

Abstract
Phomopsichalasin was isolated and assigned structure 1 over 15 years ago. Analysis of its proton NMR data led us to hypothesize that not all aspects of the relative configuration of this structure were correct. We have used both empirical and computational methods to propose an alternative structure. Diaporthichalasin was reported several years ago, and its structure was assigned as 7, a diastereomer of structure 1, and confirmed by a single crystal X-ray study. We have shown that diaporthichalasin and phomopsichalasin are identical; i.e., both have structure 7. Additional aspects of NMR interpretation that provide guidance for avoiding some of the pitfalls that can lead to incorrect structure assignments are discussed. These recommendations/reminders include i) the use of complementary solvents for acquiring NMR data that break accidental chemical shift degeneracy, ii) the importance of assigning coupling constants as extensively as possible, and iii) exercising caution when interpreting correlations in 2D spectra where overlapping resonances are involved.
In 1995 Horn and co-workers reported the isolation of a new cytochalasin-like secondary metabolite from an endophytic fungus Phomopsis sp., which they named phomopsichalasin.i The compound exhibited antibacterial activity in several disk diffusion assays. The researchers assigned structure 1 (Scheme 1) on the basis of analysis of a battery of 1D and 2D NMR data. Phomopsichalasin represents the first compound wherein the macrocyclic portion of the cytochalasin skeleton has been cyclized to an embedded A/B/C tricyclic subunit. Diaporthichalasin (7) was reported in 2007 and was deduced to have a structure diastereomeric with that of 1.ii
Scheme 1.

Biosynthetic consideration of the possible origin of structures 1 and 7, assigned to phomopsichalsin and diaporthichalasin, respectively, point to the common intermediate hexaene 4. For comparison purposes, the absolute configuration of C-18 is held constant in all structures. The absolute configuration of natural phomopsichalasin/diaporthichalasin (7) is unknown.
RESULTS AND DISCUSSION
The structure originally deduced for phomopsichalasin, 1 (Scheme 1),i is intriguing because of the possibility that it might arise via a biosynthetic pathway involving a pair of (sequential) net [4+2] intramolecular Diels-Alder (IMDA) reactions commencing from the polyene precursor 4. The double-diene/double-dienophile 4 is a reasonable biosynthetic intermediate because it could arise via polyketide synthase (PKS) and non-ribosomal peptide synthase (NRPS) pathways. In principle, either of two IMDAs within 4, leading either to octalin 2 or macrocycle 3, could be the first to occur en route from 4 to 1. We have speculated that these IMDA reactions might occur in the absence of a specific enzyme catalyst—that is, might one or both cycloadditions be sufficiently and inherently fast to occur spontaneously? If so, then the relative configuration of the nine non-carbinolamide stereocenters in 1 would be established by the relative asymmetric induction originating from the C-18 methyl-bearing stereocenter in 4. However, it would be odd if 1 were the product of a substrate-induced IMDA reaction because the relative configuration between C-18 and C-16 in 1 would be unexpected. That is, the diaxial relationship between the methyl groups attached to those stereocenters is destabilizing, and the IMDA reaction leading from 4 to 2 (or 3 to 1) would be expected to have a higher activation barrier because of the synpentane interaction partially established in the transition state geometry.
This dilemma prompted us to reexamine the data upon which the structural assignment of 1 was based. In particular, the 1H NMR chemical shifts (methanol-d4) of two protons first attracted our attention. Namely, the resonances for the axial protons at C-17 and C-19 in phomopsichalasin appear at 0.67 and 0.56 ppm, respectively. For the reasons discussed next, we surmised that these values were untenable for structure 1.
Grant (empirical) NMR Analysis
Empirical interpretation of chemical shifts has been a mainstay of NMR analysis since the infancy of NMR spectroscopy. A methyl substituent on a cyclohexane ring is known to induce significant chemical shift perturbations on various other protons on that ring. The magnitude of some of these through-bond anisotropies is surprisingly large (Figure 1A). These shift perturbations emerged from an insightful analysis of the (natural abundance deuterium) chemical shifts of numerous methyl-substituted cyclohexanes done in the Grant laboratories.iii Hence, we refer to these as the “Grant numbers” and find them to be quite broadly applicable to the interpretation of chemical shift data in the context of a variety of structural settings. Quite relevant to the case of phomopsichalasin, the effect of an axially vs. equatorially oriented methyl group on the axial proton at C-2 (see gray-highlighted data in each of structures 8eq vs. 8ax) is particularly striking—H-2ax is perturbed by over 0.5 ppm simply depending upon the dihedral relationship (anti vs. gauche) of its H—C-2 bond relative to the vicinal C-1—Me bond! That is, when the methyl group is axial, the dihedral angle to H-2ax is ca. 180° and H-2ax is relatively deshielded, whereas when the methyl is equatorial, the dihedral is ca. 60° and H-2ax is shielded.
Figure 1.

Panel A. “Grant numbers:” The incremental chemical shift perturbations imposed by a methyl substituent in the equatorial (cf. 8eq) vs. the axial (cf. 8ax) position of a chair-like cyclohexane.iii Positive values refer to downfield (and negative upfield) changes in the chemical shift of the axial or equatorial protons of cyclohexane itself. Panel B. trans-Fused octalin subunit of structure 1 (9a) vs. its equatorial 18-Me counterpart (9b).
By applying the logic of a Grant analysis to structure 1, we observed that the high field nature of the chemical shift values for H-17ax and H-19ax are inconsistent with the axial orientation of the 18-methyl substituent (cf. substructure 9a, Figure 1B), which represents the geometry of the dominant conformer expected for the trans-fused AB bicyclic octalin skeleton of 1. Instead, these shift values are much more compatible with an equatorial orientation for the 18-methyl group (cf. 9b). The diastereomeric relationship in 9b is a more reasonable stereochemical outcome for an IMDA reaction (forming the bold bonds in the cyclohexenyl B-ring in 9b) proceeding under control of relative internal asymmetric induction.
We then noticed the report from Pornpakakul and coworkers in 2007 describing the isolation (from the endophytic fungus Diaporthe sp. Bkk3) and structure determination of diaporthichalasin.ii Their thorough NMR analysis efforts led to the assignment of structure 7 (Scheme 1), which was confirmed by a single crystal x-ray structure determination, placing the assignment as 7 on secure ground. Interestingly, structure 7 shares the constitution of 1, but differs in the relative configuration of several stereocenters. The differences between structures 7 and 1 bear directly on the question of possible spontaneity of the IMDA events. That is, 7 could arise from 5a or 6 (cf. Scheme 1), either of which could be formed from 4. Of course, this presents a quandary, because 4 is the same polyene precursor that we earlier identified by retrobiosynthetic analysis of structure 1. Obviously, the uncatalyzed reaction of 4 would follow the same pathway(s) regardless of what organism may have produced that biosynthetic intermediate.
This dichotomy could be construed as evidence that the cyclization chemistry of 4 is not spontaneous but, rather, is promoted by (Diels-Alderase-like) enzymes unique to each organism. We, however and in light of this information, returned to the question of the structure assigned to phomopsichalasin—i.e., 1. In contrast to 1, diaporthichalasin (7) contains: i) an equatorial methyl substituent at C-18; ii) relative configuration of the five stereocenters within its AB bicyclic octalin subunit that is entirely consistent with the geometry expected to arise from a concerted IMDA reaction (cf. 9b in Figure 1 and 4 to 5a in Scheme 1); and iii) four stereocenters within its D-ring that would be anticipated from an IMDA cycloaddition in which the ketocarbonyl had been oriented endo to the C-5–C-8 diene (cf. 5a to 7 or 4 to 6, Scheme 1) rather than the lactam carbonyl (cf. 2 to 1 or 4 to 3, Scheme 1).
Computational (DFT) Analyses
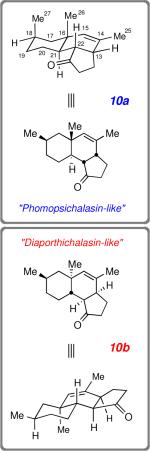
To further examine the hypothesis that structure 1 is an incorrect formulation for phomopsichalasin within (at least) the fused AB bicyclic subunit, we undertook a computational analysis. To make this study more tractable, we elected to examine two truncated structures that retained the ABC tricyclic substructure unit but lacked the additional complexity associated with the D- and E-rings and their substituents. Specifically, we computed 1H chemical shifts for the truncated substructures 10a and 10b (Table 1), which correspond to the ABC tricyclic skeleton common to both structures 1 and 7. They are tetraepimeric at carbons 16, 21, 22, and 13; that is, truncated structure 10a has the same relative configuration within its A-ring as 1 (i.e., “phomopsichalasin-like”), and 10b as 7 (“diaporthichalasin-like”). In each structure we maintained i) a trans orientation between H-21/H-22 in view of the ~12.8 Hz coupling constant between these two protons and ii) a cis-ring fusion between the hydrindenone substructure (i.e., across C-13/C-22) based on the observed NOE and ~8.1 Hz coupling constant between protons H-13 and H-22 in the spectrum of phomopsichalasin. Each of structures 10a and 10b was subjected to an initial molecular mechanics conformational search, resulting in an easily manageable number of conformational minima (two for 10a and one for 10b). DFT geometry optimization with the M062X functionaliv [6–31+G(d,p) basis set] was performed on each of these conformers. Finally, chemical shift calculations were carried out using our recently developed WP04 functional,v the polarizable continuum model (PCM) for MeOH solvation, and the pcS-2 basis set.vi
Table 1.
Comparison of the Computed Proton NMR Chemical Shifts (Δdft) for Structures 10a and 10b with the Experimentally Observed Shifts (δexp) for the Analogous Subset of Nuclei in the Spectrum Reported for Phomopsichalasin (“1”) in Methanol-d4.
| δDFTa(MeOH) | δ EXP | |Δδ| for δDFTa vs. δEXP | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Atom # | 10a | 10b | “1”b | 10a vs. “1”b | 10b vs. “1”b | |
|
|
||||||
| 13 | 2.68 | 2.70 | 2.85 | 0.17 | 0.15 |
|
| 15 | 5.49 | 5.42 | 5.36 | 0.13 | 0.06 | |
| 17eq | 1.41 | 1.40 | 1.45 | 0.04 | 0.05 | |
| 17ax | 1.42 | 0.92 | 0.67 | 0.75 | 0.25 | |
| 18 | 1.78 | 1.65 | 1.62 | 0.16 | 0.03 | |
| 19eq | 1.38 | 1.58 | 1.64 | 0.26 | 0.06 | |
| 19ax | 1.36 | 0.81 | 0.56 | 0.80 | 0.25 | |
| 20eq | 1.45 | 1.55 | 1.50 | 0.05 | 0.05 | |
| 20ax | 1.45 | 1.32 | 1.10 | 0.35 | 0.22 | |
| 21 | 1.55 | 1.49 | 1.42 | 0.13 | 0.07 | |
| 22 | 2.34 | 2.34 | 2.24 | 0.10 | 0.10 | |
| 25 | 1.73 | 1.75 | 1.88 | 0.15 | 0.13 | |
| 26 | 0.99 | 0.90 | 0.80 | 0.19 | 0.10 | |
| 27 | 1.05 | 0.81 | 0.78 | 0.27 | 0.03 | |
|
|
||||||
| MAEFULLc= | 0.25 | 0.11 | ||||
| MAELITEd= | 0.35 | 0.11 | ||||
|
|
||||||
| DP4 probabilitye = | <0.5% | >99.5% | ||||
Values for δDFT were determined by referencing the computed isotropic chemical shifts (σCOMP) for all nuclei to the isotropic chemical shift for the protons of TMS (σREF), both of which were computed at the IEFPCM-WP04/pcS-2//M06-2X/6-31+G(d,p) level of theory.
“1” = “the spectroscopic data reported for phomopsichalasin.”
Mean absolute error (|ΔδAVE|). Calculation includes all proton nuclei that are listed in the table.
Including only the subset of proton nuclei 17–19, 26, and 27.
Scaled δDFT values derived from chemical shifts computed at the B3LYP/6-31G(d,p)//MMFF level of theory were employed (see ref. viii and the Supporting Information for details).
The computed (and Boltzmann weighted) proton chemical shifts (δDFT) for 10a and 10b are listed in Table 1, as are the experimental shiftsi reported for the analogous protons in the 1H NMR spectrum of phomopsichalasin (methanol-d4, δEXP). The differences between the computed and experimental shift values (|Δδ|) are listed in the rightmost pair of columns. The bottom-line is presented as the mean absolute error (MAE) value for each of the comparisons of 10a vs. “1” vis-à-vis 10b vs. “1” (we use “1” here as a shorthand notation for “the spectroscopic data reported for phomopsichalasin”). Using the full set of all 14 numbered protons (MAEFULL, cf. the structure of 10a), substructure 10b clearly shows a substantially better match than 10a between the calculated and experimental data sets (0.11 vs. 0.25, respectively). If instead only the subset of protons that reside nearest to the point of difference in the relative configuration between 10b and 10a (i.e., protons on C-17–C-19, Me-26, and Me-27) are compared, then the “MAELITE” distinction is even larger (0.11 vs. 0.35).
Goodman (DP4) Analysis
The MAE parameter that was employed in the above analysis is a straightforward and common method for judging the relative “goodness of fit” between computed and experimental NMR data sets.vii Smith and Goodman recently introduced a more sophisticated, statistical analysis–namely, the DP4 probability,viii which was developed for the specific instance where a single experimental NMR spectrum could be assigned to one of many possible diastereomeric candidate structures. Given the relative frequency with which this dilemma is encountered in natural product structure elucidation, it is perhaps no surprise that the DP4 probability has already found use in this arena.ix The DP4 protocolviii specifies use of the MMFF force field to optimize conformer geometries, which are then subjected to single-point DFT NMR calculations (with the B3LYP functional). The Boltzmann weighted chemical shifts thus generated are then used to calculate the DP4 probability for each candidate structure. This final step is aided by use of an online applet.x This analysis was applied to each of 10a and 10b vs. δEXP. The DP4 probabilities also indicate that the truncated structure 10b (i.e., the “diaporthichalasin-like” stereoisomer) is a much better fit than 10a with the spectroscopic data reported for phomopsichalasin with a >99.5% confidence interval (Table 1).
The above analyses were based on NMR data recorded in MeOH. We felt we would gain additional confidence in our conclusions if we could analyze the experimental NMR data for diaporthichalasin (7) in the more common solvent CDCl3, with which we have considerably more experience in comparing computed with experimental proton chemical shift data sets. Professor Pornpakakul kindly provided to us a generous sample of diaporthichalasin (7). We recorded and interpreted a set of 1D and 2D NMR spectra in CDCl3 (see Table S1).
We repeated the DFT computational analysis for 10a and 10b using PCM CHCl3 solvation. Comparison of these computed δDFT values with our CDCl3 data across the same subset of protons as before (cf. Table 1) resulted in MAEFULL values of 0.25 vs. 0.10, MAELITE values of 0.34 vs. 0.09, and DP4 probabilities of <0.5% and >99.5% (Table S6). Again, the matches with the computed values for the “diaporthichalasin-like” substructure 10b are far better. In addition, the magnitudes of the MAE values for the MeOH vs. the CHCl3 analyses are strikingly similar, not only lending confidence that this methodology is applicable to a polar protic solvent, but also further (and strongly) implicating that the phomopsichalasin sample from the 1995 work has structure 7.
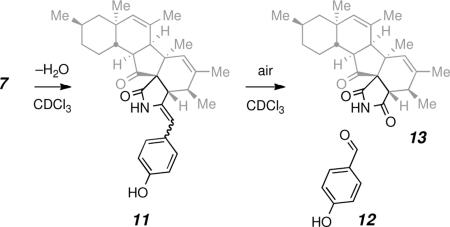
A final set of experimental data is most compelling. The published NMR data on which the analyses were performed that led to the assignments of 1 and 7 were recorded in two different solvents (methanol-d4 and DMSO-d6, respectively). We therefore also recorded spectra for the diaporthicahalasin (7) sample in each of these two solvents (Tables S2 and S3). As we now suspected would be the case, the data for 7 in methanol-d4 were essentially identical to those reported for the sample named phomopsichalasin (and incorrectly assigned structure 1) in 1995. Thus, it is clear that phomopsichalasin and diaporthichalasin are the same compound, both having structure 7. One important ramification is that the hypothesis of a spontaneous cyclization of 4 to give phomopsichalasin/diaporthichalasin (7) is still viable. Accordingly, we are continuing our efforts to synthesize polyene 4 in order to study its inherent IMDA reactivity.xi
Insights from the Initial Analysis of NMR Data of Phomopsichalasin
It is instructive to consider the analysis and logic that led to the incorrect assignment of the relative configuration of phomopsichalasin as 1.i The 1H NMR spectrum for phomopsichalasin was reported in methanol-d4 (and MeCN-d3 in order to obtain a few additional J values). Three methylene or methine proton resonances (H-19eq, H-20eq, H-18) were obscured by allylic methyl group resonances. In retrospect, dispersion of these resonances, in particular that of H-18, would have been helpful since H-18 is the methine proton at the stereogenic center whose relative configuration we now know to be incorrect. To reveal those overlapped resonances and extract essential coupling constants, we performed complementary experiments by i) recording the spectrum of 7 in methanol-d4 at a higher field strength (850 MHz) or ii) performing an NMR titration with benzene-d6 (into methanol-d4). In the former, we could decipher all of the J-values of H-18 (ddddq; J = 4.1, 4,1 12.3, 12.3, and 6.5 Hz; Figure S2). This full coupling constant analysis was integral to “walking” the coupled protons around the A-ring of the trans-fused decalin system and to assigning the orientation of H-18 to be axial rather than equatorial (e.g., the H-18 resonance includes two trans-diaxial vicinal coupling constants).
Another uncertainty was raised by the peculiar report of a doublet (δ = 0.78) with coupling constant of 2.2 Hz for Me-27. This incorrect assignment resulted from overlapped resonances for Me-11, Me-26 and Me-27 at ca. 0.8 ppm. This was clarified as well by a benzene-d6 titration (Figure 2, Panel A), which cleanly resolved all three methyl groups and allowed us to observe the coupling constant for the doublet for Me-27 (J = 6.5 Hz), which was consistent with the J value in the multiplet assigned to H-18. However, in retrospect, even simpler solutions to this problem were available: i) by straightforward line-broadening (resolution enhancement) analysis of the 1H NMR data for the three methyl resonances partially overlapped at ca. 0.8 ppm in methanol-d4 (d for Me-11, s for Me-26, and d for Me-27; Figure 2, Panel B), we could deduce directly the correct coupling values for each of the doublets; ii) upon recording the spectrum of 7 in CDCl3 (Table S1) we observed that the resonance for Me-11 (assignment confirmed by COSY) was well resolved from others, allowing the 6.5 Hz J value to be easily observed.
Figure 2.

Insets of methyl regions of 1H NMR spectra of 7 in various solvents. Panel A: in methanol-d4 containing varying percentages of added benzene-d6 in order verify the presence of two methyl doublets (with J values of 7.3 and 6.5 Hz). Panel B: in methanol-d4 with increasing levels of “resolution enhancement,” which confirmed these J values.
Finally, an unusual NOE was reported between Me-27 and H-4 in the methanol-d4 spectrum of phomopsichalasin.i Spatial proximity between these two protons appears to be inconsistent with either structure 1 or structure 7. Again, this misinterpretation of the (NOESY) NMR data can be attributed to the three overlapping methyl resonances at ca. 0.8 ppm (cf. Figure 2), which introduces uncertainty in the interpretation of the NOESY correlation peak. In fact, an NOE between Me-11 and H-4 was reported in the Pornpakakul studies,ii and that interaction is clearly consistent with 7 (and highly unlikely for structure 1).
Conclusion
Detailed NMR spectroscopic analysis and pertinent computational models were used i) to show that the structure originally proposed for phomopsichalasin (i.e., 1)i was incorrect and ii) to generate a hypothesis for an alternative structure assignment. Specifcially, we deduced that the structure of phomopsichalasin is, in fact, identical to that of the more recently reported diaporthichalasin (7).ii To validate our analyses, we then showed that the proton and carbon NMR spectra of an authentic sample of diaporthichalasin recorded in methanol-d4 were in excellent agreement with the NMR data reported for phomopsichalasin.i
Supplementary Material
ACKNOWLEDGMENTS
We thank Professor Surachai Pornpakakul for generously providing a sample of diaporthichalasin (7) and Todd Rappe and Dr. Letitia Yao for assistance and advice in collecting NMR spectroscopic data. This work was carried out in part using software and hardware resources made available through the University of Minnesota Supercomputing Institute (MSI). The research was supported by grants awarded by the DHHS (National Institute of General Medical Sciences, GM-65597) and the National Science Foundation (CHE-0911696).
Footnotes
Supporting information: Tables (S1–S4) of NMR data (chemical shift assignments; 1H/1H coupling constants; and COSY, HMQC, and HMBC correlations) for diaporthichalasin (7) in CDCl3, methanol-d4, and d6-DMSO. Computational results (including Tables S5–S6 of computed isotropic chemical shifts and computed vs. experimental chemical shifts in CDCl3). Figures (S1 and S2) of HMBC correlations of 7 and of the C-18 multiplet analysis.
REFERENCES
- (i).Horn WS, Simmonds MSJ, Schwartz RE, Blaney WM. Tetrahedron. 1995;51:3969–3978. [Google Scholar]
- (ii).Pornpakakul S, Roengsumran S, Deechangvipart S, Petsom A, Muangsin N, Ngamrojnavanich N, Sriubolmas N, Chaichit N, Ohta T. Tetrahedron Lett. 2007;48:651–655. [Google Scholar]
- (iii).Dalling DK, Curtis J, Grant DM. J. Org. Chem. 1986;51:136–142. [Google Scholar]
- (iv).(a) Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]; (b) Zhao Y, Truhlar DG. Acc. Chem. Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- (v).(a) Wiitala KW, Hoye TR, Cramer CJ. J. Chem. Theory Comput. 2006;2:1085–1092. doi: 10.1021/ct6001016. [DOI] [PubMed] [Google Scholar]; (b) Jain R, Bally T, Rablen PR. J. Org. Chem. 2009;74 doi: 10.1021/jo900482q. [DOI] [PubMed] [Google Scholar]
- (vi).Jensen F. J. Chem. Theory Comput. 2008;4:719–727. doi: 10.1021/ct800013z. [DOI] [PubMed] [Google Scholar]
- (vii).For examples, see: Braddock DC, Rzepa HS. J. Nat. Prod. 2008;71:728–730. doi: 10.1021/np0705918. Timmons C, Wipf P. J. Org. Chem. 2008;73:9168–9170. doi: 10.1021/jo801735e.
- (viii).Smith SG, Goodman JM. J. Am. Chem. Soc. 2010;132:12946–12959. doi: 10.1021/ja105035r. [DOI] [PubMed] [Google Scholar]
- (ix).For recent use of DP4 probability in the context of natural product structure elucidation, see: Paterson I, Dalby SM, Roberts JC, Naylor GJ, Guzmán EA, Isbrucker R, Pitts TP, Linley P, Divlianska D, Reed JK, Wright AE. Angew. Chem. Int. Ed. 2011;50:3219–3223. doi: 10.1002/anie.201007719. Lodewyk MW, Tantillo DJ. J. Nat. Prod. 2011;74:1339–1343. doi: 10.1021/np2000446. Wyche TP, Hou Y, Braun D, Cohen HC, Xiong MP, Bugni TS. J. Org. Chem. 2011;76:6542–6547. doi: 10.1021/jo200661n.
- (x). http://www-jmg.ch.cam.ac.uk/tools/nmr/DP4/
-
(xi).We have also seen evidence for dehydrative and subsequent oxidative degradation events in samples of 7 upon prolonged storage in a solution of CDCl3. More specifically, 1H NMR resonances for the aromatic and benzylic protons in 7 diminished, and an intermediate species we suggest to be the enamide 11 was observed by LCMS and 1H NMR spectroscopy (CDCl3, new doublets at δ 7.06 and 6.81 for aromatic resonances and new singlets at δ 5.57, 5.42, and 5.35, which are consistent with H-10, H-15, and H-7 in 11, respectively). Over additional time, resonances for the oxidative cleavage product, 4-hydroxybenzaldehyde (12), were observed (and confirmed by a doping experiment), and LCMS evidence for a new species having the mass of succinimide 13 was seen. Efficient aerobic cleavage of related enamides has been previously observed. For example, Banfi L, Andrea B, Francesco C, Giuseppe G, Naz F, Riva R, Zitoa P. Synlett. 2010:85–88.
Gunawan S, Nichol GS, Chappeta S, Dietrich J, Hulme C. Tetrahedron Lett. 2010;51:4689–4692. doi: 10.1016/j.tetlet.2010.06.131.

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.