

SUMMARY
Trypanothione is a thiol unique to the Kinetoplastida and has been shown to be a vital component of their antioxidant defences. However, little is known as to the role of trypanothione in xenobiotic metabolism. A trypanothione S-transferase activity was detected in extracts of Leishmania major, L. infantum, L. tarentolae, Trypanosoma brucei and Crithidia fasciculata, but not Trypanosoma cruzi. No glutathione S-transferase activity was detected in any of these parasites. Trypanothione S-transferase was purified from C. fasciculata and shown to be a hexadecameric complex of three subunits with a relative molecular mass of 650,000. This enzyme complex was specific for the thiols trypanothione and glutathionylspermidine, and only used 1-chloro-2,4- dinitrobenzene from a range of glutathione S-transferases substrates. Peptide sequencing revealed that the three components were the alpha, beta and gamma subunits of ribosomal eukaryotic elongation factor 1B (eEF1B). Partial dissociation of the complex suggested that the S-transferase activity was associated with the gamma subunit. Moreover, Cibacron blue was found to be a tight-binding inhibitor and reactive blue 4 an irreversible time-dependent inhibitor that covalently modified only the gamma subunit. The rate of inactivation by reactive blue 4 was increased more than 600-fold in the presence of trypanothione and Cibacron blue protected the enzyme from inactivation by 1-chloro-2,4- dinitrobenzene, confirming that these dyes interact with the active site region. Two eEF1Bγ genes were cloned from C. fasciculata but recombinant C. fasciculata eEF1Bγ had no S-transferase activity, suggesting that eEF1Bγ is unstable in the absence of the other subunits.
INTRODUCTION
Infections with parasitic protozoa of the order Kinetoplastida are a common cause of serious illness and death in the tropics. Trypanosoma brucei spp. cause sleeping sickness in Africa, Trypanosoma cruzi is the cause of Chagas’ disease in South America and infections with Leishmania spp. produce a variety of pathologies termed the leishmaniases in Asia, Africa, South America and Europe. In general, the chemotherapy of these diseases is poor, with the available drugs suffering various drawbacks such as toxicity, limited efficacy and drug resistance (1;2). For instance, the majority of cases of visceral leishmaniasis in India do not respond to antimonials, which are the first-line drugs (3). A promising target for the design of new drugs to treat these illnesses involves thiol metabolism. In these parasites, this depends upon trypanothione (T[SH]2 or N1,N8-bis(glutathionyl)- spermidine) (4), in contrast to most other organisms (including their mammalian hosts) which use glutathione as the unmodified tripeptide (Figure 1) (5). The main function of trypanothione is the maintenance of cellular redox state and consequently, studies have concentrated on its role as an antioxidant (6).
Figure 1.

Structures of glutathione (γ-L-glutamyl-L-cysteinylglycine) and trypanothione (N1,N8-bis(glutathionyl)spermidine)
Another major function for cellular thiols is the detoxification of xenobiotics, i.e. chemicals that are foreign to a particular organism. These include naturally occurring compounds, industrial chemicals, drugs, herbicides and pesticides. Hydrophobic xenobiotics can readily diffuse into cells and are usually eliminated by Phase I and Phase II biotransformation reactions. These produce generally less reactive and more polar compounds, which can be actively extruded from the cytosol. An important group of Phase II reactions are catalysed by glutathione S-transferases (GSTs) and involve conjugation with glutathione. (7). GSTs have been purified from a wide range of organisms and are almost invariably dimers of subunits with masses of approximately 25 kDa. In any one organism, many different GST isozymes are usually expressed simultaneously, with these isozymes having wide and overlapping substrate specificities. Such a broad specificity system assists an organism in the metabolism of the multitude of different reactive xenobiotics to which it may be exposed. In addition, GSTs are also important in the detoxification of endogenous reactive chemical species produced during oxidative stress, such as lipid hydroperoxides (8) or reactive aldehydes (9). The GSTs therefore have a central role in detoxification metabolism and the overexpression of these enzymes is a common mechanism of drug resistance (10).
Although the classical GSTs have been well characterised, the GST fold is also found in functionally unrelated proteins such as plant stress-induced proteins (11), β-etherases (12), ion channels (13) and the eukaryotic translation elongation factor 1B gamma (eEF1Bγ) (14;15). The functions of the GST domain in these proteins remain poorly understood, although the eEF1Bγ from rice has recently been shown to possess a GST activity (16). This result was surprising, as eEF1B was previously thought only to be involved in protein synthesis and no enzymatic activity had been proposed for the eEF1Bγ subunit. Previously, the other two subunits (eEF1Bα and β) of eEF1B were shown to function to recycle elongation factor 1A (eEF1A) complexed with GDP back to the active eEF1A.GTP form (17;18). This eEF1A.GTP complex is then able bind to an amino-acyl tRNA, forming a ternary complex that is able to enter the A-site of the ribosome, with the hydrolysis of GTP.
In contrast to the wealth of information on GSTs and related proteins in other organisms, little is known about thiol-xenobiotic conjugation in trypanosomes. Although there has been one report of low levels of GST activity in T. cruzi (19), this has been disputed (20). In addition, no GST activities have been reported in the related Leishmania spp or T. brucei. However, since the major low molecular mass thiol in these organisms is trypanothione, it has been proposed that these trypanosomatids instead possess a trypanothione S-transferase or TST (5). This activity may also be involved in the resistance of Leishmania spp. to the metalloid antimony (21), which is currently front-line drug for the treatment of leishmaniasis. It has been found that the acquisition of high-level antimony resistance in L. tarentolae requires the overproduction of trypanothione, the overexpression of metal-thiol conjugate transporters and a third unidentified trypanothione-dependent factor (22). This factor was postulated to be a TST activity involved in the formation of an antimony-trypanothione conjugate. Indeed, in an analogous system, the mechanism of resistance of mammalian cells to arsenite involves the efflux of the metalloid, which is thought to be facilitated by the overexpression of a GST-pi (23;24).
Here, we report the detection of TST activities in trypanosomatids and the purification and characterisation of the Crithidia fasciculata TST. This is identified as the ribosomal elongation factor eEF1B complex and the subunit containing the TST active site defined as eEF1Bγ.
EXPERIMENTAL PROCEDURES
Materials
All reagents were standard commercial products and of the highest available purity. Trypanothione and glutathionylspermidine were obtained from Bachem. Glutathione affinity agaroses were obtained from Sigma, all other chromatographic resins and columns were from Amersham Pharmacia Biotech. Rat liver GST was purchased as a mixture of isoforms from Sigma.
Cell culture
C. fasciculata choanomastigotes T. cruzi epimastigotes T. brucei bloodstream form and L. major, L. infantum and L. tarentolae promastigotes were grown as described (25;26).
Enzyme assays
GST was assayed at 25°C in 100 mM (Na+) phosphate pH 6.5 with 1 mM 1-chloro-2,4-dinitrobenzene (CDNB) and 1 mM GSH as substrates. The rate of formation of the glutathione S-dinitrobenzene conjugate was followed at 340 nm, using the published extinction coefficient of 9.6 mM cm−1 (27). TST activity was measured under identical conditions with T[SH]2 and CDNB being added to 400 μM. Assay reactions were initiated with thiol, which was produced immediately before addition to assays by mixing trypanothione disulphide (T[S]2) with a 2-fold excess of tris(2-carboxyethyl)phosphine (TCEP) and a five-fold excess of NaOH. The absorbance coefficient for the T[SDNB]2 conjugate was measured by allowing assays to proceed to completion and found to be 9.2 mM−1cm−1 per mM of sulphydryl group. One unit of TST activity corresponds to one micromole of sulphydryl group conjugated per minute.
Where necessary, in TST assays with alternative electrophilic substrates, the pH and substrate concentrations given for the corresponding GST assay (27) were altered to minimise the rate of the spontaneous reaction. The activities with these substrates were calculated using the absorbance coefficients for the corresponding GSH conjugates. In the case of the peroxidase, dehydroascorbate reductase and thioltransferase assays, TCEP was omitted and T[S] was reduced by 10 μg ml−1 2 trypanothione reductase and 540 μM NADPH, before addition of the second substrate.
For enzyme purification, aliquots of fractions were screened for TST activity using a Molecular Devices Thermomax plate reader. In this assay, samples were diluted in the plate with water to give a final volume of 50 μl and the reactions initiated by the addition of 200 μl of a mixture containing buffer, dithiothreitol (DTT) and substrates. The final assay mixture contained 800 μM CDNB, 1 mM DTT, 50 μM T[S]2 and 100 mM (Na+) phosphate pH 6.5. The average rate of two blank assays was subtracted from these rates. Protein concentrations were measured using the procedure of Bradford (28), using BSA as the analytical standard. All trypanosomatid extracts were produced as described below for C. fasciculata and mouse liver was extracted by disruption in a Dounce homogeniser and then processed as for C. fasciculata cells.
Purification of the C. fasciculata TST
Unless otherwise stated, all the steps in the following purification were performed at 4°C. Frozen C. fasciculata cell pellets were thawed on ice and resuspended in an equal volume of ice-cold lysis buffer, giving in the final mixture 75 mM (Na+) phosphate pH 7.2, 2 mM DTT, 1 mM EDTA, 1 mM benzamidine, 1 mM phenanthroline, 3 μg ml−1 leupeptin, 250 μM 4-(2-aminoethyl)benzenesulphonyl fluoride and 1 μM pepstatin A. The cells were lysed by sonication, (4 × 30 s pulses at 20 microns amplitude from a 19 mm end-diameter probe) in a Sanyo Soniprep 150 sonicator with cooling to <4°C in an ice-salt bath between pulses. After centrifugation for 80 min at 40,000 × g the resulting supernatant was brought to a final concentration of 4% (w/v) polyethylene glycol 6000 (PEG) over 5 min, by the dropwise addition of PEG from a 50% (w/v) stock, stirred for 30 min and centrifuged for 30 min at 30,000 × g. The supernatant was adjusted to 9% (w/v) PEG and centrifuged as before. The resulting pellet was resuspended in 150 ml of buffer A (25 mM (Na+) Bis-tris, 1 mM EDTA, 1 mM DTT, pH 6.5) and then centrifuged to remove insoluble material.
The clarified supernatant was applied at 2 ml min−1 to a 100 ml (19 × 2.6 cm) Pharmacia Q-sepharose anion exchange column equilibrated in buffer A. After washing with 2 column volumes of buffer A, bound proteins were eluted at 1 ml min−1 with a linear gradient of 200-500 mM NaCl in the same buffer and active fractions pooled.
Following overnight dialysis against two litres of 550 mM (NH4)2SO4 in buffer B (25 mM (Na+) HEPES, 1 mM EDTA, 1 mM DTT, pH 7), the sample was applied at 2 ml min−1 to a 85 ml (16 × 2.6 cm) Pharmacia phenyl sepharose (low substitution) column equilibrated with 550 mM (NH4)2SO4 in buffer B. The column was washed with 5 column volumes of the same buffer and TST activity eluted with 250 ml buffer B.
Active fractions were pooled and concentrated to 1 ml by vacuum ultrafiltration in a Sartorius collodion bag. The concentrated TST was applied to a 319 ml (2.6 × 60 cm) Superdex 200 26/60 size-exclusion column equilibrated with buffer C (50 mM (Na+) HEPES, 300 mM NaCl, 0.01% (w/v) sodium azide, pH 7.5) and eluted at a flow rate of 2 ml min−1. Fractions with TST activity were pooled and concentrated as before.
Analytical chromatography
Analytical size-exclusion chromatography was carried out using a 24 ml (1 × 30 cm) Superdex 200 HR 10/30 size-exclusion column equilibrated with buffer C, with separations performed at a flow rate of 0.5 ml min−1. This procedure was also used to calculate the relative molecular mass (Mr) of proteins, using carbonic anhydrase (29 kDa), BSA (66 kDa), alcohol dehydrogenase (150 kDa), β-amylase (200 kDa), apoferritin (443 kDa) and thyroglobulin (669 kDa) as analytical standards.
Analytical anion-exchange chromatography was carried out using a 6 ml (1.6 × 3 cm) Pharmacia Resource Q column under the same buffer conditions as the Q-sepharose step in the TST purification scheme. The sample was applied at 1 ml min−1 and the column washed with 2 column volumes of buffer. The bound proteins were then eluted with a complex gradient which went from 0- 200 mM NaCl in 6 ml, 200-500 mM NaCl in 60 ml and then 500-1000 mM NaCl in 6 ml, all in buffer A.
Analysis of products of TST reaction
Assays for mass-spectrometric analysis were performed in 200 mM ammonium acetate pH 6.5 with the other conditions as before. The reactions were followed at 340 nm and after 30 min 2-hydroxyethyl disulphide was added to 10 mM, oxidising the T[SH]2 by disulphide exchange and thus quenching the trypanothione reaction. The products were then lyophilised in glass vials, re-dissolved in water and diluted 1:500 with 1:1 acetonitrile and water. Samples were analysed on a Micromass Ultima electrospray mass spectrometer in positive mode. Several scans were combined, background subtracted and smoothed to produce the final spectra.
Cross-linking analysis
All samples and controls were dialysed overnight against 55 mM (Na+) phosphate pH 7.5 reaction buffer, protein samples were then diluted to 1 mg ml−1 in reaction buffer. The amine-reactive homobifunctional reagent bis(sulphosuccinimidyl)suberate (BS3) was added, from a freshly prepared 10 mM stock in 5 mM sodium succinate pH 5, to the required final concentration. The reactions were incubated at room temperature for 40 min and then quenched by the addition of ethanolamine to a final concentration of 5 mM. Aldolase was used as a positive control and BSA and carbonic anhydrase as negative controls. Samples from the reactions were then analysed by using either 10 % SDS-PAGE gels (29), to examine complexes less than 150 kDa or 6 % Weber SDS-PAGE (30) to examine complexes greater than 150 kDa.
Dissociation of complex
For thiocyanate disassociation, samples of TST in buffer C were incubated on ice with 2 mM DTT for 3 h in the presence or absence of 1.5 M NaSCN. The proteins were then applied to a Superdex 200 HR 10/30 column equilibrated with either buffer C or this buffer plus 500 mM NaSCN, and eluted at 0.5 ml min−1. Since NaSCN inhibits the TST assay, 200 μl samples from each column fraction were dialysed against TST assay buffer (100 mM (Na+) phosphate, pH 6.5), before analysis by enzyme assay and SDS-PAGE.
Inhibition and chemical modification of TST
Modification with N-ethylmaleimide (NEM) was carried out in a 80 μl volume at room temperature in buffer C. Samples of TST (16 μg, 61 mU) were incubated with 100 μM DTT for 5 min and then NEM added to a final concentration of 400 μM. After a 15-min incubation the reactions were quenched by the addition of DTT to 625 μM.
Inhibition of TST by Cibacron blue F3G-A was determined under standard assay conditions, in the presence or absence of 0.5% (w/v) BSA. Routinely, TST was preincubated with the inhibitor but no decrease in inhibition was observed when assays were initiated with enzyme. Dye concentrations were determined spectrophotometrically, using the absorbance coefficients of εmM = 11.6 (622 nm) for Cibacron blue (31) and εmM = 4.2 (610 nm) for reactive blue 4 (32).
The rate of time-dependent inhibition of TST by chlorotriazine dyes was measured in reaction mixtures of 100 μl containing 70 mU (18 μg) samples of TST and varying concentrations of dye in 20 mM (Na+) phosphate pH 7.5. The reactions were allowed to proceed at room temperature, with 5-μl samples being removed at intervals to determine residual TST activity. These samples were assayed by 100-fold dilution into assay mixtures containing 0.5% (w/v) BSA, to prevent inhibition by any unreacted dye. Incubations with CDNB were performed under identical conditions and samples assayed in standard assay conditions. T[SH]2 and GSH were generated in incubations by the addition of 600 μM NADPH, 1 mU TryR or glutathione reductase and 500 μM T[S]2 or GSSG. The resulting data were fitted by non-linear regression analysis to the single exponential decay function and expressed as a percentage of the activity at zero time.
Samples of proteins for mass spectrometry were desalted by dialysis against one litre of water for 3 h at 4°C, before analysis by MALDI (matrix-assisted laser desorption and ionisation) mass spectrometry on a PerSeptive Biosystems Voyager-DE STR mass spectrometer.
Cloning of eEF1Bγ genes from C. fasciculata
A partial clone of the C. fasciculata eEF1Bγ was obtained by RT-PCR from cDNA prepared using the Cells-to-cDNA kit (Ambion). The sense primer (5′-CGCTATATAAGTATCAGTTTCTGTA-3′) contained 25 nucleotides of the mini-exon sequence that is trans-spliced onto the 5′ end of all trypanosomatid mRNA transcripts (33;34). The degenerate antisense primer (5′-CCYTCCCANGCNAGGTAKTC-3′) was designed to the tryptic peptide ITDYLA(F/W)EGPTIPLPV. PCR was performed in 50 μl volume reactions containing template cDNA, 20 ng ml−1 sense and antisense primers, 250 μM dNTPs, 5 U Taq polymerase (Promega) and Taq buffer plus 1.5 mM MgCl2. The reactions were placed in a thermocycler that had been preheated to 95°C and subjected to the following: 95°C for 5 min 30 cycles of 95°C for 1 min 55°C for 1 min 72°C for 2 min and finally the reactions were heated to 72°C for 10 min. PCR products were then cloned into the pCR-Blunt II-TOPO plasmid using the Zero Blunt TOPO PCR cloning kit (Invitrogen).
Southern blotting was carried out by the standard capillary transfer method (35), using the RT-PCR clone of eEF1Bγ as a probe. The probe was labelled using the Gene Images labelling kit and hybridisation detected with the Gene Images dioxetane detection module (Amersham Biosciences).
PCR amplification of C. fasciculata eEF1Bγ intergenic regions was carried out using sense (5′-GGAGCTGTTTGACTGGGAGGAGAT-3′) and antisense primers (5′-GAAGTCCGCCGTCTC-GTTGTC-3′) as described above, with the substitution of Pfu polymerase and Pfu buffer (Promega) for Taq polymerase. Four intergenic regions were amplified, cloned and sequenced. Multiple primers were then designed to their 5′ and 3′ regions. One pair of these primers (sense 5′- GCACCGGCGTACCTGATGACTT-3′ and antisense 5′-TTAGTGGCCACTGATGCGACAGC-3′) produced a product that was cloned, sequenced and identified as an eEF1Bγ gene. This gene was then cloned into the expression vector pET15b (Novagen) and recombinant protein expressed and purified according to the manufacturer’s instructions.
Nomenclature
The terminology of eukaryotic elongation factors is somewhat confusing. In this paper we have used the IUBMB nomenclature recommendations (http://www.chem.qmul.ac.uk/iubmb/misc/trans.html), with the elongation factor 1B complex, previously named eEF-1βγδ being referred to as eEF1B. The complex’s subunits are, in order of increasing size, elongation factor 1B alpha (previously eEF-1β), now referred to as eEF1Bα elongation factor 1B beta (previously eEF-1δ) referred to as eEF1Bβ and elongation factor 1B gamma (previously eEF-1γ) referred to as eEF1Bγ. The SWISS-PROT identifier is used when referring to previously characterised elongation factors.
RESULTS
Detection and initial characterisation of trypanothione S-transferase activities in trypanosomatids
Clarified extracts of various organisms and cell types were assayed for S-transferase activities (Table 1). L. tarentolae (a lizard parasite) was included since this organism has been frequently used in studies on Leishmania antimony resistance. Mouse liver extract was assayed as a positive control for the GST assay and trypanothione reductase activity was used to confirm adequate extraction of the parasites. No GST activity was detected in the trypanosomatids. Instead a trypanothione S-transferase (TST) activity was detected in C. fasciculata, all the Leishmania spp. and T. brucei. The TST activity in mouse liver extracts is probably due to the ability of GSTs to use T[SH]2 as an alternative substrate, although with a specific activity 100-fold less than that given with GSH. Since TST activity was highest in C. fasciculata and L. major, these activities were further characterised. Activity in these extracts was proportional to the amount of protein added, heat labile, and, when analysed by size-exclusion chromatography the activity eluted close to the void volume of the column, showing Mr values greater than 400,000 (data not shown). This is in contrast to the GST activity in the mouse liver extract, which eluted with an Mr of 45,000 (this value being identical to the reported Mr of the three major mouse glutathione S-transferases) (36).
Table 1. Specific activities of TST, GST and trypanothione reductase in soluble extracts of trypanosomatids.
All enzyme activities are assayed as described in the relevant methods section and corrected for non-enzymatic background rates.
| Organism | Cell type | TST (mU mg−1) |
GST (mU mg−1) |
Trypanothione reductase (mU mg−1) |
|---|---|---|---|---|
| C. fasciculata | choanomastigotes | 16.7 | <0.1a | 920 |
| L. major | promastigotes | 6.4 | <0.1 | 580 |
| L. infantum | promastigotes | 2.9 | <0.1 | 80 |
| L. tarentolae | promastigotes | 0.4 | <0.1 | 430 |
| T. cruzi | epimastigotes | <0.1a | <0.1 | 510 |
| T. brucei | bloodstream form | 2.8 | <0.1 | 30 |
| M. musculus | liver | 23 | 1,970 | N.D. |
less than limit of detection
N.D. – not determined
A pilot study showed that C. fasciculata TST activity was not retained on S-hexylglutathione, glutathione disulfide or glutathione coupled to epoxy-activated agarose in contrast to the mouse liver GSTs, which were completely retained on S-hexylglutathione agarose (data not shown).
Purification of C. fasciculata TST
Since C. fasciculata had the highest TST activity, it was used in further studies. The C. fasciculata TST was concentrated by PEG 6000 precipitation and then purified to constant specific activity by a combination of anion-exchange, hydrophobic-interaction and size-exclusion chromatography (Table 2). Typically, TST activity can be purified 200-fold with about 10% recovery. Surprisingly, when the purification fractions were analysed by SDS-PAGE, three major polypeptides of mass 31, 38 and 45 kDa and one minor polypeptide of 65 kDa were visible. These proteins did not alter in relative abundance in the last two purification steps (Figure 2). In order to determine if these polypeptides were associated through disulphide bonds, samples of the final preparation were separated in the presence and absence of DTT. No association between the four polypeptides was seen in the oxidised sample, showing that any complex is formed by non-covalent interactions.
Table 2.
Purification of C. fasciculata TST
| Fraction | Activity (U/ml) |
Total Protein (mg) |
Total Units (mU) |
Specific Activity (mU mg−1) |
Purification factor |
Recovery (%) |
|---|---|---|---|---|---|---|
| Crude extract | 510 | 19,880 | 145,410 | 7.3 | - | 100 |
| 4-9% PEG | 640 | 2,010 | 63,110 | 31.4 | 4.3 | 43 |
| Q-sepharose | 770 | 72 | 58,239 | 815 | 112 | 40 |
| Phenyl sepharose | 2,080 | 9.1 | 13,960 | 1,530 | 210 | 9.6 |
| Superdex 200 | 4,140 | 5 | 8,290 | 1,658 | 227 | 5.7 |
Figure 2. SDS-PAGE of C. fasciculata TST purification fractions.

Samples (10 μg protein in each lane) from the purification were separated in a 10% SDS-PAGE gel. Lane 1, clarified cell extract lane 2, 2-10% redissolved PEG 6000 precipitate lane 3, Q-sepharose eluant lane 4, Phenyl sepharose eluant lane 5, Superdex 200 eluant. Lanes 6 and 7 were additional samples of the Superdex 200 pool with 1mM DTT being added to the sample in lane 6 and the sample in lane 7 containing no reducing agent.
As a further test of purity, samples of purified TST were applied to high-resolution anion-exchange and size-exclusion columns. The TST activity again eluted as a single peak from both columns, with no increase in the specific activity of the recovered enzyme. The chromatogram of the size-exclusion separation (Figure 3) shows a single major peak making up >95% of the total protein, with minor amounts of higher and lower molecular mass contaminants. The TST activity co-elutes with the main peak, which contains the four polypeptides that were visible in the last stage of the purification procedure in an unaltered stoichiometry. This stoichiometry was investigated by image analysis of multiple preparations of TST, showing a constant ratio between the 31 (α), 36 (β) and 45 kDa (γ) subunits of 1:1:2, respectively. In addition, the Mr value of 650,000 calculated for this species is consistent with the high molecular mass activities seen in the C. fasciculata and L. major crude extracts.
Figure 3. Analytical size-exclusion chromatography of purified C. fasciculata TST. (A).

Elution profiles of TST activity and protein from a Superdex 200 HR analytical size exclusion column. The absorbance at 280 nm is shown with a solid line, the fraction numbers are shown above this trace and are corrected for the delay volume between the fraction collector and the UV flowcell. The TST activity of fractions was measured using the microtitre plate assay and shown in filled circles joined with a dotted line. (B) Determination of the relative molecular mass of this species, with the sample (○) shown relative to a set of analytical standards (●). (C) SDS-PAGE analysis of 10 μl-samples taken from the 0.5 ml fractions collected during this separation. The four visible bands, subsequently identified as the eEF1Bα, β and γ subunits of the eEF1B complex and an aminoacyl-tRNA synthetase are labelled at the right-hand side of the gel.
Peptide sequencing and identification of TST subunits
The proteins in a sample of purified TST were separated by SDS-PAGE, digested in the gel with trypsin and the resulting peptides separated by reversed-phase HPLC. Selected peptides were Edman sequenced and the sequences used in database searches. These identified p31 (subunit α) to be similar to the 25 kDa ribosomal elongation factor eEF1Bα from T. cruzi (SWISS-PROT P34827). Peptides from p38 (subunit β) were likewise similar to the T. cruzi 30 kDa elongation factor eEF1Bβ (Q26914) and peptides from p45 (subunit γ) were similar to the 47 kDa eEF1Bγ from T. cruzi (P34715) and L. infantum (Q9BHZ6). Peptides from the protein of approximately 65 kDa, which was present in low and variable amounts between different TST preparations, were similar to the sequence of the predicted product of a L. major gene ID:LmjF15.0230. This is a 67 kDa protein that shows a high level of identity to amino-acyl tRNA synthetases (49% identity with a hamster lysyl-tRNA synthetase (P37879)). In combination, these data suggest that the C. fasciculata TST is an eEF1B complex, as the mammalian form of this assembly contains the eEF1Bα, β and γ subunits and has also been shown to associate with amino-acyl tRNA synthetases (37).
Quaternary structure of TST complex
The purified TST preparation contained three major polypeptides and the relative intensities of the protein bands were not altered in the last three purification steps, suggesting that these three proteins associate to form the high molecular mass TST complex. The association of these proteins was therefore investigated by chemical cross-linking. As seen in Figure 4, addition of BS3 to purified TST results in cross-linking of the eEF1B subunits, producing high molecular mass species (Panel A, lanes 2-4). In the positive control, the tetrameric enzyme aldolase is cross-linked into dimers, trimers and tetramers (Panel A, lane 7) whilst in the negative control, the monomeric proteins, BSA (68 kDa) and carbonic anhydrase (29 kDa) are unaffected (Panel A, lane 6). Significantly, the product formed in the TST cross-linking reaction (Panel B, lane 4) has a mass between 150 and 200 kDa, which is in the range of the 160 kDa mass given by the sum of the SDS-PAGE masses of the α, β and γ subunits in a 1:1:2 ratio obtained by image analysis. In addition, 170 kDa is also one quarter of the Mr given by gel filtration. These results suggest that the three subunits associate in a αβγ2 tetramer and then this tetramer itself tetramerises to form a hexadecamer. Indeed the (αβγ2)4 hexadecameric species may have been formed in the cross-linking reaction, because, despite equal amounts of protein being loaded in each lane, the total amount of protein visible in the gel decreases with increasing degrees of cross-linking. This effect may be due to species of more than 400 kDa being unable to enter these gels. These results are in good agreement with previous studies on the mammalian eEF1B, which was shown to be a Mr 670,000 complex containing the eEF1Bα, β and γ subunits in a 1:1:2 ratio (38).
Figure 4. Cross-linking of the TST complex.

(A) 10% Laemmli SDS-PAGE analysis of the products of the reaction between increasing concentrations of BS3 and TST (lane 1 - no BS3 lane 2 - 75 μM lane 3 - 200 μM and lane 4 – 1 mM. The negative control used the monomeric proteins BSA (66 kDa) and carbonic anhydrase (29 kDa), which were treated with 1 mM BS3 in the presence (lane 5) or absence (lane 6) of TST. The positive control in lane 7 shows the products from treatment of the tetrameric protein aldolase with 75 μM BS3, with the masses of the products formed indicated on the right-hand side of the gel. (B) The same samples were analysed in a 6% Weber SDS-PAGE gel. The masses on the left of the gel are from a cross-linked phosphorylase b marker and again the masses of the aldolase products are indicated on the right-hand side of the gel. Samples as described in panel A.
Catalytic activities of the C. fasciculata TST
The enzyme is highly specific for the glutathione conjugates, trypanothione and glutathionylspermidine (GspdSH). It showed only low activity with glutathione ethyl ester (H-Glu(Cys-Gly-OEt)-OH) and no detectable activity with glutathione. In contrast, when the thiol specificity of a purified mixture of rat liver GST isoforms was tested, activity was detected with T[SH]2 and GSH (data not shown). Kinetic parameters for TST with the three active thiol substrates with a fixed concentration of 400 μM CDNB were determined (Table 3). Interestingly, the Km(app) for GspdSH is twice that of trypanothione, suggesting that T[SH]2 is recognised as two independent GspdSH moieties, giving an effective concentration twice its actual concentration. TST showed no conjugation activity (<0.01 μmoles min−1 mg−1) with t-butyl hydroperoxide, cumene hydroperoxide, ethacrynic acid, 1,2-dichloro-4-nitrobenzene, p-nitrophenyl acetate, 1,2-epoxy-3-(4-nitrophenoxy)propane, trans-2-nonenal, benzyl isothiocyanate, 4-androstene-3,17-dione, butyl nitrate or trans-4-phenyl-3-buten-2-one. In addition, no thiol-transferase activity was found with GSSG or 2-hydroxyethyl disulphide as substrates and no dehydroascorbate reductase activity was detected. The only good electrophilic substrate for TST is CDNB (3300 mU mg−1), with the structurally related p-nitrobenzyl chloride showing marginal activity (70 mU mg−1).
Table 3.
Kinetic parameters of the C. fasciculata TST
| Substrate a) | Km(app) (μM) | Vmax (nmol min−1) | kcat (s−1) f) | kcat/Km (M−1 s−1) |
|---|---|---|---|---|
| CDNB b) | 190± 27 | 3.45± 0.14 | 236 | 1.2 × 106 |
| T(SH)2 c) | 159± 19 | 2.77± 0.12 | 190 | 1.2 × 106 |
| GspdSH d) | 310± 31 | 2.81± 0.13 | 193 | 0.62 × 106 |
| GSH monoethyl ester e) |
6,100± 590 | 0.59± 0.02 | 17.4 | 0.003 × 106 |
The absorbance coefficient for the T[DNB]2 conjugate of 9.2 mM cm−1 was used for both T[SH]2 and GspdSH, whilst the published value for GS-DNB of 9.6 mM cm−1 was used for GSH ester.
Measured with 400 μM T[SH]2 as the fixed substrate
Measured with 400 μM CDNB as the fixed substrate
Measured with 400 μM CDNB as the fixed substrate
Measured with 400 μM CDNB as the fixed substrate
Calculated using the calculated concentration of the [(αβγ2)]4 complex
In order to confirm that the spectrophotometric TST assay is actually measuring substrate conjugation, the products formed by TST in the CDNB/trypanothione assay were compared with both a T[SDNB]2 standard and the products of a control assay with no added TST. The UV spectrum of the TST product was identical to that of a T[SDNB]2 standard. Assay reactions performed in volatile buffer in the presence and absence of TST were analysed by electrospray mass spectrometry. This showed that significant amounts of both the bis-(M + H+ = 1056.1 Da) and the mono-(M + H+ = 966 Da) adducts with CDNB are only formed in the presence of TST (data not shown).
Denaturation of the TST complex and separation of subunits
In order to identify which subunit is responsible for the TST activity in the eEF1B complex, samples of TST were partially denatured and the subunits separated and assayed for activity. This was achieved by treatment with the chaotropic agent sodium thiocyanate (NaSCN). TST was incubated on ice in the presence or absence of NaSCN and the proteins then separated by size exclusion chromatography, again in the presence or absence of thiocyanate. As expected, a sample of the eEF1B complex that had not been exposed to NaSCN eluted as a single species (Figure 5, Panel A), with a protein peak in fraction 14 and all the subunits present in a 1:1:2 (α,β and γ respectively) ratio (Figure 5, Panel C). In contrast, when the TST was preincubated for 3 h with NaSCN and then chromatographed in the presence of NaSCN, two partially resolved protein species were observed to elute from the column (Figure 5, Panel A). Due to the incomplete separation of these complexes, their subunit compositions cannot be clearly defined. However, when the amounts of each subunit present in these fractions (Figure 5, panel D) is compared to the level of enzyme activity, a clear correlation between the levels of eEF1Bγ and TST activity can be observed. Significant TST activity is present in fractions 13-17, which corresponds well with the elution profile of eEF1Bγ, but not that of eEF1Bα or β, which elute mainly in fractions 16 and 17.
Figure 5. Separation of the subunits of the TST complex using thiocyanate.

Samples of TST were incubated in the presence and absence of 1.5 M NaSCN and then applied to a size-exclusion column. (A) Elution profiles of these samples, the untreated control is shown in red and the NaSCN-treated sample in blue. The elution positions and masses of protein standards are indicated with arrows. (B) TST activity profiles of the fractions collected from this separation, the untreated control is indicated with red triangles and the NaSCN-treated sample with blue squares. (C and D) SDS-PAGE analysis of these column fractions, with Panel C corresponding to the untreated control and Panel D to the NaSCN-treated sample.
Inhibition and affinity labelling of TST
The effects of the modification of protein thiol groups on TST activity was investigated by alkylation of a sample of reduced TST with NEM. TST was inactivated by 81 ± 2 % upon exposure to 400 μM NEM for 15 min, compared to no detectable loss of activity in a parallel negative control. Mass spectrometry of the NEM-treated TST showed the modification of 1 cysteine residue in the eEF1Bβ subunit and 5 residues in the eEF1Bγ subunit (data not shown). This result demonstrates that the eEF1Bα subunit is not responsible per se for the TST activity of eEF1B and suggests the possible involvement of protein thiol groups in the TST catalytic mechanism. Interestingly, the actual molecular masses of the eEF1Bα and β subunits, determined by mass spectrometry, are significantly different from their apparent molecular weights by SDS-PAGE, with the 23 kDa eEF1Bα migrating as a 31 kDa protein and the 27 kDa eEF1Bβ migrating with an apparent mass of 38 kDa. These effects have previously been noted for the equivalent mammalian eEF1B subunits, with for example the 25 kDa rabbit eEF1Bα (previously designated eEF-1β, P34826) migrating as a 32 kDa protein on SDS-PAGE. This anomalous migration is probably the result of the highly acidic nature of the eEF1Bα and β proteins (39).
Cibacron blue F3G-A, a monochloro triazine dye, was found to be an extremely potent inhibitor of the C. fasciculata TST, with an IC50 of 13 ± 0.8 nM (Figure 6). Inhibition constants in this range can indicate an irreversible active-site titration, and indeed no recovery of activity was observed upon prolonged dialysis of Cibacron blue-inhibited TST. However, this result was found to be due to the inability of Cibacron blue to pass through dialysis membranes, possibly as a result of dye aggregation (40). In light of the known affinity of this dye for serum albumin (41), TST inhibition was therefore determined in the presence of 0.5% (w/v) BSA. The addition of this protein caused a large shift in the IC50 to 12 ± 0.9 μM, most probably due to the sequestration of the inhibitor from the assay solution (Figure 6). This modified assay allows the accurate determination of TST activity in the presence of micromolar concentrations of Cibacron blue. Using this procedure, samples of TST were incubated with 100 μM Cibacron blue and then samples diluted into assays containing 0.5% (w/v) BSA. No change in TST activity was observed over a 30 min period, indicating that Cibacron blue does not irreversibly inactivate the enzyme (Figure 7, Panel A, inverted triangles).
Figure 6. Inhibition of TST by Cibacron blue.

Inhibition of TST by Cibacron blue in the presence (○) and absence (●) of 0.5% (w/v) BSA in the assay buffer.
Figure 7. Time-dependent inhibition of TST by reactive blue 4.

(A) Time-dependent inactivation of TST by reactive blue 4. TST was incubated with the inhibitors in 20 mM (Na+) phosphate pH 7.5 at 25°C and the residual activity determined at intervals, as described in the methods. The incubations contained either 100 μM Cibacron blue (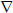 ) or reactive blue 4 at 2 μM (○), 10 μM (●), 20 μM (□), 50 μM (■), 75 μM (Δ) and 100 μM (▲). (B) Observed rates of TST inactivation by reactive blue 4 as a function of the concentration of inhibitor.
) or reactive blue 4 at 2 μM (○), 10 μM (●), 20 μM (□), 50 μM (■), 75 μM (Δ) and 100 μM (▲). (B) Observed rates of TST inactivation by reactive blue 4 as a function of the concentration of inhibitor.
However, when these experiments were repeated using reactive blue 4, a more reactive dichloro triazine dye, irreversible time-dependent inhibition was observed (Figure 7, Panel A). The inactivation reaction was saturable, with a pseudo-first order rate constant of (kobs) of 0.08 ± 0.01 min−1 and a calculated dissociation constant (Ki(app)) of 49 ± 17 μM (Figure 7, Panel B). This indicates that a reversible EI complex is formed as a reaction intermediate and therefore suggests that the inhibitor is binding at or near the TST active site. Duplicate 18 μg samples of TST were incubated at room temperature in the presence and absence of 50 μM reactive blue 4. As expected, the dye completely inactivated the enzyme (98 ± 2 %). Mass spectrometry of these samples showed no modification of the α or β subunits, whereas the eEF1Bγ subunit underwent considerable peak broadening, with a peak shift of 1,330 ± 480 Da (Figure 8). Within the precision of the method, this mass shift would be consistent with the covalent addition of two dye molecules to the eEF1Bγ subunit.
Figure 8. Covalent modification of eEF1Bγ by reactive blue 4.

Reactive blue 4-inactivated TST was analysed by MALDI-TOF mass spectrometry. The two samples were prepared as described in the text, with the TST sample shown in panel A being an untreated negative control and the TST sample shown in panel B being inactivated by incubation with 50 μM reactive blue 4 for 1 h.
More evidence as to the location of the triazine dyes’ binding site was provided by the interactions between these inhibitors and the substrates T[SH]2 and CDNB. Incubation of TST with high concentrations of the xenobiotic substrate CDNB, in the absence of T[SH]2, causes an irreversible time-dependent inactivation of the enzyme (Figure 9, Panel A). This inactivation can be prevented by addition of either the co-substrate T[SH]2 or the reversible inhibitor Cibacron blue. The protection produced by Cibacron blue suggests that the association of this inhibitor with the TST active site prevents the binding of CDNB. However, the effect of substrate on the inhibition of TST by reactive blue 4 is strikingly different. When the enzyme is incubated with 50 μM reactive blue 4 and 500 μM T[SH]2 the rate of inactivation is increased from 0.045 ± 0.003 min−1 to 29 ± 5 min−1 (Figure 9, Panel B). This corresponds to an over 600-fold increase in rate, with 80% of TST activity being lost in the first 3 seconds of the reaction. This effect is specific to T[SH]2, since addition of equal amounts of sulphydryl group in the form of the dithiol DTT or the monothiol GSH had no significant effect on the rate of inactivation, with kobs of 0.037 ± 0.005 min−1 and 0.047 ± 0.003 min−1, respectively. The specificity of this effect suggests that T[SH]2 may be causing a conformational change that increases either the affinity of the enzyme for the inhibitor or the reactivity of an active site residue. In combination, these data therefore strongly suggest that the triazine dyes bind in the active site region of the TST and that this is located within the eEF1Bγ subunit.
Figure 9. Interactions between TST substrates and triazine dyes.

Incubations were performed in 20 mM (Na+) phosphate pH 7.5 at 25°C and the residual activity determined at intervals, as described in the methods. (A) Time-dependent inactivation of TST by CDNB. The incubations contained no CDNB (○), 500 μM CDNB (●), 500 μM CDNB and 500 μM T[SH]2 (■) and 500 μM CDNB and 100 μM Cibacron blue (▲). (B) Effects of thiols on the inactivation of TST by reactive blue 4. The incubations contained no reactive blue 4 (○), 50 μM reactive blue 4 (●), 50 μM reactive blue 4 and 1 mM GSH (□), 50 μM reactive blue 4 and 500 μM T[SH]2 (■) and 50 μM reactive blue 4 and 500 μM DTT (Δ).
Cloning of C. fasciculata eEF1Bγ genes
The tryptic peptides produced from the digestion of purified C. fasciculata eEF1Bγ were aligned with the sequence of the L. infantum eEF1Bγ (Q9BHZ6). A peptide with the sequence ITDYLA(F/W)EGPTIPLPV aligned with the C-terminus of this protein and was therefore used to design a degenerate 3′-primer. This allowed the cloning of a partial C. fasciculata eEF1Bγ gene from cDNA by RT-PCR, using the spliced leader sequence as the 5′ primer (Figure 10, C. f. RT-PCR, and data not shown ‡). This clone was then used as a probe in a Southern blot analysis of C. fasciculata gDNA, which indicated that multiple copies of the eEF1Bγ gene were present as tandem repeats (data not shown ‡). Primers were therefore designed to sequences in the 5′ and 3′ regions of the L. infantum eEF1Bγ that were conserved between the known trypanosomatid eEF1Bγ genes. This allowed the PCR cloning of four intergenic regions that were flanked in the C. fasciculata genome by copies of eEF1Bγ genes, confirming that the C. fasciculata eEF1Bγ genes are organised in a non-identical tandem array. Primers were then designed to these sequences and a full-length eEF1Bγ sequence cloned (Figure 10, C. f. full, and data not shown ‡). Of the thirteen peptide sequences isolated from the purified C. fasciculata eEF1Bγ protein, seven can be aligned with the predicted sequence of the full-length gene, confirming the identity of the clone. The predicted products of the two C. fasciculata eEF1Bγ genes are 88% identical to each other and the full-length C. fasciculata eEF1Bγ is 32%, 68%, 77% and 63% identical to the human, T. brucei, L. infantum and T. cruzi eEF1Bγ proteins, respectively. The full-length and RT-PCR C. fasciculata eEF1Bγ proteins contain eight and six cysteine residues, respectively. These values are in agreement with the five NEM-accessible cysteine residues detected in the purified C. fasciculata eEF1Bγ by mass-spectrometry. Interestingly, only one of these residues (Cys53 in the full-length eEF1Bγ) is conserved in all the trypanosomatid proteins and this residue may therefore be the site where alkylation by NEM inactivates the enzyme. The full-length C. fasciculata eEF1Bγ was expressed and the recombinant protein purified, however this protein showed no TST activity (data not shown‡).
Figure 10. Alignment of the sequences of trypanosomatid eEF1Bγ proteins.

The predicted amino-acid sequences of the C. fasciculata eEF1Bγ genes isolated by RT-PCR (RT-PCR C.f.) and intergenic PCR (Full-length C.f.) are shown aligned with the sequences of the Homo sapiens (P26641), T. brucei (temporary name - TRYPtp_ends-17b11.p1k_188), L. infantum (Q9BHZ6) and T. cruzi (P34827) proteins. Gaps are indicated with dashes and the alignment shaded according to similarity, the colours indicate black background, white text - over 80% identity dark grey background, white text - over 60% identity and light grey background, black text – over 40% identity.
DISCUSSION
The novel S-transferase activity detected in trypanosomatids has a unique substrate specificity for trypanothione and glutathionylspermidine. The C. fasciculata enzyme appears to recognise the GspdSH moiety as a substrate, with trypanothione probably being used as two independent GspdSH substrate molecules. This requirement for a glutathione amide is in complete contrast with the substrate requirements of classical GSTs. Indeed, rat liver GSTs’ glutathione-binding sites have been probed with a large number of glutathione derivatives and it was found that the least important part of the substrate for enzyme recognition is the glycyl moiety (42). Our finding that rat liver GSTs can utilise T[SH]2 is in agreement with these results. As discussed later, the specificity of the C. fasciculata TST for CDNB and the comparatively low specific activity with this substrate appears to be a common feature of the S-transferase activities of eEF1Bγ subunits.
The assignation of the TST activity of the C. fasciculata eEF1B to eEF1Bγ is supported by several independent studies. From sequence analysis and homology modelling Koonin et al. predicted that the N-terminal domain found in eEF1Bγ proteins would form a GST-like fold, which was expected to possess GST activity (15). Subsequent structural studies on the Saccharomyces cerevisiae eEF1B gamma-1 (P29547) N-terminal domain confirmed that this domain’s fold is remarkably similar to that of the GSTs, but no GST activity was detected in this truncated protein (14). However, an unusual 50 kDa GST from the yeast Yarrowia lipolytica has been purified (43) and we note that the published N-terminal sequence shows high similarity to the S. cerevisiae eEF1B gamma-2 (P36008 61% identity and 80% similarity in 29 residues). In common with the C. fasciculata TST, this unusual GST was highly specific for CDNB as an electrophilic substrate, and did not bind to glutathione-affinity resins. These data suggest that native forms of yeast eEF1Bγ proteins should be examined for S-transferase activities.
More recently, GST activity was detected in recombinant rice eEF1Bγ (Q9ZRI7) and preparations of the native eEF1ββ′γ complex (16). The eEF1Bγ had a low specific activity, with a kcat value of approximately a fiftieth of a classical rice GST. Moreover, unlike classical GSTs from rice, this eEF1Bγ lacked glutathione peroxidase activity. The S-transferase activity of the C. fasciculata eEF1Bγ therefore appears to be similar to that of the rice subunit, with no activity towards organic peroxides and a comparatively low specific activity with CDNB. Interestingly, in common with the T. cruzi and Bombyx mori (Q9BPS3) eEF1Bγ subunits (44;45), and in contrast to the C. fasciculata protein, the rice eEF1B can be purified by glutathione-affinity chromatography. This indicates that the eEF1Bγ of these organisms can bind GSH and may indicate that the T. cruzi eEF1Bγ has a different substrate specificity to those of the other kinetoplastids.
We were unable to detect TST or GST activity in crude extracts of T. cruzi, in agreement with another report (20). However, another study did detect trace amounts of GST activity (70 mU mg−1) in T. cruzi proteins purified on a glutathione-affinity resin (46). Interestingly, the major components of this preparation were later identified as the three subunits of the T. cruzi eEF1B complex (47;48). Purified recombinant T. cruzi eEF1Bγ was found to be devoid of GST activity (49) and activity with trypanothione was not examined in either study (46;49). However, the authors proposed that the GST activity of the γ subunit may require association with the other subunits of the eEF1B complex. This hypothesis is consistent with the instability of the isolated rice eEF1Bγ protein, in comparison to the native complex (16) and our failure to detect TST activity in the recombinant C. fasciculata eEF1Bγ. Indeed, further studies in our laboratory have shown that no S-transferase activity is detectable in the recombinant L. major eEF1Bγ protein either, whereas the L. major eEF1B holocomplex possesses TST activity. These findings will be described in a subsequent paper.
The diseases caused by Leishmania spp. are usually treated by antimonial compounds and Grondin et al. have hypothesised that a TST activity may be important in the development of drug resistance (22). This was suggested by their finding that the overexpression of thiol-metal conjugate transporters and increased T[SH]2 levels are necessary, but not sufficient for high-level metalloid resistance in L. tarentolae. They therefore proposed that overexpressed TST was the missing trypanothione-dependent factor in the resistance phenotype. Indeed, the mechanism of resistance in Leishmania may be similar to that of mammalian cells, where increased GSH levels and transporter expression are coupled to the overexpression of GSTs (23;24). Interestingly, L. tarentolae is hypersensitive to metals in comparison with pathogenic species (50;51) and this correlates with a low level of extractable TST activity, compared to the pathogens L. major and L. infantum (Table 1). However, many other factors such as thiol levels differ between these species and more work is required to investigate if the TST activity in leishmania is involved in metal resistance.
The trypanosomatids appear to be unusual amongst eukaryotes in lacking a classical low-molecular mass S-transferase activity (52). However, as Moutiez et al. (53) suggested, at physiological pH the increased reactivity of T[SH]2 over GSH may make the non-enzymatic conjugation of xenobiotics far more efficient in the trypanosomatids than other organisms. Thus, high cytosolic T[SH]2 concentrations may efficiently substitute for a high enzymatic TST activity and at cytosolic pH the eEF1B TST activity may represent only a small fraction of the non-enzymatic S-transferase activity in the cell. A minor contribution of eEF1B to total cellular S-transferase activity is also consistent with the low specific activity of the rice eEF1B (16) and the failure to detect significant S-transferase activity in T. cruzi, without partial purification of the eEF1B complex (46). This suggests that either the appropriate xenobiotic substrate of eEF1Bγ has not yet been identified, or that this activity has a distinct physiological role unrelated to the conjugation and detoxification of xenobiotics. Importantly, the evolution of thiol substrate specificity of the trypanosomatid eEF1Bγ proteins towards trypanothione appears to have paralleled the evolution of trypanothione as these organisms’ major thiol metabolite. This demonstrates that a specific interaction with cellular thiols is required for the eEF1B complex’s function.
An alternative activity of GSTs is the binding of ligands. These ligands include hydrophobic compounds such as haemin and glutathione S-conjugates (54). In addition, glutathione can form a mixed disulphide with a cystine residue in the active site of the omega class of GSTs (55). It is therefore possible that the eEF1B complex may be involved in regulating protein synthesis in response to oxidative or toxic stress. Tight control of protein synthesis by the redox state of the GSH/GSSG couple has been observed by Kosower et al. (56), and in vivo studies have indicated that it is the elongation step of protein synthesis which is most sensitive to oxidative stress (57). Significantly, translational control may be especially important in trypanosomes as they lack RNA polymerase II promoters and are thus unusually dependent on post-transcriptional regulation, for review see (58).
One mechanism for this response is suggested by the finding that the valyl-tRNA synthetase activity (ValRS) in yeast is controlled by an uncharacterised high molecular weight oxido-reductive regulatory apparatus which is activated by reduced glutathione (59-61). ValRS has since been found to be present in mammalian cells exclusively as a complex with eEF1B (62) and to associate with the elongation factor complex through an N-terminal extension similar in sequence to eEF1Bγ (63). Furthermore, several other aminoacyl-tRNA synthetases (ARSs) have recently been shown to associate with eEF1B in vivo (64). Our finding that the C. fasciculata eEF1B co-purifies with a probable ARS indicates that this interaction is conserved in the trypanosomatids. The binding of eEF1B to ARS is a functional interaction, as the ValRS activity of the ValRS.eEF1B complex, but not the free ValRS is activated by eEF1A and GTP (62). This activation could be reversed by the addition of anti-eEF1A and eEF1Bγ antibodies (64). Translational control by eEF1B could therefore be due to either effects upon tRNA synthetases, or by ligands directly modulating the complex’s GDP/GTP exchange activity.
The identification of this unique activity within the trypanosomatid protein synthesis machinery has revealed a potential drug target in what was thought to be a highly conserved part of the eukaryotic cell. Furthermore, the investigation of the role of the eEF1B complex in drug resistance, thiol metabolism and translational control promises to yield important new insights into these parasites’ biochemistry.
Acknowledgements
Tim Vickers was funded by a research studentship from the Medical Research Council and Alan Fairlamb is funded by the Wellcome Trust. We thank Dr Nick Morrice and Dr Douglas Lamont for protein sequencing and Dr Angela Mehlert for mass-spectrometry.
Abbreviations
- T[SH]2 and T[S]2
trypanothione and trypanothione disulphide, respectively
- GSH and GSSG
glutathione and glutathione disulphide, respectively
- GspdSH
glutathionylspermidine
- CDNB
1-Chloro-2,4-dinitrobenzene
- T[SDNB]2
trypanothione bis-dinitrobenzene
- TCEP
tris(2- carboxyethyl)phosphine
- BS3
bis(sulfosuccinimidyl)suberate
- GST
glutathione S-transferase
- TST
trypanothione S-transferase
- eEF1A
eukaryotic elongation factor 1A (formerly eEF-1α)
- eEF1B
the eukaryotic elongation factor 1B complex (formerly eEF-1βγδ)
- eEF1Bα
was formerly eEF-1β
- eEF1Bβ
was formerly eEF-1δ
- eEF1Bγ
was formerly eEF-1γ
- ARS
amino-acyl tRNA synthetase
- PCR
polymerase chain reaction
Footnotes
Vickers, T.J. (2003) ‘Trypanothione S-transferase in the Trypanosomatidae’, PhD thesis, University of Dundee.
REFERENCES
- 1.Fairlamb AH. Trends Parasitol. 2003;19:488–494. doi: 10.1016/j.pt.2003.09.002. [DOI] [PubMed] [Google Scholar]
- 2.Croft SL, Coombs GH. Trends Parasitol. 2003;19:502–508. doi: 10.1016/j.pt.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Zilberstein D, Ephros M. Clinical and laboratory aspects of Leishmania chemotherapy in the era of drug resistance. In: Black SJ, Seed JR, editors. World Class Parasites. Vol. 4. Leishmania Kluwer Academic Press; London: 2002. [Google Scholar]
- 4.Fairlamb AH, Blackburn P, Ulrich P, Chait BT, Cerami A. Science. 1985;227:1485–1487. doi: 10.1126/science.3883489. [DOI] [PubMed] [Google Scholar]
- 5.Fairlamb AH, Cerami A. Annu.Rev.Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- 6.Flohé L, Hecht HJ, Steinert P. Free Radic.Biol.Med. 1999;27:966–984. doi: 10.1016/s0891-5849(99)00172-0. [DOI] [PubMed] [Google Scholar]
- 7.Sies H, Ketterer B, editors. Glutathione Conjugation: Mechanisms and Biological Significance. Academic Press; London: 1988. [Google Scholar]
- 8.Hurst R, Bao YP, Jemth P, Mannervik B, Williamson G. Biochem.J. 1998;332:97–100. doi: 10.1042/bj3320097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berhane K, Widersten M, Engstrom A, Kozarich JW, Mannervik B. Proc.Natl.Acad.Sci.U.S.A. 1994;91:1480–1484. doi: 10.1073/pnas.91.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hayes JD, Wolf CR. Role of glutathione transferase in drug resistance. In: Sies H, Ketterer B, editors. Glutathione Conjugation: Mechanisms and Biological Significance. Academic Press Limited; London: 1988. [Google Scholar]
- 11.Czarnecka E, Nagao RT, Key JL, Gurley WB. Mol.Cell.Biol. 1988;8:1113–1122. doi: 10.1128/mcb.8.3.1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Masai E, Katayama Y, Kubota S, Kawai S, Yamasaki M, Morohoshi N. FEBS Lett. 1993;323:135–140. doi: 10.1016/0014-5793(93)81465-c. [DOI] [PubMed] [Google Scholar]
- 13.Dulhunty A, Gage P, Curtis S, Chelvanayagam G, Board P. J.Biol.Chem. 2001;276:3319–3323. doi: 10.1074/jbc.M007874200. [DOI] [PubMed] [Google Scholar]
- 14.Jeppesen MG, Ortiz P, Shepard W, Kinzy TG, Nyborg J, Andersen GR. J.Biol.Chem. 2003;278:47190–47198. doi: 10.1074/jbc.M306630200. [DOI] [PubMed] [Google Scholar]
- 15.Koonin EV, Mushegian AR, Tatusov RL, Altschul SF, Bryant SH, Bork P, Valencia A. Protein Sci. 1994;3:2045–2054. doi: 10.1002/pro.5560031117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi S, Kidou S, Ejiri S. Biochem.Biophys.Res.Commun. 2001;288:509–514. doi: 10.1006/bbrc.2001.5799. [DOI] [PubMed] [Google Scholar]
- 17.van Damme HT, Amons R, Karssies R, Timmers CJ, Janssen GM, Moller W. Biochim.Biophys.Acta. 1990;1050:241–247. doi: 10.1016/0167-4781(90)90174-z. [DOI] [PubMed] [Google Scholar]
- 18.Janssen GM, Moller W. Eur.J.Biochem. 1988;171:119–129. doi: 10.1111/j.1432-1033.1988.tb13766.x. [DOI] [PubMed] [Google Scholar]
- 19.Yawetz A, Agosin M. Comp.Biochem.Physiol. 1981;68B:237–243. [Google Scholar]
- 20.Moutiez M, Meziani-Cherif D, Aumercier M, Sergheraert C, Tartar A. Chem.Pharm.Bull.(Tokyo) 1994:2641–2644. [Google Scholar]
- 21.Legare D, Richard D, Mukhopadhyay R, Stierhof YD, Rosen BP, Haimeur A, Papadopoulou B, Ouellette M. J.Biol.Chem. 2001;276:26301–26307. doi: 10.1074/jbc.M102351200. [DOI] [PubMed] [Google Scholar]
- 22.Grondin K, Haimeur A, Mukhopadhyay R, Rosen BP, Ouellette M. EMBO J. 1997;16:3057–3065. doi: 10.1093/emboj/16.11.3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu J, Chen H, Miller DS, Saavedra JE, Keefer LK, Johnson DR, Klaassen CD, Waalkes MP. Mol.Pharmacol. 2001;60:302–309. doi: 10.1124/mol.60.2.302. [DOI] [PubMed] [Google Scholar]
- 24.Wang HF, Lee TC. Biochem.Biophys.Res.Commun. 1993;192:1093–1099. doi: 10.1006/bbrc.1993.1529. [DOI] [PubMed] [Google Scholar]
- 25.Ariyanayagam MR, Fairlamb AH. Mol.Biochem.Parasitol. 2001;115:189–198. doi: 10.1016/s0166-6851(01)00285-7. [DOI] [PubMed] [Google Scholar]
- 26.Haimeur A, Brochu C, Genest PA, Papadopoulou B, Ouellette M. Mol.Biochem.Parasitol. 2000;108:131–135. doi: 10.1016/s0166-6851(00)00187-0. [DOI] [PubMed] [Google Scholar]
- 27.Habig WH, Jakoby WB. Methods Enzymol. 1981;77:398–405. doi: 10.1016/s0076-6879(81)77053-8. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. Anal.Biochem. 1976;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- 29.Laemmli UK. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 30.Davies GE, Stark GR. Proc.Natl.Acad.Sci.U.S.A. 1970;66:651–656. doi: 10.1073/pnas.66.3.651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashton AR, Gideon GM. Biochem.J. 1978;175:501–506. doi: 10.1042/bj1750501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Small DAP, Lowe CR, Atkinson T, Bruton CJ. Eur.J.Biochem. 1982;128:119–123. [PubMed] [Google Scholar]
- 33.Perry KL, Watkins KP, Agabian N. Proc.Natl.Acad.Sci.U.S.A. 1987;84:8190–8194. doi: 10.1073/pnas.84.23.8190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabriel A, Sisodia SS, Cleveland DW. J.Biol.Chem. 1987;262:16192–16199. [PubMed] [Google Scholar]
- 35.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: a Laboratory Manual. 2 Ed Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- 36.Warholm M, Jensson H, Tahir MK, Mannervik B. Biochemistry. 1986;25:4119–4125. doi: 10.1021/bi00362a020. [DOI] [PubMed] [Google Scholar]
- 37.Bec G, Kerjan P, Zha XD, Waller JP. J.Biol.Chem. 1989;264:21131–21137. [PubMed] [Google Scholar]
- 38.Sheu GT, Traugh JA. J.Biol.Chem. 1997;272:33290–33297. doi: 10.1074/jbc.272.52.33290. [DOI] [PubMed] [Google Scholar]
- 39.Graceffa P, Jancso A, Mabuchi K. Arch.Biochem.Biophys. 1992;297:46–51. doi: 10.1016/0003-9861(92)90639-e. [DOI] [PubMed] [Google Scholar]
- 40.Datta J, Samanta TB. Mol.Cell.Biochem. 1992;118:31–38. doi: 10.1007/BF00249692. [DOI] [PubMed] [Google Scholar]
- 41.Naval J, Calvo M, Lampreave F, Pineiro A. Comp.Biochem.Physiol.B. 1982;71:403–407. doi: 10.1016/0305-0491(82)90401-1. [DOI] [PubMed] [Google Scholar]
- 42.Adang AE, Brussee J, Vandergen A, Mulder GJ. Biochem.J. 1990;269:47–54. doi: 10.1042/bj2690047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foley V, Sheehan D. Biochem.J. 1998;333:839–845. doi: 10.1042/bj3330839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Plumas-Marty B, Taibi A, Pessoa H, Verwaerde C, Loyens M, Pommier V, Velge P, Capron A, Ouaissi A. Res.Immunol. 1993;144:553–563. doi: 10.1016/s0923-2494(05)80002-1. [DOI] [PubMed] [Google Scholar]
- 45.Kamiie K, Nomura Y, Kobayashi S, Taira H, Kobayashi K, Yamashita T, Kidou S, Ejiri S. Biosci.Biotechnol.Biochem. 2002;66:558–565. doi: 10.1271/bbb.66.558. [DOI] [PubMed] [Google Scholar]
- 46.Plumas-Marty B, Verwaerde C, Loyens M, Velge P, Taibi A, Cesbron M-F, Capron A, Ouaissi MA. Parasitol. 1992;104:87–98. doi: 10.1017/s0031182000060832. [DOI] [PubMed] [Google Scholar]
- 47.Billaut-Mulot O, Pommier V, Schoneck R, Plumas-Marty B, Taibi A, Loyens M, Capron A, Ouaissi MA. Nucleic Acids Res. 1993;21:3901. doi: 10.1093/nar/21.16.3901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plumas-Marty B, Schoneck R, Billaut-Mulot O, Taibi A, Capron A, Ouaissi MA. Parasitol.Res. 1994;80:626–628. doi: 10.1007/BF00933014. [DOI] [PubMed] [Google Scholar]
- 49.Billaut-Mulot O, Fernandez-Gomez R, Ouaissi A. Gene. 1997;198:259–267. doi: 10.1016/s0378-1119(97)00323-5. [DOI] [PubMed] [Google Scholar]
- 50.Légaré D, Papadopoulou B, Roy G, Mukhopadhyay R, Haimeur A, Dey S, Grondin K, Brochu C, Rosen BP, Ouellette M. Exp.Parasitol. 1997;87:275–282. doi: 10.1006/expr.1997.4222. [DOI] [PubMed] [Google Scholar]
- 51.Callahan HL, Beverley SM. J.Biol.Chem. 1991;266:18427–18430. [PubMed] [Google Scholar]
- 52.Fahey RC, Sundquist AR. Adv.Enzymol.Relat.Areas Mol.Biol. 1991;64:1–53. doi: 10.1002/9780470123102.ch1. [DOI] [PubMed] [Google Scholar]
- 53.Moutiez M, Aumercier M, Parmentier B, Tartar A, Sergheraert C. Biochim.Biophys.Acta. 1995;1245:161–166. doi: 10.1016/0304-4165(95)00085-p. [DOI] [PubMed] [Google Scholar]
- 54.Mannervik B, Danielson UH. C.R.C. Crit.Rev.Biochem. 1988;23:283–337. doi: 10.3109/10409238809088226. [DOI] [PubMed] [Google Scholar]
- 55.Board PG, Coggan M, Chelvanayagam G, Easteal S, Jermiin LS, Schulte GK, Danley DE, Hoth LR, Griffor MC, Kamath AV, Rosner MH, Chrunyk BA, Perregaux DE, Gabel CA, Geoghegan KF, Pandit J. J.Biol.Chem. 2000;275:24798–24806. doi: 10.1074/jbc.M001706200. [DOI] [PubMed] [Google Scholar]
- 56.Kosower NS, Vanderhoff GA, Benerofe B, Hunt T, Kosower EM. Biochem.Biophys.Res.Commun. 1971;45:816–821. doi: 10.1016/0006-291x(71)90490-6. [DOI] [PubMed] [Google Scholar]
- 57.Ayala A, Parrado J, Bougria M, Machado A. J.Biol.Chem. 1996;271:23105–23110. doi: 10.1074/jbc.271.38.23105. [DOI] [PubMed] [Google Scholar]
- 58.Clayton CE. EMBO J. 2002;21:1881–1888. doi: 10.1093/emboj/21.8.1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Black S. Biochem.Biophys.Res.Commun. 1993;191:95–102. doi: 10.1006/bbrc.1993.1189. [DOI] [PubMed] [Google Scholar]
- 60.Black S. Science. 1986;234:1111–1114. doi: 10.1126/science.3535073. [DOI] [PubMed] [Google Scholar]
- 61.Black S. J.Biol.Chem. 1983;258:2112–2114. [PubMed] [Google Scholar]
- 62.Negrutskii BS, Shalak VF, Kerjan P, El’skaya AV, Mirande M. J.Biol.Chem. 1999;274:4545–4550. doi: 10.1074/jbc.274.8.4545. [DOI] [PubMed] [Google Scholar]
- 63.Yang DCH. Curr.Top.Cell.Regul. 1996;34:101–136. doi: 10.1016/s0070-2137(96)80004-5. [DOI] [PubMed] [Google Scholar]
- 64.Sang LJ, Gyu PS, Park H, Seol W, Lee S, Kim S. Biochem.Biophys.Res.Commun. 2002;291:158–164. doi: 10.1006/bbrc.2002.6398. [DOI] [PubMed] [Google Scholar]