by interleukin-4 and interleukin-13. Pharmacol Ther. 2000;88:143–51. doi: 10.1016/s0163-7258(00)00088-7. [DOI] [PubMed] [Google Scholar]
- 36.Doucet C, Brouty-Boye D, Pottin-Clemenceau C, Canonica GW, Jasmin C, Azzarone B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts implication in asthma. J Clin Invest. 1998;101:2129–39. doi: 10.1172/JCI741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenzel SE, Trudeau JB, Barnes S, et al. TGF-beta and IL-13 synergistically increase eotaxin-1 production in human airway fibroblasts. J Immunol. 2002;169:4613–9. doi: 10.4049/jimmunol.169.8.4613. [DOI] [PubMed] [Google Scholar]
- 38.Lee CG, Homer RJ, Zhu Z, et al. Interleukin-13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1) J Exp Med. 2001;194:809–21. doi: 10.1084/jem.194.6.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wen FQ, Kohyama T, Liu X, et al. Interleukin-4- and interleukin-13-enhanced transforming growth factor-beta2 production in cultured human bronchial epithelial cells is attenuated by interferon-gamma. Am J Respir Cell Mol Biol. 2002;26:484–90. doi: 10.1165/ajrcmb.26.4.4784. [DOI] [PubMed] [Google Scholar]
- 40.Hoshino M, Nakamura Y, Sim JJ. Expression of growth factors and remodelling of the airway wall in bronchial asthma. Thorax. 1998;53:21–7. doi: 10.1136/thx.53.1.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang S, Smartt H, Holgate ST, Roche WR. Growth factors secreted by bronchial epithelial cells control myofibroblast proliferation: an in vitro co-culture model of airway remodeling in asthma. Lab Invest. 1999;79:395–405. [PubMed] [Google Scholar]
- 42.Chakir J, Shannon J, Molet S, et al. Airway remodeling-associated mediators in moderate to severe asthma: effect of steroids on TGF-beta, IL-11, IL-17, and type I and type III collagen expression. Allergy Clin Immunol. 2003;111:1293–8. doi: 10.1067/mai.2003.1557. [DOI] [PubMed] [Google Scholar]
- 43.Corrigan CJ, Kay AB. CD4 T-lymphocytes activation in acute severe asthma. Am Rev Respir Dis. 1990;141:970–7. doi: 10.1164/ajrccm/141.4_Pt_1.. [DOI] [PubMed] [Google Scholar]
- 44.Cembrzynska-Nowak M, Szklarz E, Inglot AD, Teodorczyk-Injeyan JA. Elevated release of tumor necrosis factor-alpha and interferon-gamma by bronchoalveolar leukocytes from patients with bronchial asthma. Am Rev Respir Dis. 1993;147:291–5. doi: 10.1164/ajrccm/147.2.291. [DOI] [PubMed] [Google Scholar]
- 45.Hartnell A, Robinson D, Kay AB, Wardlaw AJ. CD69 is expressed by human eosinophils activated in vivo in asthma and in vitro by cytokines. Immunology. 1993;80:281–6. [PMC free article] [PubMed] [Google Scholar]
- 46.Wills-Karp M, Luyimbazi J, Xu X, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 47.Grunig G, Warnock M, Wakil AE, et al. Requirement for IL-13 independently of IL-4 in experimental asthma [see comments] Science. 1998;282:2261–3. doi: 10.1126/science.282.5397.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Grootendorst DC, Dahlen SE, van den Bos JW, et al. Benefits of high altitude allergen avoidance in atopic adolescents with moderate to severe asthma, over and above treatment with high dose inhaled steroids. Clin Exp Allergy. 2001;31:400–8. doi: 10.1046/j.1365-2222.2001.01022.x. [DOI] [PubMed] [Google Scholar]
- 49.Foster PS, Hogan SP, Ramsay AJ, Matthaei KI, Young IG. Interleukin 5 deficiency abolishes eosinophilia, airways hyperreactivity, and lung damage in a mouse asthma model. J Exp Med. 1996;183:195–201. doi: 10.1084/jem.183.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]