

Abstract
All cancers carry somatic mutations in their genomes. A subset, known as driver mutations, confer clonal selective advantage on cancer cells and are causally implicated in oncogenesis1, and the remainder are passenger mutations. The driver mutations and mutational processes operative in breast cancer have not yet been comprehensively explored. Here we examine the genomes of 100 tumours for somatic copy number changes and mutations in the coding exons of protein-coding genes. The number of somatic mutations varied markedly between individual tumours. We found strong correlations between mutation number, age at which cancer was diagnosed and cancer histological grade, and observed multiple mutational signatures, including one present in about ten per cent of tumours characterized by numerous mutations of cytosine at TpC dinucleotides. Driver mutations were identified in several new cancer genes including AKT2, ARID1B, CASP8, CDKN1B, MAP3K1, MAP3K13, NCOR1, SMARCD1 and TBX3. Among the 100 tumours, we found driver mutations in at least 40 cancer genes and 73 different combinations of mutated cancer genes. The results highlight the substantial genetic diversity underlying this common disease.
The coding exons of 21,416 protein coding genes and 1,664 microRNAs were sequenced and copy number changes examined in 100 primary breast cancers, 79 of which were oestrogen receptor positive (ER+) and 21 of which were oestrogen receptor negative (ER−) (Supplementary Table 1). We sequenced normal DNAs from the same individuals to exclude inherited sequence variation. We identified 7,241 somatic point mutations: 6,964 were single-base substitutions, of which 4,737 were predicted to generate missense; 422, nonsense; 158, an essential splice site; 8, stop codon read-through; and 1,637, silent changes in protein sequence. Two substitutions were found in microRNAs. There were 277 small insertions or deletions (71 and 206, respectively), of which 231 introduced translational frameshifts and 46 were in-frame (Supplementary Table 2). Analyses of copy number yielded 1,712 homozygous deletions and 1,751 regions of increased copy number (amplification) (Supplementary Table 3).
Somatic driver substitutions and small insertions/deletions (indels) were identified in cancer genes previously implicated in breast cancer development, including AKT1, BRCA1, CDH1, GATA3, PIK3CA, PTEN, RB1 and TP53 (Supplementary Table 4; see also http://www.sanger.ac.uk/genetics/CGP/Census). Likely drivers were also found in cancer genes involved in other cancer types, including APC, ARID1A, ARID2, ASXL1, BAP1, KRAS, MAP2K4, MLL2, MLL3, NF1, SETD2, SF3B1, SMAD4 and STK11.
To identify new cancer genes, we searched for non-random clustering of somatic mutations in each of the 21,416 protein-coding genes2,3 and sequenced a subset of genes highlighted by this analysis in a followup series of 250 breast cancers (Supplementary Tables 5 and 6). Persuasive evidence was found for nine new cancer genes (Fig. 1a and Supplementary Fig. 1). Of these ARID1B, CASP8, MAP3K1, MAP3K13, NCOR1, SMARCD1 and CDKN1B had the truncating mutations and often biallelic inactivation characteristic of inactivated, potentially recessive cancer genes (Supplementary Table 4). AKT2 is probably an activated, dominantly acting cancer gene. The effects of TBX3 mutations on its function are unclear.
Figure 1. New cancer genes established in this study and involvement of the JUN kinase signalling pathway.

a, Representations of the protein-coding sequences and major domains in cancer genes established in this study. Somatic mutations are shown as circles: truncating (red), essential splice site (blue), missense (green) and in-frame indel (yellow). The red lines indicate the positions of large homozygous deletions. aa, amino acids. b, Pathways regulating the JUN kinases MAP2K7 and MAP2K8, indicating genes with mutations in this series. Genes in green are activated by mutations, whereas genes in red are inactivated.
MAP3K1 encodes a serine/threonine protein kinase that regulates the activity of the ERK MAP kinase (the extracellular signal-regulated mitogen-activated protein kinase), JUN kinase and p38 signalling pathways implicated in control of cell proliferation and death4. Somatic mutations in MAP3K1 were observed in 6% of breast cancers, predominantly in ER+ cases. Most were protein truncating. MAP3K1 phosphorylates and activates the protein encoded by MAP2K4, a known recessive cancer gene with inactivating mutations in breast and other cancers5. In turn, MAP2K4 phosphorylates and activates the JUN kinases MAPK8 (also known as JNK1) and MAPK9 (also known as JNK2), which phosphorylate JUN, TP53 and other transcription factors mediating cellular responses to stress4. Truncating mutations and other non-synonymous mutations were also found in MAP3K13, which encodes a kinase that phosphorylates and activates MAP2K7. MAP2K7 phosphorylates and activates MAPK8 and MAPK9 (ref. 4). Thus, in breast cancer, inactivating mutations in MAP3K1, MAP2K4 and MAP3K13 are predicted to abrogate signalling pathways that activate JUN kinases (Fig. 1b).
In the serine/threonine kinase gene AKT2, we identified a single somatic missense mutation, Glu 17 Lys, that is identical to the recurrent, activating mutation in AKT1 previously reported in breast cancer6. Thus, AKT2 is also probably a cancer gene, albeit one infrequently implicated in breast cancer development. Because AKT phosphorylates and inhibits MAP2K4 (ref. 7) and mutations in PIK3CA and PTEN can result in AKT activation8, about half of breast cancers may have abrogation of JUN kinase signalling (Fig. 1b). The biological consequences of the reduction in JUN kinase activity are likely to be diverse and complex, but may include destabilization and consequent inactivation of TP53 with disruption of pro-apoptotic cellular signalling in response to stress9.
We observed truncating mutations and homozygous deletions of NCOR1. In addition to mediating repression of thyroid-hormone and retinoic-acid receptors by promoting chromatin condensation and preventing access of the transcription machinery10, NCOR1 participates in ligand-dependent transcriptional repression by oestrogen receptor alpha11. We also identified inactivating mutations in SMARCD1 and ARID1B, further implicating aberrant chromatin regulation. The encoded proteins of both are components of the SWI/SNF chromatin modelling complex, which incorporates the products of several established recessive cancer genes, including PBRM1, ARID1A, SMARCB1 and SMARCA4 (refs 3, 12-14).
We found three truncating mutations and a missense mutation in CDKN1B. Two truncating mutations in CDKN1B in cancer have previously been reported15,16, and collectively the results confirm that CDKN1B is a cancer gene. CDKN1B (also known as p27 or KIP1) normally inhibits activation of cyclin E/CDK2 and cyclin D/CDK4 complexes, thus preventing cell cycle progression at phase G117.
Three truncating mutations were observed in CASP8. CASP8 is a member of the cysteine/aspartic acid protease family that forms a complex with the FAS cell surface receptor to promote programmed cell death. Inactivation of CASP8 in these cancers is therefore predicted to abrogate apoptosis in response to a variety of signals.
Six tumours had mutations in TBX3, which encodes a T-box transcription factor that regulates stem cell pluripotency-associated and reprogramming factors and is involved in normal breast development18,19. Constitutional inactivating mutations in TBX3 cause ulnar-mammary syndrome, in which there is failure of breast and apocrine development coupled with abnormalities of limb morphogenesis20. Three breast cancers had in-frame deletions, one of Thr 210 and the other two of Asn 212, a residue through which the T-box domain binds to DNA. Despite the presence of truncating mutations in three further cases, the recurrent and clustered in-frame deletions and the finding that all mutations were heterozygous suggests that they may not simply result in loss of function. Indeed, recent reports suggest that increased activity of TBX3 is likely to contribute to oncogenesis. The proportion of stem-like cells in breast cancers is increased by oestrogen-dependent activation of the TBX3 pathway21. Moreover, TBX3 overexpression increases the efficiency of the derivation of induced pluripotent stem cells18 and the ability of cancer cells to form tumours21.
Further supporting their role in oncogenesis, three of the nine newly identified somatically mutated cancer genes, MAP3K1, CASP8 and TBX3, carry inherited common variants, identified by genome-wide association studies, that confer small increased risks of breast cancer22,23. Several additional genes showed truncating mutations and are biologically plausible candidate cancer genes contributing infrequently to breast cancer development. Some, including ASXL2, ARID5B, KDM3A, SETD1A, CHD1, NCOR2, HDAC9 and CTCF, encode proteins that regulate chromatin structure, whereas others, including FANCA and ATR, are involved in DNA repair.
Cancers arise through successive waves of clonal expansion dependent on the sequential acquisition of driver mutations. A central parameter of cancer development is therefore the number of driver mutations required for conversion of a normal cell into a symptomatic cancer. Estimates based on cancer age–incidence curves have indicated that approximately five rate-limiting steps underlie the development of common adult solid tumours24. Experimental studies have similarly indicated that a limited number of key genetic changes are required for neoplastic transformation of human cells25. Our systematic genome analysis now provides a direct survey of the landscape of driver mutations in breast cancer.
Somatic driver point mutations and/or copy number changes in at least 40 cancer genes were implicated in the development of the 100 breast cancers (Fig. 2, Supplementary Tables 3 and 4, and Supplementary Methods). The maximum number of mutated cancer genes in an individual cancer was 6, but 28 cases only showed a single driver. Thus, there seems to be substantial variation in the number of drivers. In some cases, the presence of multiple drivers was associated with subclonal evolution of the cancer (Supplementary Statistical Analyses). However, in others multiple drivers were in the root cancer clone. Seven of the 40 cancer genes (TP53, PIK3CA, ERBB2, MYC, FGFR1/ZNF703, GATA3 and CCND1) were mutated in more than 10% of cases. Collectively these contributed 58% of driver mutations (144 of 250). Therefore, 33 mutated cancer genes, each contributing relatively infrequently, were responsible for the remaining 42% of driving genetic events. We observed 73 different combinations of mutated cancer genes. Thus, most breast cancers differed from all others (Fig. 2 and Supplementary Fig. 2). This assessment of the genetic diversity of breast cancer is probably conservative because, for several reasons, it underestimates the number of mutated cancer genes in each case.
Figure 2. The landscape of driver mutations in breast cancer.

Each of the 40 cancer genes in which a driver mutation or copy number change has been identified is listed down the left-hand side. The number of mutations in each gene in the 100 tumours is shown (rows), as is the number of driver mutations in each breast cancer (columns). Point mutations and copy number changes are coloured red and blue, respectively.
At present, we know little about the mutational processes responsible for the generation of somatic mutations in breast and other cancers. In the 100 breast cancers analysed here, there was substantial variation in the total numbers of base substitutions and indels between individual cases (Fig. 3a). There was also considerable diversity of mutational pattern, ranging from cases in which C•G → T•A transitions predominated to cases in which all transitions and transversions made equal contributions (Fig. 3b and Supplementary Fig. 3). Taken together, the results suggest that multiple distinct mutational processes are operative. For most of these processes, the underlying mechanism is unknown.
Figure 3. The variation in numbers and types of mutation between individual breast cancers.

a, Numbers of small indels and base substitutions in the protein-coding exons of each of the 100 breast cancers studied. The cases are ranked according to the number of base substitutions. *Breast cancer PD4120 (see main text). b, Mutation spectrum of four primary tumours with diverse mutational patterns.
To illustrate one mutational signature in detail, we selected the ER+ breast cancer with the largest number of base substitutions in the series, PD4120 (Fig. 3a, asterisk; Fig. 4). The mutation spectrum of this case was distinctive, featuring C•G → T•A, C•G → G•C and C•G → A•T mutations and very few mutations at A•T base pairs (Fig. 4a). To characterize this process further, we examined the sequence context in which the mutations occurred (in the following discussion, mutations at C•G base pairs are represented as the change at the C base) and found pronounced overrepresentation of thymine immediately 5′ to the mutated cytosines. Thus, in PD4120 the large majority of mutations were of cytosine at TpC dinucleotides (Fig. 4b).
Figure 4. The mutational signature of ER1 breast cancer PD4120.

a, The mutational spectrum. b, The sequence context of C → T, C → G and C → A mutations. The central blue bar indicates the position of the mutated cytosine and the bases 5′ and 3′ are numbered on the horizontal axis. c, Strand bias of mutations showing substitutions at C bases and at T bases according to whether they are on the transcribed (T) or untranscribed (U) strands of the genes screened.
To obtain further insight into the underlying mechanism in this case, we looked for differences in mutation prevalence between the transcribed and untranscribed strands of the 21,416 genes analysed (‘strand bias’) and found a higher prevalence of C→T, C→G and C→A mutations on transcribed strands (P = 0.02) (Fig. 4c and Supplementary Table 7). This strand bias raises the possibility that transcription-coupled nucleotide excision repair (NER) has been operative. NER removes bulky DNA adducts that distort the DNA double helix, notably pyrimidine dimers due to ultraviolet light exposure or adducts due to mutagens in tobacco smoke26. There is a form of NER, recruited by RNA polymerase II, that is operative only on the transcribed strand of each gene and thus introduces a strand bias for mutations27. Therefore, one hypothesis to account for the strand bias in PD4120 is past involvement of NER, in turn implicating exposure to a bulky DNA-damaging agent, either of endogenous or exogenous origin. However, we cannot exclude the possibility that other DNA damage or repair processes generate a strand bias. At least eight additional cancers in this series had a very similar mutational spectrum, sequence context and strand bias (Supplementary Fig. 4 and Supplementary Statistical Analysis). None had been treated before excision of the cancer.
The somatic mutations in a cancer genome accumulate over a patient’s lifetime, during the lineage of mitotic divisions from the fertilized egg to the cancer cell. Some are acquired while cells in the lineage are biologically normal, whereas others are acquired after acquisition of the neoplastic phenotype. However, the relative proportions accumulated in these two phases are unknown. To explore this question, we examined the relationship between the total numbers of somatic base substitutions and the age at diagnosis in the 100 tumours (Fig. 5). In both ER+ and ER− cancers, no correlation was observed (P = 0.33 and 0.14 respectively). If most somatic mutations in a cancer genome are acquired in normal tissues before neoplastic transformation, the later the onset of the cancer the longer this part of the lineage is likely to have been and, consequently, the higher the number of mutations. The absence of a correlation there fore suggests that most mutations in breast cancer genomes occur after the initiating driver event.
Figure 5. The relationship between age at breast cancer diagnosis and all substitutions, and for C → T substitutions at CpG sites.
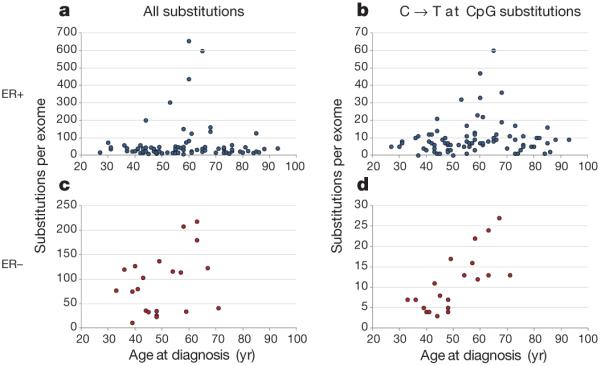
a, b, Data from the 79 ER+ breast cancers. c, d, Data from the 21 ER− breast cancers.
We then considered separately the subset of somatic mutations constituted by C•G → T•A substitutions at CpG dinucleotides, because this mutational pattern is observed in non-diseased tissues, manifesting prominently in normal germline variation. This subset showed a strong positive correlation with the age at cancer diagnosis in ER− cancers (P = 1.2 × 10−7), supporting the proposition that it is enriched in mutations occurring in normal tissues and that, overall, other mutation classes occur later. By contrast, ER+ cancers showed no correlation between C•G → T•A substitutions at CpG dinucleotides and age at diagnosis (P = 0.27). The basis for this pronounced difference is unclear, but potentially highlights a profound divergence in the dynamics of mutation acquisition between these two major subclasses of breast cancer.
In clinical practice, breast cancers are graded microscopically on the basis of mitotic counts, pleomorphism of cancer cell nuclei and extent of tubule formation, which are then collected into an overall grade score. High scores indicate large numbers of mitoses, substantial tumour cell pleomorphism and little tubule formation, and are generally associated with more rapid progression. Significant correlations were not observed between numbers of driver mutations and grade scores (Supplementary Statistical Analysis). However, there were strong positive correlations between the total number of substitutions (that is, drivers and passengers) and mitosis and tubule scores (P = 0.0002 and 0.002 respectively), which remained significant after multiple testing corrections. The causal relationships between these features are unclear. However, because most substitutions are likely to be biologically inert passengers, it is possible that the biological state of high-grade breast cancers may be responsible for generating increased numbers of mutations, rather than the converse.
The panorama of mutated cancer genes and mutational processes in breast cancer is becoming clearer, and a sobering perspective on the complexity and diversity of the disease is emerging. Driver mutations are operative in many cancer genes. A few are commonly mutated, but many infrequently mutated genes collectively make a substantial contribution in myriad different combinations. Multiple somatic mutational processes have been operative. Ultimately, characterization of the genomes of breast cancer, and others, will provide a robust and biologically meaningful classification generating insights into the clinical heterogeneity of the disease and influencing strategies to find new modes of prevention and treatment.
METHODS
Patient samples
Informed consent was obtained from all subjects and ethical approval obtained from Cambridgeshire 3 Research Ethics Committee (ref 09/H0306/36). Collection and use of patient samples were approved by the appropriate IRB of each Institution. In addition, this study and usage of its collective materials had specific IRB approval.
Exome enrichment and sequencing
Genomic libraries were prepared using the Illumina Paired End Sample Prep Kit following the manufacturer’s instructions. Enrichment was performed as described previously31, using the Agilent SureSelect Human All Exon 50Mb kit following the manufacturer’s recommended protocol but excluding pre-enrichment PCR amplification. Each exome was sequenced using the 75 or 76-bp paired-end protocol, on an Illumina GAII or HiSeq DNA Analyser, to produce approximately 10 Gb of sequence per exome. Sequencing reads were aligned to the human genome (NCBI build 37) using the BWA algorithm on default settings32. Reads which were unmapped, PCR-derived duplicates or outside the targeted region of the genome were excluded from the analysis. The remaining uniquely mapping reads (~60%) provided 60–80% coverage over the targeted exons at a minimum depth of ×30.
Sequencing of pooled PCR amplimers
Selected genes were targeted for followup investigations in 250 additional breast cancers by sequencing of pooled PCR products. An 8-bp index was introduced during amplification to enable sequence data from individual tumours to be identified in downstream analyses.
For each amplimer, a primary PCR was performed using gene-specific primers modified with the inclusion of a common upstream adaptor sequence. A secondary PCR was performed using primers complementary to the common adaptor sequences. The reverse secondary primer contained the internal index, and 96 different indexed primers were used to enable 96 different DNAs to be pooled before sequencing. The primary and secondary PCR amplifications were performed as a simultaneous multiplex reaction. Primer sequences are available on request.
For each amplimer, PCR was performed in batches of 96 DNA samples. Following amplification, the 96 PCR products were pooled, purified using a QiaQuick column (Qiagen) and quantified on a Bioanalyser (Agilent). Pooled reactions from different amplimers (up to 50) were normalized for concentration and subsequently also pooled to produce the final template used for sequencing on a single lane of an Illumina GAII DNA Analyser (~5,000 amplimers per lane). Amplimers which failed PCR were excluded from the pooling experiments. The subsequent sequence reads were aligned with BWA and resulted in coverage typically exceeding ×500 per individual sample amplimer.
Variant detection
The CaVEMan (cancer variants through expectation maximization) algorithm was used to call single nucleotide substitutions31. This uses a naive Bayesian classifier to estimate the posterior probability of each possible genotype (wild type, germline, somatic mutation) at each base. We applied several post-processing filters to the set of initial CaVEMan mutation calls to remove variants reported in poor-quality sequence and increase the specificity of the output.
To call insertions and deletions, we used split-read mapping implemented as a modification of the Pindel algorithm33. This algorithm searches for reads where one end is anchored on the genome and the other end can be mapped with high confidence in two (split) portions, spanning a putative indel. Post-processing filters were applied to the output to improve specificity.
Mutations were annotated to Ensembl version 58.
Variant validation
Validation of all 7,241 putative somatic variants in the primary screen of 100 tumours and all variants found in the follow-up of 250 cases was attempted by either capillary resequencing or 454 pyrosequencing of PCR products spanning the mutation in the tumour and the normal pair. Where independent validation failed (approximately 20%) variants were reported to be somatic if manual inspection of the aligned sequence reads provided strong evidence to support their validity.
Identification of likely driver base substitutions and indels
A subset of the 7,241 substitution and indel somatic mutations identified in the exome screen were classified as ‘likely driver mutations’ using conservative criteria. To do this, we identified the established cancer genes from the Cancer Gene Census (http://www.sanger.ac.uk/genetics/CGP/Census/) that are known to be mutated by base substitutions and indels to contribute to cancer development. We then classified as likely driver mutations those that conformed to the known patterns of cancer-causing mutation for each cancer gene. Thus, for recessive cancer genes truncating mutations, essential splice site mutations and homozygous deletions were included. Missense mutations were also included where they had been seen previously or conformed to the known pattern of missense mutation in each gene (COSMIC database; http://www.sanger.ac.uk/genetics/CGP/cosmic/). For established, dominantly acting cancer genes, we included mutations that had been previously registered in COSMIC. For the new cancer genes established in this study, we applied essentially the same rules. However, for the recessive cancergenes, to be conservative we did not include missense variants (other than the single variant in MAP3K1, which is almost certainly disruptive to the function of the protein). We included the variant in AKT2 because it is identical in nature to the recurrent variant in AKT1, and we included all TBX3 mutations. As indicated in the main text, we may have both underestimated and overcalled some somatic variants as drivers using this approach. However, the number of erroneous calls is likely to be small and overall we have probably underestimated the number of driver mutations. For the calling procedure for likely driver copy number variants, see below.
Detection of copy number variation
Single nucleotide polymorphism (SNP) array hybridization on the SNP6.0 platform was done according to Affymetrix Protocols and as described at http://www.sanger.ac.uk/cgi-bin/genetics/CGP/cghviewer/CghHome.cgi.
Copy number analysis was performed using ASCAT (version 2.1) taking into account non-neoplastic cell infiltration and tumour aneuploidy34, and resulted in integral allele-specific copy number profiles for the tumour cells. Amplifications in the 100 samples analysed were called if copy number was ≥5 (for diploid tumours, with ASCAT ploidy <2.7 n) or ≥9 (for tumours with evidence of a whole-genome duplication, with ASCAT ploidy ≥2.7 n). Homozygous deletions were called if there were zero copies in the tumour cells.
Identification of likely driver copy number variants
To identify likely driver copy number variants, we derived a conservatively generated list of frequently amplified regions in breast cancer from a previous study35. From the amplified regions in breast cancer obtained by GISTIC analysis of that study, those with a GISTIC Q-value of less than 10−5 were selected. Regions within 40 Mb of amplified regions with more significant Q-values were excluded, as many of these probably point to the same amplified target gene. This process generated seven focal, highly significantly amplified regions. These regions were annotated with their putative target genes where additional biological studies have indicated that they are the likely targets (ERBB2, CCND1, MYC, FGFR1/ZNF703, ZNF217, MDM2). Only the amplified region on chromosome 15 was not annotatable. Driver amplification of these seven focal regions in the 100 samples was called using the criteria above. Driver homozygous deletions were called if part or all of a homozygous deletion overlapped with a known recessive cancer gene from the Cancer Gene Census36 or a newly discovered gene from this study.
Estimation of the number of mutated copies
Allele-specific copy number estimates for point mutations and indels were obtained by integrating copy number and sequencing data. In a sample containing only tumour cells, the number of reads, r, with a mutation can be expressed as
| 1 |
In equation (1), nlocus is the copy number of the locus, nmut is the number of mutated copies and R is the total number of reads from that locus. In case of a tumour sample consisting of a fraction of tumour cells ρ, infiltrated with a fraction of normal cells 1–ρ (assumed to have two copies), equation (1) becomes
Hence, allele-specific copy number estimates for point mutations and indels can be obtained as
| 2 |
In equation (2), fs = r/R is the frequency of mutated reads observed in the sequencing data, and ρ and nlocus can be obtained from the ASCAT copy number analysis.
These copy number estimates of mutations were used to determine which mutations are likely subclonal: if nmut ≥ 0.8, the mutation is called likely clonal and if nmut < 0.8, the mutation is called likely subclonal.
In the case of indels, reads with an insertion or deletion may not map as well as reads without insertions and deletions. Therefore, a procedure was followed to estimate fs for indels that was independent of ease of mapping. Reads were obtained by matching flanking sequence (10 bp on each side) around the indel, further filtered to exclude spurious matches. The mutated read frequency was subsequently calculated, accounting for the difference in sequence lengths with and without the indel:
| 3 |
In equation (3), rindel and rnormal are the respective numbers of reads with and without the indel, ls is the read length (76 bp), and lindel and lnormal are the respective lengths of the matching fragment in sequences with and without the indel.
Detection of selection and oncogenicity in protein-coding genes
The overall significance of an excess of non-silent mutations was determined using the methods previously described37. The ranking of gene significances was determined using the following model. We let denote the number of silent mutations, where k indexes mutation type (C•G → A•T, C•G → T•A, C•G → G•C, T•A → A•T, T•A → C•G or T•A → G•C) in gene g, where i = 1 for the primary screen and i = 2 for the follow-up screen. We also have counts and of missense and nonsense mutations, respectively. Finally we have counts of indels. The numbers of screened bases, , and , in each gene for each mutation type were also calculated. The total number of screened bases was . We let ρk represent the per-base passenger mutation prevalence and use γ to denote the per-base passenger rate of indels.
Next we assume that genes can be neutral to cancer, oncogenically triggered by missense mutations or inactivated by truncating mutations. Genes are not precluded from belonging to both of the last two categories. We assume that proportions α and β of genes belong to the missense group and truncating group, respectively. Genes that belong to these groups have mutation rates that increase by factors λ and μ, respectively. These terms quantify the selection pressure for missense and truncating variants, respectively. This results in a mixture model with the following likelihood:
Here α1 = 1–α, α2 = α, β1 = 1–β, β2 = β, λ1 = 1, λ2 = l, μ1 = 1 and μ2 = μ, and Poc(r) indicates the Poisson probability of obtaining value c from a Poisson process with rate parameter r. GF denotes the set of genes in the follow-up study. The parameters for this model were then estimated with the expectation-maximization algorithm. Confidence intervals for these parameters were obtained using parametric bootstrapping. Conditional on these parameter estimates, we can then use Bayes’ law to calculate the probability that each gene belongs to the neutral, the missense or the truncating group. Specifically, if  g, ψg
g, ψg
 {1, 2} index whether the gene g does or does not belong to the missense or truncating group, respectively, we have
{1, 2} index whether the gene g does or does not belong to the missense or truncating group, respectively, we have
The probability of belonging to either the missense or the truncating group, 1–Pr(g = 1, ψg = 1), was then used to rank the genes.
Generalized linear models
Generalized linear models (GLMs) are extensions to ordinary linear regression that model underlying distributions using members of the exponential family38. The response variable is related to the linear model by a link function using maximum-likelihood estimates of the parameters. Because they are not restricted to modelling normally distributed data, GLMs have particular utility in modelling count data such as, in this manuscript, the number of mutations.
If mutations were generated by a random process, with a constant probability of occurring at any point throughout an individual’s life, we would expect the number of mutations to have a Poisson distribution, dependent only on the (unknown) rate of mutation and the age of the individual. Where goodness-of-fit tests indicated that the Poisson distribution was an appropriate model for the number of mutations, we used this distribution. However, in the models where goodness-of-fit tests indicated that mutation numbers were overdispersed, we used negative binomial distributions in place of Poisson distributions, as the negative binomial distribution incorporates an additional parameter that allows the adjustment of the variance of the distribution independently of its mean.
GLMs were implemented using the glm and glm.nb functions in R. The predictor variables were {age, tumour grade, tubule score, pleomorphism score, mitotic score, mitotic count}, each of which was used within a two-factor model, with oestrogen receptor status as the second predictor variable. The response variable was the number of mutations of a particular type, from the set {substitutions 1 indels, substitutions, indels, copy number amplifications, C → T at CpG mutations, all driver mutations}.
Evaluation of strand bias in tumours displaying the mutator phenotype
To assess whether there was a strand bias of C → X (C → T, C → G and C → A) mutations in PD4120 and the other tumours showing the mutator phenotype, we first estimated the expected ratio of cytosines found in transcribed and untranscribed strands, by random sampling of 20,000 CCDS exons from Ensembl version 61. A χ-squared test was then used to examine whether the C → X mutations observed in each sample differed significantly from this ratio. Similar tests were conducted on the combined mutations from all mutator phenotype samples and on all mutator phenotype samples except PD4120.
Acknowledgements
This work was supported by the Wellcome Trust (grant reference 077012/Z/05/Z) and Breakthrough Breast Cancer. P.J.C. is personally funded through a Wellcome Trust Senior Clinical Research Fellowship (grant reference WT088340MA). P.V.L. is a postdoctoral researcher at the Research Foundation - Flanders (FWO) and is a visiting scientist at the Wellcome Trust Sanger Institute, supported by a travel grant from the FWO. I.V. is supported by a fellowship from The International Human Frontier Science Program Organization. A.-L.B.-D. and A.L. are funded by the Norwegian Research Council, The Norwegian Cancer Society, The Radium Hospital Foundation and Health Region SØ. A.V.S. was supported by an ‘Interface INSERM’ grant. J.S.R.-F. is funded in part by Breakthrough Breast Cancer and is a recipient of the 2010 CRUK Future Leaders Prize. D.E. is a Principal Research Fellow of Cancer Research UK. A.T. receives financial support from the Department of Health via the National Institute for Health Research comprehensive Biomedical Research Centre award to Guy’s and St Thomas’ NHS Foundation Trust in partnership with King’s College London, and from King’s College Hospital NHS Foundation Trust in conjunction with The Experimental Cancer Medicine Centre Initiative jointly funded by Cancer Research UK, the National Institute for Health Research, the Welsh Assembly Government, the HSC R&D Office for Northern Ireland and the Chief Scientist Office, Scotland. C.D. and C.S. received partial funding from the MEDIC foundation and the Fonds National de Recherche Scientifique. J.M. and J.F. are funded in part by a research grant from the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research. The INCa-Synergie facility received support from the Institut National du Cancer, the Fondation Synergie-Lyon-Cancer, the Canceropole Lyon Auverge Rhone Alpes and the Centre Leon Berard. A.C.V. is funded by The Ludwig Institute for Cancer Research. L.v.’t.V. and A. Broeks receive funding from the Dutch Genomics Initiative-Cancer Genomics Center. We also acknowledge support for sample collection, banking and processing from the Biological Resource Center of Institut Curie; the Breakthrough Breast Cancer Unit; P. Watson and the BCCA Tumour Tissue Repository; the Centre for Translational Genomics; A. Lane and P. T. Simpson; the Australian Biospecimens Network; the Breast Unit at Royal Brisbane and Women’s Hospital; the Dana-Farber/Harvard SPORE in breast cancer (reference CA089393); A. M. Sieuwerts; and the Singhealth Tissue Repository, Singapore. We are grateful also for the support of T. B. Tean, and acknowledge the input and guidance of P. Spellman and A. Ashworth.
The Oslo Breast Cancer Consortium (OSBREAC)
Rolf Karesen1,2, Ellen Schlichting1, Bjorn Naume2,3, Torill Sauer2,4 & Lars Ottestad3
1Department of Breast and Endocrine Surgery, Oslo University Hospital, O424 Oslo, Norway.
2Medical Faculty, University of Oslo, O424 Oslo, Norway.
3Department of Oncology, Oslo University Hospital, O424 Oslo, Norway.
4Department of Pathology, Oslo University Hospital, O424 Oslo, Norway.
Footnotes
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Author Contributions P.J.S., P.S.T. and H.D. performed analysis of the sequence data, aided by S.N.Z., I.V., G.R.B., L.R.Y., E.P., D.J.M., M.S.-C. and R.R. P.V.L. performed analysis of the SNP6 data. C.G., D.C.W., K.W.L. and D.E. performed the statistical investigations. S. Martin coordinated sample acquisition and pathology review. S. McLaren coordinated sample processing. D.B., A. Butler, J.G., J.H., M.J., A.J., D.J., A.M., K.R. and J.T. performed informatics investigations. A.C., C.L. and L.M performed technical investigations. A.L., OSBREAC, M.T.M.L., C.-Y.S., B.T.K.T., B.W.H., A. Broeks, A.C.V., G. Turashvili, J.M., A.F., P.M., S.-F.C., G. Thomas, S.B., O.M., S.R.L., M.v.d.V., L.v.’t.V., J.F., C.D., C.S., A.T., C.C., J.S.R.-F., S.A.J.R.A., A.V.S., A.-L.B.-D. and A.R. contributed samples, clinical data and scientific advice. P.J.C. and P.A.F. directed the research and contributed to the manuscript. M.R.S. directed the research and wrote the manuscript.
Author Information Genome sequence data have been deposited at the European Genome-phenome Archive under accession number EGAD00001000133. Affymetrix SNP6 data have been deposited under accession number E-MTAB-1110. Reprints and permissions information is available at www.nature.com/reprints. The authors declare no competing financial interests. Readers are welcome to comment on the online version of this article at www.nature.com/nature. Correspondence and requests for materials should be addressed to M.R.S. (mrs@sanger.ac.uk).
References
- 1.Stratton MR, Campbell PJ, Futreal PA. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006;173:2187–2198. doi: 10.1534/genetics.105.044677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keshet Y, Seger R. The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol. Biol. 2010;661:3–38. doi: 10.1007/978-1-60761-795-2_1. [DOI] [PubMed] [Google Scholar]
- 5.Su GH, et al. Alterations in pancreatic, biliary, and breast carcinomas support MKK4 as a genetically targeted tumor suppressor gene. Cancer Res. 1998;58:2339–2342. [PubMed] [Google Scholar]
- 6.Carpten JD, et al. A transforming mutation in the pleckstrin homology domain of AKT1 in cancer. Nature. 2007;448:439–444. doi: 10.1038/nature05933. [DOI] [PubMed] [Google Scholar]
- 7.Park HS, et al. Akt (protein kinase B) negatively regulates SEK1 by means of protein phosphorylation. J. Biol. Chem. 2002;277:2573–2578. doi: 10.1074/jbc.M110299200. [DOI] [PubMed] [Google Scholar]
- 8.Carnero A, Blanco-Aparicio C, Renner O, Link W, Leal JF. The PTEN/PI3K/ AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Targets. 2008;8:187–198. doi: 10.2174/156800908784293659. [DOI] [PubMed] [Google Scholar]
- 9.Wu GS. The functional interactions between the MAPK and p53 signaling pathways. Cancer Biol. Ther. 2004;3:146–151. doi: 10.4161/cbt.3.2.614. [DOI] [PubMed] [Google Scholar]
- 10.Horlein AJ, et al. Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature. 1995;377:397–404. doi: 10.1038/377397a0. [DOI] [PubMed] [Google Scholar]
- 11.Merrell KW, et al. Differential recruitment of nuclear receptor coregulators in ligand-dependent transcriptional repression by estrogen receptor-alpha. Oncogene. 2010;30:1608–1614. doi: 10.1038/onc.2010.528. [DOI] [PubMed] [Google Scholar]
- 12.Jones S, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–231. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 14.Wiegand KC, et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010;363:1532–1543. doi: 10.1056/NEJMoa1008433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spirin KS, et al. p27/Kip1 mutation found in breast cancer. Cancer Res. 1996;56:2400–2404. [PubMed] [Google Scholar]
- 16.Tigli H, Buyru N, Dalay N. Molecular analysis of the p27/kip1 gene in breast cancer. Mol. Diagn. 2005;9:17–21. doi: 10.2165/00066982-200509010-00003. [DOI] [PubMed] [Google Scholar]
- 17.Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nature Rev. Cancer. 2008;8:253–267. doi: 10.1038/nrc2347. [DOI] [PubMed] [Google Scholar]
- 18.Han J, et al. Tbx3 improves the germ-line competency of induced pluripotent stem cells. Nature. 2010;463:1096–1100. doi: 10.1038/nature08735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howard B, Ashworth A. Signalling pathways implicated in early mammary gland morphogenesis and breast cancer. PLoS Genet. 2006;2:e112. doi: 10.1371/journal.pgen.0020112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bamshad M, et al. Mutations in human TBX3 alter limb, apocrine and genital development in ulnar-mammary syndrome. Nature Genet. 1997;16:311–315. doi: 10.1038/ng0797-311. [DOI] [PubMed] [Google Scholar]
- 21.Fillmore CM, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc. Natl Acad. Sci. USA. 2010;107:21737–21742. doi: 10.1073/pnas.1007863107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghoussaini M, et al. Genome-wide association analysis identifies three new breast cancer susceptibility loci. Nature Genet. 2012;44:312–318. doi: 10.1038/ng.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Varghese JS, Easton DF. Genome-wide association studies in common cancers-what have we learnt? Curr. Opin. Genet. Dev. 2010;20:201–209. doi: 10.1016/j.gde.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 24.Miller D. On the nature of susceptibility to cancer. Cancer. 1980;46:1307–1318. doi: 10.1002/1097-0142(19800915)46:6<1307::aid-cncr2820460602>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 25.Schinzel AC, Hahn WC. Oncogenic transformation and experimental models of human cancer. Front. Biosci. 2008;13:71–84. doi: 10.2741/2661. [DOI] [PubMed] [Google Scholar]
- 26.Shuck SC, Short EA, Turchi JJ. Eukaryotic nucleotide excision repair: from understanding mechanisms to influencing biology. Cell Res. 2008;18:64–72. doi: 10.1038/cr.2008.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fousteri M, Mullenders LH. Transcription-coupled nucleotide excision repair in mammalian cells: molecular mechanisms and biological effects. Cell Res. 2008;18:73–84. doi: 10.1038/cr.2008.6. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Varela I, et al. Exome sequencing identifies frequent mutation of the SWI/SNF complex gene PBRM1 in renal carcinoma. Nature. 2011;469:539–542. doi: 10.1038/nature09639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ye K, Schulz MH, Long Q, Apweiler R, Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Van Loo P, et al. Allele-specific copy number analysis of tumors. Proc. Natl Acad. Sci. USA. 2010;107:16910–16915. doi: 10.1073/pnas.1009843107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beroukhim R, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Futreal PA, et al. A census of human cancer genes. Nature Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nelder JA, Wedderburn R. Generalized linear models. J. R. Stat. Soc. A. 1972;135:370–384. [Google Scholar]