

Abstract
Trypanothione reductase (TR), an enzyme that buffers oxidative stress in trypanosomatid parasites, was screened against commercial libraries containing approximately 134,500 compounds. After secondary screening, four chemotypes were identified as screening positives with selectivity for TR over human glutathione reductase. Thirteen compounds from these four chemotypes were purchased, and their in vitro activity against TR and Trypanosoma brucei are described.
Trypanosomatid protozoan parasites cause several devastating diseases in large parts of the developing world. The three most important are human African trypanosomiasis (sleeping sickness) caused by Trypanosoma brucei, Chagas disease caused by T. cruzi, and leishmaniasis caused by members of the genus Leishmania. These three diseases are estimated to cause 113,000 deaths annually and more than 4 million disability adjusted life years – the healthy life years lost due to disability and death.1 Currently available therapies are inadequate due to issues involving safety, efficacy, resistance, toxicity, ease of administration in poor conditions and cost. Despite the urgent need for new drugs, these diseases have been neglected, largely due to market forces, by most pharmaceutical companies.
As part of a concerted campaign to discover new treatments organized by DNDi (Drugs for Neglected Diseases Initiative), we undertook a high-throughput screen for inhibitors of trypanothione reductase (TR).2 Trypanosomatids use a unique biosynthetic pathway that conjugates glutathione and spermidine to form trypanothione (1, Figure 1) to perform the same antioxidant role as glutathione (2) in mammalian cells.3 TR, the parasite analog of glutathione reductase (GR), plays an essential role in maintaining the intracellular redox balance by returning trypanothione disulfide to its reduced state. Genetic knockouts have illustrated the importance of TR for parasite viability,6 and therefore as a target for drug development for all three diseases. Comparison of TR and human GR (hGR) crystal structures reveals significant differences between their active sites, and these differences are consistent with the selectivity shown by GR and TR for their respective substrates.4 TR possesses a more open active site with an overall negative charge to accommodate the positively charged spermidine, while the more compact GR active site has a positive charge to compensate the glycine carboxylate.5 These active site differences could be exploited in the development of TR-selective inhibitors.
Figure 1.
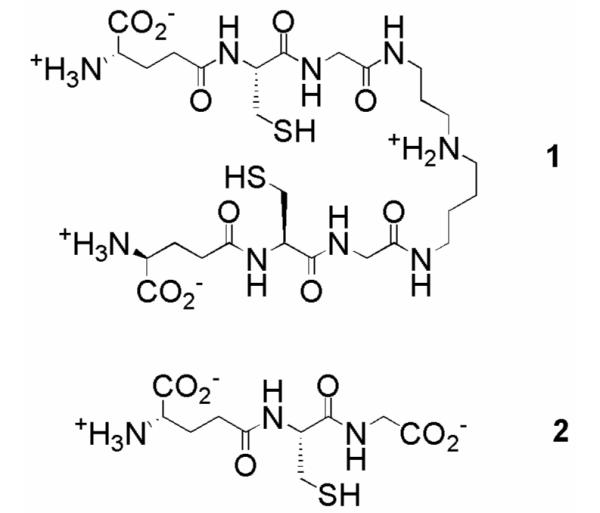
Structures of trypanothione (1) and glutathione (2).
The HTS program utilized an assay developed by Fairlamb et al7 that measures TR activity by coupling the formation of 1 to the reduction of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), regenerating trypanothione disulfide and forming the yellow thionitrobenzoate ion (TNB), which absorbs strongly at 405 nm. Minor alterations to the assay procedure made it amenable to the 384-well format (see ref. 8 for details). The primary screening of approximately 134,500 compounds from commercial libraries was performed. A Z factor of 0.71 was calculated for the entire data set. At the nominal compound concentration of 11.1 μg/mL present in the assay, 90% inhibition was used as an arbitrary cutoff point to identify 89 compounds (0.07% “hit rate”) from 24 chemotypes (based on a shared central scaffold) as having a level of activity sufficient for further analysis. The distribution of compound activities by percent inhibition is summarized in Figure 2.
Figure 2.
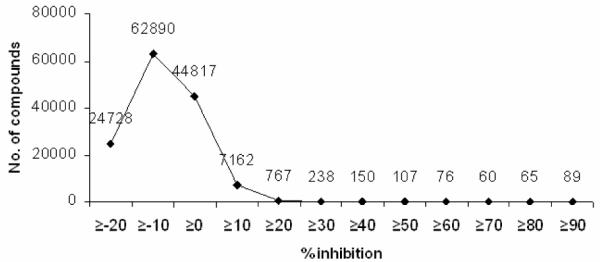
Distribution of compounds from primary screen by percent inhibition.
Within the 24 chemotypes, we identified several classes of compounds, including the maleimides represented by 3 in Figure 3. These chemotypes all had an explicit enone functionality that could react with the free thiols present in the coupled assay (TNB, reduced 1, or TR active site cysteines). We adapted the classical assay9 for measuring TR activity to the 384-well format (see ref. 10 for details), resulting in the elimination of 12 chemotypes that were either inactive (i.e. reacted with TNB in the coupled assay) or weak inhibitors of TR. Maleimides, while active in the classical assay, were eliminated due to the high proportion of this chemotype displaying >75% inhibition during primary screening (65% of tested compounds). Additionally, succinimide-based screening positives were represented at a relatively high rate (20% of tested compounds gave >75% inhibition), and this chemotype was eliminated from further consideration due to concerns regarding selectivity.
Figure 3.
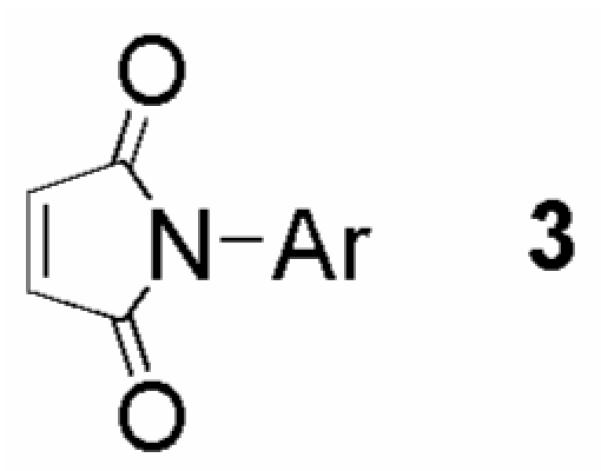
Representative structure of maleimide Michael acceptors.
ChemBank11 was used to perform substructure similarity searches on the screening positives from the remaining 10 chemotypes (represented by the general structures 4-13 in Figure 4) that displayed TR inhibition in the classical assay. Between 6 and 19 compounds from each chemotype were selected on the basis of structural similarity‡ and percent TR inhibition generated during primary screening, and then selected from the compound library stock plates for additional assays. These 115 compounds were tested in the DTNB-coupled assay in a concentration range of 1.91 ng/mL to 31.2 μg/mL, and IC50 values were determined.
Figure 4.

Representative structures 4-13 of cherry-picked chemotypes, split into active chemotypes and false positives.
Data analysis revealed that chemotypes 10-13, which had few representatives, were false positives, and were therefore eliminated from future consideration (data not shown). The remaining six chemotypes had selected examples reselected, which were tested for activity against hGR. Compound classes 4 and 9 did not display any selectivity for TR over hGR, and were not investigated further (data not shown).
Solid samples of selected compounds from the remaining chemotypes (5-8), which displayed selective inhibition of TR over hGR, were purchased to confirm their activity (Table 1). Purity was determined by LCMS and 1H NMR, and by these methods all compounds were ≥95% pure. In general, the nitrobenzenes 6a-e gave the greatest selectivity for TR over hGR, and they afforded the most potent inhibitor tested (6a, IC50 = 0.34 μM). Replacing the dimethylamino group of 6a with an arylamino group lowered activity, although 6b-e still gave an IC50 <10 μM. The trisubstituted phenols 7a and 7b displayed moderate selectivity for TR, and activity decreased with the incorporation of an N-homopiperidyl group into 7c. Replacement of the dimethylamino moiety possessed by the phenylketone 8a with a bulkier group led to a substantial loss of activity. For all four chemotypes, the most potent compound in each class was the hydrochloride salt. Three of these compounds possess the dimethylamino functionality found in previously described TR inhibitors, such as clomipramine.2
| No.a | Structureb | TR IC50 (μM)c |
GR IC50 (μM)c |
Selectivity for TRe |
T. brucei EC50 (μM)f |
||
|---|---|---|---|---|---|---|---|
|
| |||||||
| R1 | R2 | R3 | |||||
| 5a | NMe2.HCl | - | - | 2.9 (±1.4) | 20.4 (±6.3) | 7 | 0.55 (±0.04) |
| 5b | N-piperidyl | - | - | 7.8 (±3.7) | 105 (±36) | 13 | 0.95 (±0.09) |
| 6a | NMe2.HCl | - | - | 0.34 (±0.13) | 20.3 (±8.4) | 59 | 0.68 (±0.10) |
| 6b | NH-4-(CO2-nBu)Ph | - | - | 2.4 (±0.92) | > 135d | >56 | 0.96 (±0.11) |
| 6c | NHPh | - | - | 1.7 (±0.78) | 58.0 (±16) | 35 | 1.01 (±0.07) |
| 6d | NH-2,3-diMePh | - | - | 1.5 (±0.71) | 86.3 (±41) | 58 | 5.87, 0.68, 0.15g |
| 6e | NH-3-FPh | - | - | 3.3 (±1.1) | > 173 | >52 | 0.82 (±0.16) |
| 7a | tBu | N((CH2)2OH).HCl | - | 2.0 (±0.21) | 24.9 (±6.0) | 13 | 3.01 (±0.30) |
| 7b | iPr | NMe2 | - | 11.8 (±3.2) | 79.3 (±25) | 6.7 | > 50 |
| 7c | tBu | N-homopiperidyl | - | 38.8 (±5.0) | > 155 | >4 | 3.30 (±0.30) |
| 8a | Cl | Cl | R3 = NMe2.HCl | 3.3 (±2.2) | 64.8 (±18) | 20 | 1.00 (±0.21) |
| 8b | Cl | H | R3 = N-morpholinyl | 148 (±31) | > 212 | >1.4 | 0.98 (±0.07) |
| 8c | Cl | Cl | R3 = NH-4-EtPh | 107 (±0.71) | > 157 | >1.5 | 1.65 (±0.18) |
All compounds were purchased from ChemBridge, except for 5a and 5b (Peakdale).
Dash signifies that this chemotype did not possess variable groups at this position.
Values are means of two experiments, standard deviation is given in parentheses.
IC50 value greater than the highest tested inhibitor concentration.
GR IC50/TR IC50.
Values are weighted means of three independent experiments, standard errors are given in parentheses. IC50 values for pentamidine are 4.6 (± 0.2) nM.
Compound 6d gave highly variable EC50 results, all three are listed.
All compounds in Table 1 were then tested for their ability to restrict Trypanosoma brucei proliferation using an in vitro assay that determines a compound’s activity via the conversion of a redox sensitive dye to a fluorescent product by viable cells12,13. Six compounds (5a-b, 6a-b, 6e, 8b) gave submicromolar EC50 values, and with the exception of 6a, these values were moderately lower than their IC50 against TR. Compounds 7c and 8b-c were noticeably more active against the parasite than TR, perhaps indicating some off-target effects. The presence of a dimethylamino group at R2 of 7b, and conversion of the R1 group from tert-butyl to iso-propyl, resulted in a substantial loss of activity compared to 7a and 7c.
It should be noted that all of the compounds that emerged from this set of screens have the potential to eliminate an amine to generate a compound that could undergo a conjugate addition. In the case of 5, 6, and 8, an α,β-unsaturated ketone could be formed, and in the case of 7, a para-quinone methide could be formed. Conversely, amino groups could be selected because they mimic the positively charged central amine in trypanothione’s spermidine moiety, increasing an inhibitors affinity for the negatively charged binding site of TR. In any case, the compounds all show selectivity for TR over hGR and activity in a whole organism assay. A ChemBank search for compounds that possessed the 3-(dimethylamino)-1-arylpropan-1-one motif shared by 5a, 6a and 8a found ten compounds, of which five showed negligible (<15%) TR inhibition.
In conclusion, we have performed a HTS program against TR to identify novel inhibitors. Secondary screening revealed four chemotypes that displayed selectivity for TR over hGR, and selected representatives all displayed activity against T. brucei in a whole organism assay. Focused library synthesis of some of these compound classes will be undertaken.
Acknowledgements
Financial support was provided by the Drugs for Neglected Diseases Initiative (DNDi). AHF is a Wellcome Trust Principal Research Fellow. We wish to thank the National Cancer Institute’s Initiative for Chemical Genetics (contract no. N01-CO-12400), who provided support for this publication, and the Chemical Biology Platform of the Broad Institute of Harvard and MIT for their assistance in this work.
Footnotes
The screening positive itself gives a similarity score of 1.0, and only compounds with a score 0.7 were considered.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1. http://www.who.int/tdr/diseases/
- 2.Fries DS, Fairlamb AH. In: Burger’s Medicinal Chemistry and Drug Discovery. 6th Abraham DJ, editor. Vol. 5. John Wiley & Sons, Inc.; New York: 2003. pp. 1033–1087. [Google Scholar]
- 3.Fairlamb AH, Cerami A. Annu. Rev. Microbiol. 1992;46:695. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham ML, Fairlamb AH. Eur. J. Biochem. 1995;230:460. doi: 10.1111/j.1432-1033.1995.tb20583.x. [DOI] [PubMed] [Google Scholar]
- 5.Bailey S, Smith K, Fairlamb AH, Hunter WN. Eur. J. Biochem. 1993;213:67. doi: 10.1111/j.1432-1033.1993.tb17734.x. [DOI] [PubMed] [Google Scholar]
- 6.Krieger S, Schwarz W, Ariyanayagam MR, Fairlamb AH, Krauth-Siegel RL, Clayton C. Mol. Microbiol. 2000;35:542. doi: 10.1046/j.1365-2958.2000.01721.x. [DOI] [PubMed] [Google Scholar]
- 7.Hamilton CJ, Saravanamuthu A, Eggleston IM, Fairlamb AH. Biochem. J. 2003;369:529. doi: 10.1042/BJ20021298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An assay solution containing potassium HEPES (40 mM, pH 7.4), EDTA (1 mM), trypanothione disulfide (4.22 μM), DTNB (70.3 μM) and TR (2.81 mU/mL) was prepared. The positive control contained the above reagents plus the TR inhibitor clomipramine hydrochloride (200 μM). 80 μL per well of assay solution or positive control is added to the appropriate columns of clear Nunc 384-well Polysorb polystyrene plates using a BioTek μFill liquid handler. A CyBio liquid handler was used to transfer 200 nL of library compound, dissolved in DMSO at a concentration of 5 mg/mL, to each well. Enzymatic activity is initiated by the addition of 10 μL of an NADPH solution (135 μM in potassium HEPES, 40 mM, pH 7.4 and EDTA, 1 mM). The NADPH solution is added four plates at a time, with 4 minutes 20 seconds delay between each set of plates. Plates are incubated for 60 minutes at 30°C, before being removed and read in batches of four on a Perkin Elmer Envision plate reader at 405 nm. Raw data is processed using Microsoft Excel.
- 9.Borges A, Cunningham ML, Tovar J, Fairlamb AH. Eur. J. Biochem. 1995;228:745. doi: 10.1111/j.1432-1033.1995.tb20319.x. [DOI] [PubMed] [Google Scholar]
- 10.An assay solution containing potassium HEPES (40 mM, pH 7.4), EDTA (1 mM), NADPH (130 μM) and TR (10 mU/mL) was prepared. The positive control does not use enzyme. 20 μL per well of reagent A or positive control is added to the appropriate columns of clear Nunc 384-well Polysorb polystyrene plates. 100 nL of library compound, dissolved in DMSO at a concentration of 5 mg/mL, was added to each well. Enzymatic activity is initiated by the addition of 20 μL of a trypanothione disulfide solution (110 μM in potassium HEPES, 40 mM, pH 7.4 and EDTA, 1 mM). The trypanothione disulfide is added four plates at a time, with 4 minutes 20 seconds delay between each set of plates. Incubation, plate reading and data analysis is performed as in ref. 9, except that the plates are read at 355 nm.
- 11. http://chembank.broad.harvard.edu/
- 12.Raz B, Iten M, Grether-Buhler Y, Kaminsky R, Brun R. Acta Trop. 1997;68:139. doi: 10.1016/s0001-706x(97)00079-x. [DOI] [PubMed] [Google Scholar]
- 13.EC50 determinations were performed as in ref 12 with the following modifications. Test compounds (10 mM in DMSO) were serially diluted in HMI9 culture medium in 96-well plates (Greiner). Bloodstream T.b.brucei (S427) were added to give a final concentration of 103 ml−1 cells in 0.5% DMSO with inhibitor ranging from 50 to 0.2 μM in a total volume of 200 μl. Wells containing media only and media plus cells were used as controls. Pentamidine (250 – 1 nM) was included as a standard drug control on each test plate. Plates were incubated at 37 °C in a 5% CO2 humidified atmosphere for 72h, then 45 μM resazurin (Sigma) added. After a further 4 h incubation fluorescence due to formation of resorufin was measured at λ excitation 528 nm, λ emission 590 nm. Each inhibitor set was done in triplicate and the experiments repeated on three separate occasions. Data are presented as the weighted means and standard errors of the mean.