

Abstract
Understanding and treatment of spinal cord pathology is limited in part by a lack of longitudinal in vivo imaging strategies at the cellular level. We developed a chronically implanted spinal chamber and surgical procedure suitable for time-lapse in vivo multiphoton microscopy of mouse spinal cord without the need for repeat surgical procedures. Repeated imaging was routinely achieved for more than five weeks post-operatively with up to ten separate imaging sessions. We observed neither motor function deficit nor neuropathology in the spinal cord as a result of chamber implantation. Using this chamber we quantified microglia and afferent axon dynamics following a laser-induced spinal cord lesion and observed massive microglia infiltration within one day along with a heterogeneous dieback of axon stumps. By enabling chronic imaging studies over timescales ranging from minutes to months, our method offers an ideal platform for understanding cellular dynamics in response to injury and therapeutic interventions.
Introduction
In vivo optical imaging in the live rodent cortex has been achieved at depths of more than 1 mm with sub-micron resolution using nonlinear microscopy coupled with cranial window preparations1. This imaging approach has particular value for the study of neuropathology. For example, longitudinal studies in murine models have used 2-photon excited fluorescence (2PEF) microscopy to study the appearance and evolution of amyloid-β (Aβ) plaques in Alzheimer’s disease2-4, the rate and extent of Aβ clearance in response to antibody therapy5, the diapedesis of circulating cancer cells and formation of metastatic tumors6, as well as the long-term effects of occlusion and reperfusion of cerebral blood vessels7. In all cases, a surgical protocol enabling repeated optical access to the relevant tissue is prerequisite to longitudinal studies. Existing protocols include an open-skull protocol8 for a cranial window in which a small portion of the mouse skull is replaced with glass, thinned skull preparations9, and recently, a glass-reinforced, thinned-skull preparation10.
Chronic optical imaging could offer comparable experimental advantages to the study of spinal cord, including studies of spinal cord injury (SCI), spinal tumors, syringeomyelia, myelitis and spinal cord ischemia. For example, time-lapse in vivo studies of axon trajectories following SCI would enable unambiguous identification of spared, injured, and regenerating axons and provide an ideal platform for evaluating therapies aimed at spurring axon growth. This approach has been demonstrated in vivo in zebrafish11, where injured axons as well as axons that regenerated in response to therapy were imaged over several days after SCI. Others have performed time-lapsed fluorescence imaging in the spinal cord of mice expressing green fluorescent protein (GFP) in a subset of dorsal root ganglia (DRG) neurons for up to 72 hours after SCI12. However, this imaging requires artificial ventilation and the suppression of breathing during image acquisition, limiting the timescale of cellular dynamics accessible. Similarly, 2PEF imaging in the spinal cord of mice expressing GFP in microglia has been demonstrated13 with up to two imaging sessions in the same animal separated by several days. Heavy sedation, whole-body suspension, and local clamping are used to reduce remaining motion artifacts due to breathing and heartbeat. Similar techniques have been used to study microglia dynamics following a laser-induced micro-injury14, calcium signaling of dorsal horn neurons15, the effects of peripheral nerve lesioning on sprouting of severed DRG axons16, and experimental autoimmune encephalomyelitis17. Finally, imaging time points as long as four months post-SCI with as many as six imaging sessions in one subject has been achieved without interfering with animal breathing during imaging18.
In all of these studies, obtaining images on days following the initial surgery is possible only by repeated surgical opening of the animal for each imaging session. Repeat surgeries increase the risk of infection, add a potentially confounding source of inflammation, cause tissue damage, cause additional pain and distress to the animal, and severely limit the number of imaging time points possible. However, unlike the cranium, the relevant portions of the spine undergo routine flexion and sit in close proximity to the heart and lungs, requiring any chronic preparation to maintain the mechanical stability of the spine and minimize motion artifact during imaging.
In an effort to provide a surgical preparation that enables repeated optical imaging of the mouse spinal cord without requiring repeated surgeries, we developed an implantable spinal chamber that provides continuous optical access to the mouse spinal cord. Using this chamber, we performed 2PEF imaging in transgenic mice expressing fluorescent proteins in microglia and afferent axons, and found axon morphology to be stable over eight weeks of imaging. Standard tests of locomotor function did not show deficits in mice with the implant as compared to control animals. Post-mortem histology did not show evidence of damage to the spinal cord from the surgery, but did show increases in microglial numbers in the dorsal portion of the spinal cord, consistent with mild inflammation. Finally, we imaged blood vessels, afferent axons, and microglia following laser-induced microlesioning of the spinal cord using 2PEF microscopy and quantified microglial infiltration and axon dieback dynamics on time scales ranging over four orders of magnitude. We found that the increase in microglial density at the lesion site was fastest within the first 24 hours after injury. Axon dieback rates were highly heterogeneous among axons, were fastest immediately after injury, and slowed over five weeks after SCI.
Results
The imaging chamber
The chamber consisted of two metal bars that attach to the spine together with a top plate that attaches to the bars and holds a glass window (Fig. 1a). The implantation procedure entailed retracting muscles and scraping clean the dorsal laminae over the imaging site, then fusing three vertebrae by clamping them on either side with small metal bars (Fig. 1b) held magnetically on fixed posts, and finally performing a dorsal laminectomy with vanna scissors. We trimmed the lateral edges of the bone as close to the bars as possible, and sealed the bone using a combination of cyanoacrylate and dental acrylic. We maintained clamping pressure as the chamber assembly was completed by bolting a top plate onto the metal bars, applying a silicone elastomer over the spinal cord, sealing the chamber with a glass coverslip on top and with additional glue around the edges, and finally gluing the skin to the base of the top plate (Supplementary Figure 1; see Online Methods and Supplementary Protocol for details). During 2PEF imaging, motion artifacts were reduced by securing the mouse to tapped holding posts twisted onto the set screws in the exposed wings of the top plate thereby stabilizing the spine and elevating the mouse off of the surgical table (Supplementary Figure 2) to permit free expansion of the chest cavity during inspiration. Mechanical drawings for custom parts are provided as Supplementary Notes 1-6. The chamber enabled continuous optical access to the spinal cord (Fig. 1c) without repeat surgeries. Animals behaved normally over the weeks after surgery, with no signs of lordosis or kyphosis (Fig. 1d). Micro-CT imaging of an animal six days after implantation confirmed alignment of the spine and showed no signs of vertebral damage (Supplementary Video 1).
Figure 1. An imaging chamber for longitudinal optical access to mouse spinal cord without the need for repeated surgeries.
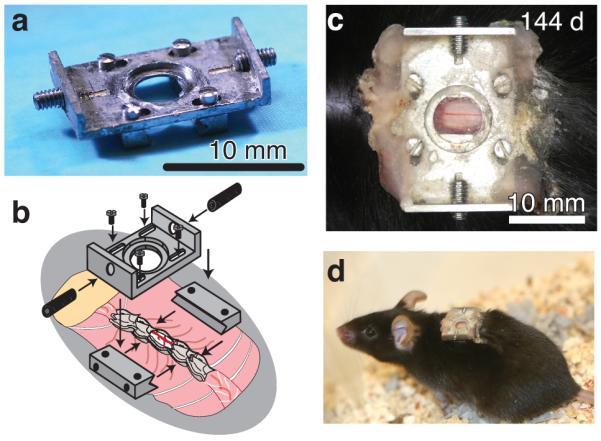
(a) Photograph of the imaging chamber. Scale bar, 10 mm. (b) Schema showing the implantation of the imaging chamber in mice at the T11-T12 vertebra, just below the dorsal fat pad (taupe). (c), Photograph showing the spinal cord imaged through the implanted chamber 144 days after the surgery. Scale bar, 10 mm (d) Photograph of a mouse with an implanted chamber (same mouse from panel (c)).
Long-term imaging of spinal cord axons and blood vessels
We implanted our chamber in transgenic mice expressing yellow fluorescent protein (YFP) in afferent axons from a subset of DRG neurons. We imaged mice at up to eight separate time points out to as long as 67 days post-operatively, revealing that axon morphologies and numbers were stable (Fig. 2a). We used blood vessels labeled with Texas Red dextran and obvious features of the axons to navigate to the same region of interest across different imaging sessions. At some time points, blood vessels showed signs of increased or decreased diameter compared to baseline, but with stable morphology. Image contrast visibly diminished over time (Fig. 2b), which we quantified by fitting projections of axon segments to a simple model (Fig. 2c; see Online Methods). We found a decrease in image contrast (Fig. 2d) and a loss of lateral resolution (Fig. 2e) over time, with the greatest change occurring over the first 14 days due to the formation of a fibrous growth over the surface of the spinal cord. The duration over which we could resolve individual axons expressing fluorescent proteins using 2PEF imaging varied between as few as five and as many as 140 days (Supplementary Figure 3). We found that in animals where only minimal fibrosis was present at one to two weeks after surgery, axons could routinely be resolved out to more than five weeks. We achieved long time point imaging (> five weeks) in approximately 50% of the implanted animals.
Figure 2. Longitudinal 2PEF imaging of axons and blood vessels over many weeks post-surgery.

(a) Projections of 2PEF image stacks of afferent axons expressing YFP (teal) and blood vessels labeled with intravenously-injected Texas Red dextran (red) taken over nine weeks after chamber implantation. The asterisks indicate the location of red autofluorescence from invading, likely inflammatory, cells located above the spinal cord at later time points. Arrows denote landmark features of the axons that were visible at all time points. Scale bar, 100 μm (b) High-resolution 2PEF imaging of YPF-expressing axons from the same region imaged in a. Scale bar, 50 μm (c) Fit of a profile (Equation (S3)) across maximum intensity projections (c, inset) of selected axon segments (b, red box). (d) Image contrast and (e) lateral spatial resolution as functions of time after surgery from the fits for all axon segments for two mice (separate curves for each animal, ~10 axons measured at each time point for each animal). Error bars represent the standard deviation.
At all time points, wherever fibrous tissue growth was minimal, imaging depth was limited to 30-50 μm by the short scattering length of myelin19. In cases where fibrous growth was dense, we could not resolve axons at any depth. In some animals, a myelin-poor region was observed between adjacent dorsal roots that enabled imaging up to depths of 300 μm, well into the dorsal horn (Supplementary Figure 4).
Locomotor function is preserved after implantation
We used two behavioral assays to assess the impact of the implant on motor function. We first tested mice for gait abnormalities during normal movement20. Mice with inked paws ran the length of a narrow enclosure, after which the patterns of their footprints were analyzed to measure base of support (lateral distance between hindlimb placement), stride length, and running speed for mice with spinal chamber implants as well as sham controls (n = 3 mice per group). We found no significant differences in gait attributable to the implant at any time point (Supplementary Figure 5).
In the second assay, we assessed spontaneous activity, rearing behaviors, and movement speed by video-monitored open-field testing. The cumulative time spent immobile, grooming, rearing and speed distributions were determined from post hoc video analysis. Grooming time in implanted mice was significantly higher at days 1-3 post-surgery than in sham controls, but this difference did not persist over time. We observed no differences in immobility or rearing times. Nominal top speeds were derived from speed distributions as the average speed above the 75th percentile. While we observed a slight reduction in top speed in animals with implants, as compared to shams, it was not statistically significant. Results are summarized in Supplementary Figure 5. Finally, implanted mice did not appear to exhibit any difficulty in grooming hindquarters or climbing (Supplementary Video 2).
Inflammation but no focal trauma to the spinal cord
We performed histological analysis using mice expressing GFP in microglia (CX3CR1-GFP; Fig. 3a) or in astrocytes (GFAP-GFP; Fig. 3b), or via standard hematoxylin and eosin (H&E) staining (Fig. 3c).
Figure 3. Histological analysis of reactive microglia and astrocytes, and tissue morphology after chamber implantation.

(a,b) Wide-field fluorescence images of 30 μm-thick coronal tissue sections from the laminectomy site and adjacent vertebral segments one day and one week after implantation and in control animals for mice expressing GFP in microglia (a) or astrocytes (b). Scale bar, 100 μm. (c) Hematoxylin and eosin stained tissue section taken seven days post-implantation. Scale bar, 3 mm. Boxed regions are shown at higher magnification below. Green boxed inset shows the fibrous connective tissue that covered the dorsal aspect of the spinal cord underneath the implant. The red boxed inset shows the neural tissue. Scale bar, 200 μm. (d) Microglial and (e) astrocyte density in spinal cord sections one and seven days after implantation for sections one vertebrae rostral to the surgical site (rost.), at the surgical site (surg.), and one vertebrae caudal to the surgical site (caud.), and in controls (* P = 0.012; ** P = 0.0010; *** P = 0.0098; # P < 0.0001; n >= 15 measurements per segment per time point; 3 mice per time point). Error bars denote the standard error of the mean.
We implanted the spinal chamber in the transgenic mice and later perfused for histology at time points of one day and one week post-surgery. We used mice not undergoing surgery as controls (n = 3 mice per group). In mice receiving implants, we analyzed sections from the site of laminectomy as well as the immediately rostral and caudal vertebrae. At time points of both one day and one week post-surgery, microglia in sections under the surgical site showed a more condensed structure with fewer processes compared to controls, but no ameboid structures indicative of phagocytic microglia21 were observed (Fig. 3a). Microglia densities (Fig. 3d) in the dorsal aspect of the rostral segment of the spinal cord at one day post-surgery and in the dorsal aspect of all segments at one week post-surgery were elevated compared to controls. Astrocytes showed no obvious changes in morphology (Fig. 3b) or cell density (Fig. 3e) across all groups.
For standard histopathology studies, we euthanized and perfused mice at time points of one day, one week, and one month post-operatively (n = 2 mice per group). In all cases, control regions showed normal tissue in nerves, bone, muscle, and spinal cord. Mild dermatitis was present at the skin-implant junction in all mice. Focal meningitis at the caudal edge of the window occurred in one mouse in the one-week group. We observed no signs of meningitis or disruption of neural tissue in the other mice (Fig. 3c) or in other regions of the mouse exhibiting focal meningitis. Neutrophils were absent in spinal cord tissue of all mice at all time points. The fibrous tissue (Fig. 3c) over the dorsal surface of the cord progressively thickened over time and was the limiting factor on the longitudinal extent of imaging. We observed indications of muscle injury including myositis, myodegeneration, and myoregeneration along with epidural neutrophilic fascitis at the site of implantation at the one-day and one-week time points. Reactive bone growth and fibroplasia occurred at the lateral edges of the window at the one-month time point. The silicone elastomer showed no signs of cellular infiltration.
Microglia numbers increase over seven days after SCI
We used double transgenic mice expressing YFP in a subset of DRG axons and GFP in microglia to evaluate the dynamics of microglial scar formation following a ~200-μm long, 35-μm deep laser-induced transection injury to the dorsal column produced using tightly-focused femtosecond laser pulses (Fig. 4a-c, four lesions in two mice). We intentionally spared blood vessels in the creation of the lesion. Microglial cell counts increased dramatically within the first day and continued to grow more slowly over the following six days (Fig. 4d). The spatial extent of the densely packed microglial scar grew steadily over seven days (Fig. 4e).
Figure 4. Imaging and quantification of microglial scar formation at the site of a laser-induced spinal cord injury.

(a.b) Projections of 2PEF image stacks of YFP-labeled axons (teal) and GFP-labeled microglia (mauve) before(a), one day (b), and one week (c) after producing a ~200-μm long laser-induced microlesion in the spinal cord. Scale bar, 100 μm. (d,e) Boxplots of the number of microglia (d) and the microglial scar size (e) within the 300-μm field of view over time (four lesions across two mice). Horizontal red lines denote the median, blue boxes bound the 25th and 75th percentiles of the data, and the whiskers denote non-outlier extrema (defined as outside the box by less than 1.5 times the interquartile range).
2PEF imaging of axon dieback after SCI
We evaluated axonal response to injury in nine laser lesions across five mice expressing YFP in DRG afferent axons (Fig. 5a). We imaged two mice out to five weeks post-injury (4 lesions). Supplementary Video 3 shows several image stacks at a lesion site taken at different times after SCI. We used characteristic features of spared axons to define a common point of origin across different imaging sessions (Fig. 5b). We used manual tracking both from 3D stacks and 2D projections to determine the distance of individual axon tips from the lesion along the rostral-caudal direction (Fig. 5b,c). Owing to axon density and insufficient spatial extent of the image stacks, we were not able to distinguish ascending axons from descending branches of the DRG neurons. Due to loss of image contrast immediately after injury, tracing was possible for most but not all (95 of 107 traced) axons on acute timescales (0-2 hours). The average response of axons was to die back from the lesion site, but the extent and rate of axon dieback was highly heterogeneous (Fig. 5b,c). Some axons exhibited the rapid acute axonal degeneration that has been previously described12. Other axons persisted near the lesion site for several days or even weeks, with some mounting an abortive growth response (Fig. 5b,c, red). Axon tip morphology (Fig. 5b) also varied from day to day. The average dieback speed of the axons (defined as the change in an axon tip’s axial position between successive imaging sessions divided by the elapsed time, with growth phases excluded) declined by nearly three orders of magnitude over the five weeks following injury (Fig. 5d).
Figure 5. 2PEF imaging and quantification of axon dieback after a laser-induced spinal cord injury.

(a) Projection of a 2PEF image stack from mice expressing YFP (teal) in a subset of DRGs with the vasculature labeled with Texas Red dextran (red). The yellow box denotes the location of the high-resolution images shown in panel (b). Scale bar, 200 μm. (b) Projections of 2PEF image stacks of YFP-expressing axons before, shortly after, and over time up to five weeks after a lesion produced by translating high-energy, tightly-focused femtosecond laser pulses through the cord. Mauve circles indicate easily-recognizable patterns in spared axons that are identified at all time points and provide a point of origin. The yellow arrow shows an axon that exhibited rapid degeneration, the blue arrow indicates an axon that died back more slowly, and the red arrow indicates an axon that persisted near the lesion site and made an ultimately aborted growth response (the morphology of this axon’s tip is shown in the red boxed insets). The yellow asterisk indicates the location of early sprouting responses that did not persist over time. Scale bar, 100 μm. (c) Position of axon endings over time after the lesion, with positive and negative values corresponding to positions rostral and caudal to the lesion site, respectively (see inset) (107 individual axon trajectories over nine lesions in five mice). Axon trajectories highlighted in color correspond to the colored arrows in the time-lapse images in panel (b). (d) Speed of axon tip dieback for axons remaining within the field of view over time after the lesion. Black circles denote measurements of dieback speed from individual axon tips, horizontal red lines represent the median, the blue boxes bound the 25th to 75th percentage of the data and the whiskers extend 1.5 times the interquartile range beyond the boxes. Points outside the whiskers were considered outliers and have a red cross through them. Because axons died back beyond the imaging field over time, the dieback speed at early times includes data from ~100 axons, while the last time point includes data from only 16 axons.
Discussion
Our study of microglial invasion following laser-induced SCI reveals that while the number and spatial extent of microglia continues to grow over time, the largest fractional change occurs within the first 24 hours post-injury, consistent with previous studies14. Microglia are known to phagocytose growth-inhibitory axon debris22 in white matter tracts23, suggesting that the chronic, gradual recruitment of microglia may be related to the progressive degeneration of the axons involved. Thus, imaging of microglia infiltration and the clearance of axon debris will be critical for developing optimal therapeutic strategies for manipulating the microglial response.
One of the key challenges in studies of regenerative strategies for SCI is the establishment of an optimal therapeutic time window24-27. While delay of treatment has been discussed at length with respect to the glial scar25, our observation of heterogeneity in axon dieback, instances of early but transient sprouting, and decrease in dieback rate at longer timescales may suggest an optimal therapeutic window based on axon dynamics. Our axon-by-axon characterization of axon dieback rates is in agreement with previous studies of acute12 and longer term28,29 measurements, but provides details on this transition and allows the classification of subpopulations of axons by response: ~15% are stable (remain within 400 μm of the lesion for at least four weeks), ~15% rapidly disintegrate (die back beyond the field of view within the first day), and ~70 % exhibit progressive degeneration over the first month. This type of dynamic data is inaccessible to post-mortem histology or analysis of gross lesion size. Future studies will attempt to correlate these classes of heterogeneity in axon dieback with heterogeneity in regenerative responses to therapy.
Previous studies12,13,16,18 have demonstrated time-lapse imaging of the spinal cord via repeated surgical opening of the skin above the spinal cord. Due to the inherent stresses and risks of repeated surgeries or restrictions placed by the Institutional Animal Care and Use Committee, the number of times these procedures may be performed places severe limitations on longitudinal studies. For example, in one long-term longitudinal study, only six imaging sessions were possible in four months18. To effectively study disease dynamics and especially response to therapeutic agents, the longitudinal extent of imaging must span a sufficiently long period of time to establish therapeutic limits with an imaging frequency capable of capturing transient responses. The chamber we have developed requires only a single surgery and grants continuous optical access, with the frequency of imaging being limited only by the ability of the animal to endure multiple rounds of anesthesia. We have demonstrated acute imaging after SCI up to two hours post injury, followed by imaging every 12 hours for one day, then daily for three days, and finally weekly for five weeks, for a total of 13 different imaging timepoints after SCI.
We observed microglia at the surgical site to have higher densities and fewer processes one week after surgery compared to control animals, suggesting an activated but not phagocytic phenotype21. This result is consistent with an analogous study of cranial windows30, and care will need to be taken when using our spinal cord window in studies sensitive to even mild inflammatory responses. However, we have observed that even a minimal laser injury to the spinal cord results in an order of magnitude increase in microglia density near the injury, including those showing phagocytic ameboid morphologies. This increase far exceeds the less than twofold increase in microglia density observed due to the surgery, suggesting that our chamber does not significantly confound studies where microglial responses are more drastic.
When combined with 2PEF imaging of transgenic mice expressing fluorescent proteins in axons, microglia, astrocytes, oligodendrocytes, endothelial cells, and immune cells and with nonlinear microscopy techniques to visualize myelin (e.g. third harmonic generation19), the spinal chamber developed here is an ideal tool for longitudinal studies of healthy and diseased-state spinal cord, including pathologies such as multiple sclerosis, implanted spinal cord tumors, or the establishment of meningitis after bacterial challenge. Since our chamber does not lead to motor deficits, functional loss or recovery may be straightforwardly correlated with cellular-level images.
Methods
Surgical Procedure
We anesthetized mice under 5% isoflurane on a custom-built surgery table (Supplementary Figure 2) and then maintained on ~1.5% isoflurane in 100% oxygen. We injected 0.05 mg of glycopyrrolate (an anticholinergic) per 100-g mouse intramuscularly. We also injected 1 mL per 100-g mouse of 5% glucose in normal saline subcutaneously hourly. We used a rectal thermometer and feedback-controlled heating blanket to maintain body temperature at 37.5 °C. We shaved the dorsal surface above the thoracic spine and applied three alternating washes each of 70% ethanol and iodine to the skin to reduce the likelihood of infection. We gave a subcutaneous injection of 0.1 mL of 0.125% bupivicaine at the site of skin incision.
We made a small incision in the skin at the T11-T13 level of the spine and the skin was held back with retractors. We made an incision three-vertebrae long on either side of T12 and scraped the bone clean on the top and the sides. We severed tendons attached to the three vertebrae using surgical scissors. We trimmed all incongruous tissue to reduce necrosis. We used sterile cotton applicators to control bleeding. We clamped the three vertebrae by magnetic stainless steel bars with a notched groove and held under pressure on 30 mm stainless steel posts with a 3-pronged plug, consisting of two pins to prevent rotation and a central magnet to hold the bar. We removed the dorsal lamina of T12 using vanna scissors, and used sterile gel foam (Pharmacia & Upjohn Co.) or cotton applicators along with sterile saline to control bleeding and keep blood off the surface of the cord. We trimmed the lateral edges of the bone back as close as possible to the edges of the bars and the surface of the bone sealed with dental acrylic and cyanoacrylate. Where possible, we left the dura intact.
Keeping the cord irrigated with normal saline, we positioned a top plate and screws inserted into the metal bars. We injected Kwik-Sil® silicone elastomer into the space between the cord and the top plate and sealed the chamber with a 5-mm diameter coverslip. We used cyanoacrylate glue and dental acrylic to seal the chamber at the rostral and caudal vertebrae. With pressure maintained by the screws, we removed the 3-pronged steel posts. We pulsed the skin to the edge of the implant and secured it with cyanoacrylate glue and dental acrylic. We inserted setscrews into the wings of the top plate. An illustration of the procedure with accompanying photographs is given in Supplementary Figure 1. We again injected bupivicaine (0.1 mL, 0.125%) around the edge of the implant. During recovery, we placed the animal on a heated surface, and administered ketoprofen (5 mg/kg/day) and dexamethasone (0.2 mg/kg/day) every 24 hours for 72 hours. A detailed, step-by-step description of how to perform the procedure, with suggested solutions to common problems, is provided in the Supplementary Protocol. We performed all surgery under a stereomicroscope (Leica MZ12.5; Leica Microsystems).
All animal procedures performed have the approval of the Cornell Institutional Animal Care and Use Committee (IACUC) and under the guidance of the Cornell Center for Animal Resources and Education.
Mice
For imaging purposes, we used mice of the YFP-H line (Jackson Labs), which expresses YFP in a subset of pyramidal neurons and dorsal root ganglia. In addition we used CX3CR1-GFP mice (Jackson Labs), which express GFP in microglia. We also used crosses between the YFP-H and CX3CR1-GFP lines.
For histopathological analysis, we used mice of the YFP-H line and mice of the Emx-1-cre (Jackson Labs) lines. We used CX3CR1-GFP mice and GFAP-GFP (Jackson labs), which express GFP in astrocytes, to study reactive gliosis in microglia and astrocytes, respectively.
In all cases, mice were of both sexes and 4 to 8 months in age when the device was implanted and they were heterozygous for each transgene. Mice were group housed prior to chamber implantation and were singly housed in rat-size cages after implantation to minimize the risk of the animal bumping the implanted chamber against the cage lid.
Histology
We deeply anesthetized mice and perfused transcardially with phosphate buffered saline (PBS) (Sigma-Aldrich) to clear the blood, followed by fixation with 4% paraformaldehyde (PFA) (ThermoFisher Scientific) in PBS.
For gliosis studies in GFAP-GFP and CX3CR1-GFP mice, we immersed whole spines in PFA for one day post-perfusion followed by removal of the spinal cord from the vertebral canal by microsurgical dissection. We immersed dissected spinal cords in 30% sucrose in PBS until saturated. We froze spinal cords in optimal cutting temperature (OCT) compound (Tissue-Tek) and cut sections at a nominal thickness of 30 μm on a Microm HM550 cryotome (ThermoFisher Scientific). We took sections at the rostral and caudal edges of the implant, underneath the glass, and control regions located one vertebrae in the rostral and caudal direction from the edges of the implant. We examined tissues under an Olympus BX41 wide-field fluorescence microscope (Olympus America).
For hematoxylin and eosin (H&E) histopathology studies, we examined whole fixed mice for gross pathology and the skin surrounding the chambers was gently freed and removed. We separated the chambers from their attachment to the subjacent vertebral bodies starting at one end by gentle dorsal traction. Once freed from the vertebral bodies, we carefully separated the chambers from subjacent soft tissues (epaxial muscle and connective tissues). We removed any material (mostly injected silicone) that had adhered to the ventral surface of the glass windows using a scalpel and reserved it for staining. After removing ribs and organs from the vertebral column, we collected 3-mm cross sections of the vertebral column–including surrounding muscle–from approximately 1 cm rostral, 1 cm caudal and directly below the center of the window by making cuts perpendicular to the vertebral column using a broad tissue blade. We inserted tissues into cassettes and immersed them again in PFA. We decalcified tissues by rinsing tissue cassettes under running water for 15 minutes, followed by placement in a vacuum jar containing equal volumes of 20% sodium citrate dihydrate and 50% formic acid. Tissues were held under vacuum at room temperature with constant stirring using a magnet for approximately 24 hours. Following this procedure, we rinsed tissues under running water for 10 minutes and put in a solution of 70% ethyl alcohol. We then embedded tissues in paraffin wax using an automated tissue processor (Tissue-Tek VIP), sectioned (4-μm thick sections), and stained them with H&E using an automated stainer (Shandon Varistain 24-4; Thermo Scientific, Waltham, MA). We sealed slides with a cover slip and examined using an Olympus BX40 microscope (Olympus America).
2PEF Microscopy
To image (or reimage) the spinal cord, we anesthetized mice with isoflurane and placed on the custom-surgery table described previously for the laminectomy procedure. Mice also received glycopyrrolate and glucose as described above. We used tapped 30-mm posts secured in an optical post holder to screw finger-tight onto the set screws of the wings of the top plate of the implant to locally immobilize the spine (Supplementary Figure 1g). We elevated mice slightly by the implant to allow room for chest expansion and contraction during breathing. After imaging, we twisted off the posts and the animal was allowed to recover on a heated surface.
We performed imaging using a custom-designed multiphoton microscope with a 20x water immersion objective lens (NA = 1.0; Carl Zeiss MicroImaging, Inc.), a 40x water immersion objective (NA = 0.8; Olympus America), or a 4x objective (NA = 0.28; Olympus). We performed 2PEF imaging using 1043-nm wavelength, 1-MHz, 300-fs pulses from a fiber laser (FCPA μJewel D-400; IMRA America, Inc) and/or 920-nm, 87-MHz, 100-fs pulses from a Ti:Sapphire laser oscillator (MIRA HP; Coherent). We used emission filters at 645/65 nm (center wavelength/bandwidth), 550/50 nm, and 517/65 nm (Chroma Technology) to isolate fluorescence from Texas Red dextran, YFP, and GFP, respectively.
Spinal cord lesioning
We made lesions measuring 100–300-μm long, 5–10-μm wide and 30–40-μm deep in the dorsal spinal cord by femtosecond laser ablation using ~100-nJ pulses from a regenerative amplifier (800-nm wavelength, 50-fs pulse duration, 1-kHz repetition rate; Legend, Coherent Inc.). We used custom software in MATLAB (MathWorks) to define a two-dimensional trajectory by tracing a pattern on a z-projection of a 3D image stack. To minimize the loss of image contrast caused by excessive bleeding, we intentionally avoided cutting blood vessels where possible. To execute the pattern, the animal was translated at 500 μm/s along the traced trajectory in the xy-plane while a shutter controlling the femtosecond pulses was opened, producing a cut about 2-3 μm deep. The animal was then translated by 1 μm in the z-direction and the cut pattern was repeated. This procedure was iterated until a cut depth of 30–40 μm was achieved. The shutter was closed during translation in z. When deemed necessary, we repeated the cut to ensure complete transection of axons. Because the damage is mediated by an electron-ion plasma formed by nonlinear optical absorption and there is very little thermal energy deposited, the damage is largely confined to the focal volume.
Image Processing
We computed image projections by taking the standard deviation along the z-axis of three-dimensional image stacks. For contrast and resolution measurements, we used maximum pixel intensity projections of isolated axon segments. Since we oversampled image stacks, we manually removed frames with significant motion artifact due to breathing without loss of information. Due to the high density of microglia observed after SCI, there was ambiguity in distinguishing cell bodies from densely packed processes. We manually identified microglia cell bodies as fluorescent ameboid structures with visible boundaries, traced, and counted them in 2-D projections using custom MATLAB software. We defined microglia scar size as the mean square radius in manual traces of the boundary of the largest contiguous aggregate of microglia. For axon tracing, we Fourier filtered high-resolution (0.59 μm/pixel) 3-D image stacks, took the standard deviation projection, and stitched the images together using PanaVue stitching software (PanaVue). We used spared axons that were stable over the duration of the experiments to define a common point of origin among imaging sessions. We marked axon endings and tracked them using custom software in MATLAB. We resolved ambiguities by examining trajectories in the 3-D stacks.
In double-transgenic mice expressing YFP in axons and GFP in microglia, we used emission filters with 517/65 nm and 550/50 nm (center wavelength/bandwidth) with 920-nm excitation for 2PEF imaging. We linearly unmixed images in custom software written in MATLAB. Briefly, we manually selected image features corresponding to axons (YFP) or microglia (GFP) in both imaging channels and generated a mixing matrix. We then solved for the inverse matrix, and calculated the resultant unmixed images containing separate fluorescent species.
Contrast and resolution fitting
To characterize the contrast and resolution, we first used ImageJ to isolate axon segments of approximately 40-50 μm in length from image stacks the same region across multiple days. We performed subsequent analysis in MATLAB. We computed the maximum projection along the z-axis and median filtered the resulting image with a 1-pixel filter radius. To orient the segment so that the axis ran parallel to the y-axis, we used a radon transform to find the angle of orientation and the image was rotated accordingly.
Since we considered the maximum intensity projection across the axon volume, the intensity profile is equal to the value of the intensity profile taken immediately through the center of the axon, where, to good approximation, the excited fluorescence within the axon is approximately constant in the x-direction. For an axon of radius R, and displaced from the origin by an amount δ, the fluorophore concentration profile, C, in a single scan at the center is:
| (S1) |
where C0 is the axonal fluorophore concentration and θ(x) is the Heaviside step function. For a Gaussian excitation beam with intensity profile:
| (S2) |
where I0 is the peak laser intensity and a is a measure of the beam waist, the image intensity, F(x), is given by the convolution of Equation S1 and S2 with the addition of a background noise floor, B:
| (S3) |
where
| (S4) |
We fit Equation (S3) to each line of the axon profile (see Fig. 2c) and averaged the results. Failure to converge by nonlinear least squares fitting or an R2 value less than 0.85 was used to exclude data points. We defined contrast as:
| (S5) |
which ranges between 0 (no contrast) and 1 (noiseless contrast), where F(-δ) is the intensity peak in the axon. We took resolution as the parameter a.
Behavioral Assays
We subjected mice to open field and runway assays. We first made measurements one day prior to implantation of an imaging window, followed by measurements each day for the first seven days and a final time point at 14 days post-implantation. We made all measurements at the same time of day to avoid circadian variability. We tested animals that were shaved and anesthetized, but not operated on, simultaneously as sham controls.
We constructed analysis of footprints from the runway assay in which mice with inked paws traversed the length of a Plexiglass enclosure (76 cm long x 8 cm wide x 20 cm high) to enter a dark goal box at the end of the runway. We placed mice on an inked pad in a 15 cm long staging area separated from the main runway by a sliding insert. We placed paper tape on the floor of the runway to collect ink pawprints. Removal of the insert marked the beginning of the trial. All trials were video recorded from above. We performed three trials at each time point, and took footprint measurements from 5 consecutive steps in each trial. We then returned mice to their home cage between trials to minimize the effects of fatigue. Mice received 10-14 days of behavioral training prior to surgery. During training, we encouraged mice to traverse the length of the runway without pausing, receiving prompting from the experimenter where necessary. We deemed training complete when mice traversed the length of the runway without pausing or prompting. We assessed hindlimb base of support as the lateral distance between hindlimbs, and stride length as the distance between the central pads of two consecutive hindlimb prints on the left or right. We determined average speed by dividing the runway length by the total time of the trial, as determined by the video clock.
We assessed rearing, grooming, mobility and top speeds in open field measurements. We placed mice in the center of a Plexiglass enclosure (46 cm long x 46 cm wide x 47 cm high) with black sides and a white base. We recorded mice from above for 5 minutes under red light illumination. Video tracking analysis was performed based on the videos using ANY-maze software (Stoelting Company) and MATLAB. We defined rearing as any period during which the animal lifted both of its forelimbs off the ground simultaneously. We defined grooming as any period during which the animal licked its fur or moved its forelimbs over the head. Top speeds were determined as the mean of the speeds greater than the 75th percentile.
Statistical Analysis
We compared grooming time, time spent immobile, rearing time, base of support, stride length and average speed using the analysis of variance (ANOVA) test. Where the null hypothesis was rejected, we performed post hoc analyses using Tukey’s honestly significant difference for pairwise comparisons. We compared top speeds of mice receiving surgery and shams each day using a Mann-Whitney-U test.
We compared microglia and astrocyte densities using ANOVA. Where the null hypothesis was rejected, we performed post hoc analyses using Tukey’s honestly significant difference on mean to compare groups.
We performed statistical tests in Kaelidograph (Synergy) and MATLAB. We set the criterion for significance in all cases to be α = 0.05.
Supplementary Material
Acknowledgements
We would like to thank the National Institutes of Health (DP OD006411 to J.R.F., R01 EB002019 to C.B.S) and the National Science and Research Council of Canada (to M.J.F.) for financial support, and IMRA America, Inc. for the loan of their FCPA μJewel D-400 laser. We thank J. Siebert for his critical reading of this manuscript and M. Riccio for his assistance with the MicroCT imaging.
Footnotes
Author contributions MJF, TAC, JRF, and CBS conceived and designed the experiments. MJF performed surgeries and imaging experiments, IMB performed behavioral assays, and DHS performed histopathology. MJF, IMB, JRF, and CBS analyzed the data. JRF, TAC, and CBS contributed reagents and materials. MJF, JRF, and CBS wrote the paper.
Competing Financial Interests The authors declare no competing financial interests.
References
- 1.Kobat D, et al. Deep tissue multiphoton microscopy using longer wavelength excitation. Opt. Express. 2009;17:13354–13364. doi: 10.1364/oe.17.013354. [DOI] [PubMed] [Google Scholar]
- 2.Christie RH, et al. Growth arrest of individual senile plaques in a model of Alzheimer’s disease observed by in vivo multiphoton microscopy. J. Neurosci. 2001;21:858–864. doi: 10.1523/JNEUROSCI.21-03-00858.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan P, et al. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J. Neurosci. 2009;29:10706–10714. doi: 10.1523/JNEUROSCI.2637-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer-Luehmann M, et al. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature. 2008;451:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prada CM, et al. Antibody-mediated clearance of amyloid-beta peptide from cerebral amyloid angiopathy revealed by quantitative in vivo imaging. J. Neurosci. 2007;27:1973–1980. doi: 10.1523/JNEUROSCI.5426-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kienast Y, et al. Real-time imaging reveals the single steps of brain metastasis formation. Nat. Med. 2010;16:116–122. doi: 10.1038/nm.2072. [DOI] [PubMed] [Google Scholar]
- 7.Lam CK, Yoo T, Hiner B, Liu Z, Grutzendler J. Embolus extravasation is an alternative mechanism for cerebral microvascular recanalization. Nature. 2010;465:478–482. doi: 10.1038/nature09001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holtmaat A, et al. Long-term, high-resolution imaging in the mouse neocortex through a chronic cranial window. Nat. Protoc. 2009;4:1128–1144. doi: 10.1038/nprot.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang G, Pan F, Parkhurst CN, Grutzendler J, Gan WB. Thinned-skull cranial window technique for long-term imaging of the cortex in live mice. Nat. Protoc. 2010;5:201–208. doi: 10.1038/nprot.2009.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drew PJ, et al. Chronic optical access through a polished and reinforced thinned skull. Nat. Methods. 2010;7:981–984. doi: 10.1038/nmeth.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhatt DH, Otto SJ, Depoister B, Fetcho JR. Cyclic AMP-induced repair of zebrafish spinal circuits. Science. 2004;305:254–258. doi: 10.1126/science.1098439. [DOI] [PubMed] [Google Scholar]
- 12.Kerschensteiner M, Schwab ME, Lichtman JW, Misgeld T. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 2005;11:572–577. doi: 10.1038/nm1229. [DOI] [PubMed] [Google Scholar]
- 13.Davalos D, et al. Stable in vivo imaging of densely populated glia, axons and blood vessels in the mouse spinal cord using two-photon microscopy. J. Neurosci. Methods. 2008;169:1–7. doi: 10.1016/j.jneumeth.2007.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dibaj P, et al. NO Mediates Microglial Response to Acute Spinal Cord Injury Under ATP Control In Vivo. Glia. 2010;58:1133–1144. doi: 10.1002/glia.20993. [DOI] [PubMed] [Google Scholar]
- 15.Johannssen HC, Helmchen F. In vivo Ca2+ imaging of dorsal horn neuronal populations in mouse spinal cord. J. Physiol. 2010;588:3397–3402. doi: 10.1113/jphysiol.2010.191833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ylera B, et al. Chronically CNS-Injured Adult Sensory Neurons Gain Regenerative Competence upon a Lesion of Their Peripheral Axon. Curr. Biol. 2009;19:930–936. doi: 10.1016/j.cub.2009.04.017. [DOI] [PubMed] [Google Scholar]
- 17.Kim JV, et al. Two-photon laser scanning microscopy imaging of intact spinal cord and cerebral cortex reveals requirement for CXCR6 and neuroinflammation in immune cell infiltration of cortical injury sites. J. Immunol. Methods. 2010;352:89–100. doi: 10.1016/j.jim.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dray C, Rougon G, Debarbieux F. Quantitative analysis by in vivo imaging of the dynamics of vascular and axonal networks in injured mouse spinal cord. Proc. Natl. Acad. Sci. USA. 2009;106:9459–9464. doi: 10.1073/pnas.0900222106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Farrar MJ, Wise FW, Fetcho JR, Schaffer CB. In Vivo Imaging of Myelin in the Vertebrate Central Nervous System Using Third Harmonic Generation Microscopy. Biophys. J. 2011;100:1362–1371. doi: 10.1016/j.bpj.2011.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Crawley JN. What’s wrong with my mouse? : behavioral phenotyping of transgenic and knockout mice. Wiley-Liss; New York: 2000. [Google Scholar]
- 21.Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog. Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka T, Ueno M, Yamashita T. Engulfment of axon debris by microglia requires p38 MAPK activity. J. Biol. Chem. 2009;284:21626–21636. doi: 10.1074/jbc.M109.005603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aldskogius H, Kozlova EN. Central neuron-glial and glial-glial interactions following axon injury. Prog. Neurobiol. 1998;55:1–26. doi: 10.1016/s0301-0082(97)00093-2. [DOI] [PubMed] [Google Scholar]
- 24.Garcia-Alias G, et al. Therapeutic time window for the application of chondroitinase ABC after spinal cord injury. Exp. Neurol. 2008;210:331–338. doi: 10.1016/j.expneurol.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 25.Rolls A, Shechter R, Schwartz M. The bright side of the glial scar in CNS repair. Nat. Rev. Neurosci. 2009;10:235–241. doi: 10.1038/nrn2591. [DOI] [PubMed] [Google Scholar]
- 26.Tator CH. Review of treatment trials in human spinal cord injury: issues, difficulties, and recommendations. Neurosurgery. 2006;59:957–982. doi: 10.1227/01.NEU.0000245591.16087.89. discussion 982-957. [DOI] [PubMed] [Google Scholar]
- 27.Thuret S, Moon LD, Gage FH. Therapeutic interventions after spinal cord injury. Nat. Rev. Neurosci. 2006;7:628–643. doi: 10.1038/nrn1955. [DOI] [PubMed] [Google Scholar]
- 28.Tator CH, Seif GI, Nomura H. Retrograde axonal degeneration (“Dieback”) in the corticospinal tract after transection injury of the rat spinal cord: A Confocal Microscopy study. J. Neurotraum. 2007;24:1513–1528. doi: 10.1089/neu.2007.0323. [DOI] [PubMed] [Google Scholar]
- 29.Silver J, Horn KP, Busch SA, Hawthorne AL, van Rooijen N. Another barrier to regeneration in the CNS: Activated macrophages induce extensive retraction of dystrophic axons through direct physical interactions. J. Neurosci. 2008;28:9330–9341. doi: 10.1523/JNEUROSCI.2488-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu HT, Pan F, Yang G, Gan WB. Choice of cranial window type for in vivo imaging affects dendritic spine turnover in the cortex. Nat. Neurosci. 2007;10:549–551. doi: 10.1038/nn1883. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.