

Abstract
Background
Clinical application of adoptive T cell therapy (ACT) has been hindered by an inability to generate adequate numbers of non-tolerized, functionally active tumor-specific T cells which can persist in vivo. In order to address this, we evaluated the impact of IL-12 signaling during tumor-specific CD8+ T cell priming in terms of persistence and anti-tumor efficacy using an established B16 melanoma tumor adoptive therapy model.
Study Design
B6 mice were injected subcutaneously with B16 melanoma tumor cells. On day 12 of tumor growth, mice were preconditioned with cyclophosphamide (4mg dose, i.p.), and one day later, treated by adoptive transfer of tumor-specific pmel-1 CD8+ T cells primed ex vivo 3 days earlier with (i) both IL-12 and antigen (hGP10025–33 peptide) or (ii) antigen only. Tumors were measured biweekly and infused donor T cells were analyzed for persistence, localization to the tumor, phenotype, and effector function.
Results
Adoptive transfer of tumor-specific CD8+ T cells primed with IL-12 was significantly more effective in reducing tumor burden in mice preconditioned with cyclophosphamide compared with transfer of T cells primed without IL-12. This enhanced anti-tumor response was associated with increased frequencies of infused T cells in the periphery and tumor as well as elevated expression of effector molecules including granzyme B and interferon-γ (IFNγ).
Conclusions
Our findings demonstrate that ex vivo priming of tumor-specific CD8+ T cells with IL-12 dramatically improves their in vivo persistence and therapeutic ability upon transfer to tumor-bearing mice. These findings can be directly applied as novel clinical trial strategies.
Introduction
The adoptive transfer of ex vivo expanded lymphocytes for the treatment of cancer holds great promise. In a seminal study in 1988, Rosenberg and colleagues achieved objective responses in about one third of metastatic melanoma patients treated with tumor-infiltrating lymphocytes in combination with IL-2 (1–3). Today, significant advances in adoptive cellular therapy (ACT) have allowed the successful application of this treatment to a much larger group of patients. In metastatic melanoma, the addition of lymphodepletion prior to therapy with tumor infiltrating lymphocytes (TIL) appears to increase the objective response rate to over 50% of patients (4). The transfer of Epstein-barr virus-specific T cells can dramatically reduce the post-transplant lymphoproliferative diseases associated with this virus (5). And the gene transfer of T-cell receptor (TCR) genes as well as chimeric antibody receptors into peripheral blood potentially allows for the treatment of patients that might otherwise lack isolatable tumor-specific lymphocytes of therapeutic value (6–9).
While there has been much progress in the development of more effective adoptive T cell therapy strategies, relatively little is known about optimal T cell culture conditions for T cell expansion. Current approaches suffer from an inability to generate adequate numbers of non-tolerized, functionally active tumor-specific T cells which can persist in vivo. Traditionally, T cells have been expanded with a combination of TCR engagement (either tumor or anti-CD3 mAb) and high dose IL-2 (10,11). It is, however, becoming increasingly clear that the differentiation, function, and phenotype of the adoptively transferred T cells are critical parameters that can be manipulated ex vivo in order to achieve adequate short-term function and long-term immunologic memory (12–21). Furthermore, recent studies suggest that optimal T cell priming may require stimulation with a unique third signal, which can include IL-12 (22,23). Thus, mouse CD8+ T cells primed in vitro with antigen and in the presence of only IL-12 exhibit enhanced functional ability as measured by cytotoxicity and survival in vivo, and have been shown to be protective against tumor challenge (24–26). Finally, we have demonstrated that CD8+ T cells primed with IL-12 and sorted on CD62Lhi expression survive much better in the periphery after cyclophosphamide-mediated lymphodepletion (25). These properties suggest that tumor-specific CD8+ T cells primed in the presence of IL-12 will mediate more effective anti-tumor immunity in the context of cyclophosphamide-mediated lymphodepletion (27–29).
To directly assess the anti-tumor efficacy of CD8+ T cells primed with IL-12, we isolated naive tumor-specific CD8+ T cells, which recognize the gp100 (Db-restricted) shared/self tumor antigen expressed on mouse melanoma tumor cells, from pmel-1 TCR transgenic mice (30). T cells were activated with peptide for three days with (pmelIL-12) or without (pmelsham) IL-12. T cells primed with IL-12 exhibited a unique phenotypic and functional signature including elevated expression of the IL-2Rα and enhanced ability to produce IFNγ. Importantly, unlike pmelsham cells, adoptively transferred pmelIL-12 cells accumulated in both the periphery and the tumor. Finally, to directly assess anti-tumor efficacy, B6 mice were injected subcutaneously with B16 tumor cells. At day 12, when tumors were palpable, mice were treated with cyclophosphamide and/or the adoptive transfer of pmelIL-12 or pmelsham T cells. A substantial impact upon tumor growth was only observed in mice treated with both cyclophosphamide and pmelIL-12 CD8+ T cells. These findings are the first to show a significant enhancement of efficacy via ex vivo IL-12 priming in an adoptive T cell transfer model that could be directly incorporated into current clinical practice.
Methods
Cell cultures and flow cytometry
B16-F1 tumor cells (ATCC, Manassas, VA) were cultured in complete media as previously described (31). Spleen cells from pmel-1 TCR transgenic mice (1×106 cells/well in 1.5ml, unless otherwise indicated) were stimulated in complete media with H-2Db-restricted human gp10025–33 epitope (KVPRNQDWL, 1μg/ml (American Peptide Company)) for 3 days with or without mIL-12 (10ng/ml) to generate pmelIL-12 or pmelsham T cells, respectively. For flow cytometric analysis, cells were analyzed as previously described (32). In some experiments, cells were fixed with a 4% paraformaldehyde solution prior to analysis. For intracellular flow cytometry, cells were stained using the protocol and reagents from the BD Cytofix/Cytoperm with Golgistop kit (BD Biosciences, San Jose, CA). The antibodies used in this study include: B220 (RA3-6B2), CD8 (53-6.7), CD25 (PC61.5), CD28 (37.51), CD69 (H1.2F3), CD122 (TM-β1), CD127 (SB/199), CD132 (4G3), GATA3 (TWAJ), Granzyme B (GB12), IFNγ (XMG1.2), ICOS (C398.4A), OX40 (OX-86), PD-1 (J43), RORγt (AFKJS-9), and T-bet (4B10). These were purchased from BD Bioscience, Biolegend (San Diego, CA), Invitrogen (Carlsbad, CA), and/or Ebioscience (San Diego, CA). Flow cytometry was performed on a BD FACSCalibur, BD FACSVerse, or BD FACSAccuri (BD biosciences). Data were analyzed using FlowJo software (TreeStar, San Carolos, CA).
Mice
C57BL/6 (B6), B6.PL (Thy1.1), and pmel-1 TCR transgenic mice (30) were obtained from the Jackson Laboratory (Bar Harbor, ME). Pmel-1 mice were maintained by crossing a pmel-1 (male) to a Thy1.1 (female) generating hemizygous offspring. All animals were housed under specific pathogen-free conditions in accordance with institutional and federal guidelines at the Medical University of South Carolina.
Tumor challenge, CTX preconditioning, and adoptive T cell transfer
For tumor experiments, B6 mice were challenged (s.c.) with 2.5 × 105 B16-F1 tumor cells. Tumor growth was measured by caliper every 2–4 days and the tumor surface area (mm2) was calculated by length × width. For treatment with CTX, mice were given 4 mg/mouse CTX as previously described (27,28). For adoptive T cell transfer experiments, pmelIL-12 or pmelsham T cells (at day 3 of culture) were injected intravenously.
Assay of intracellular IFNγ production by T cells
Day 3 CD8+ pmelIL-12 and pmelsham T cells were co-cultured with 1×105 B6 splenocytes in 200μl of a flat 96-well plate with GolgiStop (BD catalog #554724). Soluble hgp100 peptide (1μg/ml) was added as indicated. After 6 hours, cells were analyzed for expression of intracellular IFNγ using the BD Cytofix/Cytoperm Fixation/Permeabilization Kit (BD catalog #555028).
Results
Phenotype and function of activated CD8+ T cells stimulated with IL-12
To assess the phenotype of tumor-specific CD8+ T cells primed with IL-12, naïve pmel-1 TCR transgenic CD8+ T cells were stimulated with peptide with or without IL-12 for three days. Cells were analyzed by flow cytometry for expression of cytokine receptors, costimulatory molecules, and transcription factors. In comparison with pmelsham, pmelIL-12 cells showed elevated levels of IL-2Rα (CD25) but no significant changes in other cytokine receptor subunit components (Figure 1A), consistent with previous studies (26,33). We also observed an elevation in ICOS and OX40 expression, but not other costimulatory molecules (Figure 1B). Finally, pmelIL-12 cells exhibited enhanced T-bet expression and reduced GATA-3 expression (Figure 1C), consistent with polarization to a Tc1 phenotype (34).
Figure 1.

Phenotypic analysis of CD8+ T cells primed with or without IL-12. After priming for three days, pmelIL-12 or pmelsham T cells were analyzed by flow cytometry for the markers indicated on the far left: (A) shows cytokine receptor subunits and CD69, (B) shows costimulatory and inhibitory molecules, and (C) shows transcription factors. All results are representative of at least 2 independent experiments.
In terms of functional markers, pmelIL-12 cells had elevated levels of intracellular granzyme B (Figure 2A). Interestingly, the elevation in granzyme B was more pronounced at lower cell concentrations suggesting that while IL-12 was directly impacting granzyme B, other molecules likely elevated with increased cell density were also able to impact granzyme B expression. In addition to granzyme B, pmelIL-12 and pmelsham cells both produced IFNγ specifically upon antigen stimulation in either flat or round bottom 96-well plates (Figure 2B&C). The maximal level of IFNγ trended higher with pmelIL-12 cells, and there was also a higher level of basal IFNγ in pmelIL-12 cells without antigen stimulation. Cumulatively, these properties suggest that pmel-1 CD8+ T cells primed in the presence of IL-12 express elevated levels of molecules associated with functional ability, and thus, may be more efficacious in mediating anti-tumor immunity.
Figure 2.
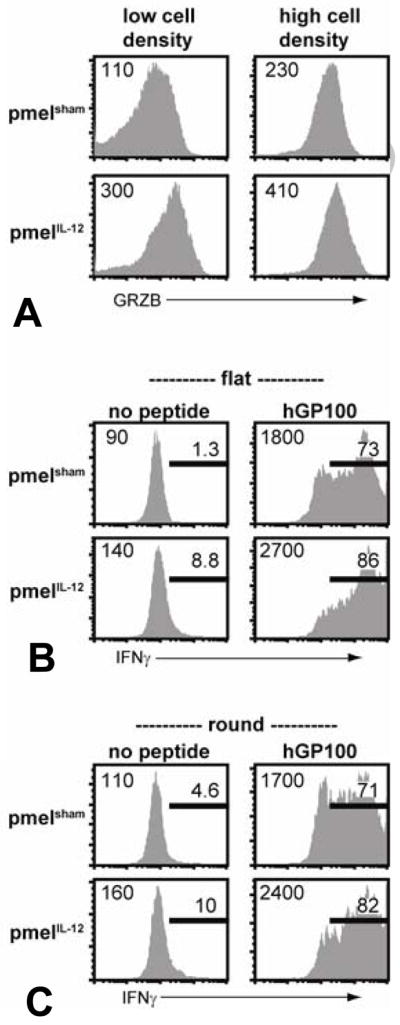
Priming pmel-1 CD8+ TCR transgenic T cells with IL-12 leads to enhanced granzyme B and IFNγ expression. (A) After priming for three days, pmelIL-12 or pmelsham T cells were directly analyzed for granzyme B expression. (B) PmelIL-12 or pmelsham T cells were cocultured with B6 splenocytes with or without relevant peptide in a flat 96-well plate for 6 hours. (C) as in ‘B’ except in a round 96-well plate. All results are representative of at least 3 independent experiments.
Tumor localization of CD8+ T cells primed with IL-12
To assess whether priming with IL-12 affected the ability of CD8+ T cells to migrate to the tumor, pmelIL-12 and pmelsham cells were adoptively transferred into B6 mice with day 10 established B16 (s.c.) tumors. Five days later, spleens and tumors were harvested and analyzed for the presence of donor cells. As we and others have previously observed (23–25), CD8+ T cells preferentially accumulated in the spleen when primed with IL-12 (Figure 3A). Strikingly, pmelIL-12 cells also preferentially accumulated in the tumor (Figure 3B). Even the adoptive transfer of a 5-fold excess of pmelsham cells was not nearly equivalent to the transfer of cells primed with IL-12. These results strongly suggest that priming with IL-12 dramatically enhances the migratory and/or survival properties of tumor-specific CD8+ T cells.
Figure 3.
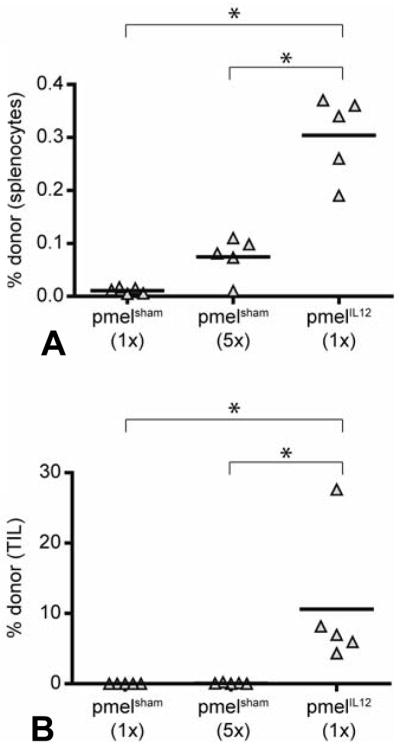
IL-12 primed CD8+ T cells exhibit improved localization to the tumor. PmelIL-12 or pmelsham T cells were adoptively transferred into B6 mice with day 7 established B16 tumors. Mice received either 5×106 (1×) or 2.5×107 T cells. One week later, spleens and tumors were harvested. Shown is the percentage of donor T cells (Thy1.1+) among total lymphocytes in the (A) spleen and (B) tumor. Each triangle represents one mouse. *, p < 0.05, indicate statistical analysis by Student’s t test.
Enhanced anti-tumor immunity depends on both priming with IL-12 and pretreatment with cyclophosphamide
To determine whether priming of tumor-specific CD8+ T cells with IL-12 would lead to more effective anti-tumor immunity, B6 mice with palpable, day 12, B16 tumors (s.c.) were treated by adoptive transfer of pmelIL-12 cells. Despite the ability of pmelIL-12 cells to effectively migrate into the tumor, we failed to observe significant anti-tumor efficacy (Figure 4). We hypothesized that competition with host T cells and/or regulatory cells may be inhibiting the function of the adoptively transferred cells, and thus, evaluated whether host lymphodepletion with CTX could enhance the efficacy of the adoptively transferred T cells. Treatment of mice with CTX alone did induce a delay in tumor growth, but this was transient and tumor growth reoccurred quickly (Figure 4). However, upon the adoptive transfer of pmelIL-12 cells into mice treated one day earlier with CTX, we observed dramatic reduction in measurable tumor (Figure 4). This effect was dependent on IL-12 during ex vivo T cell priming, as the combination of pmelsham cells and CTX was not efficacious (Figure 4). Taken together, these results demonstrate that ex vivo IL-12 priming of adoptively transferred cells in combination with cyclophosphamide-mediated host lymphodepletion yielded synergistic enhancement of anti-tumor immunity.
Figure 4.
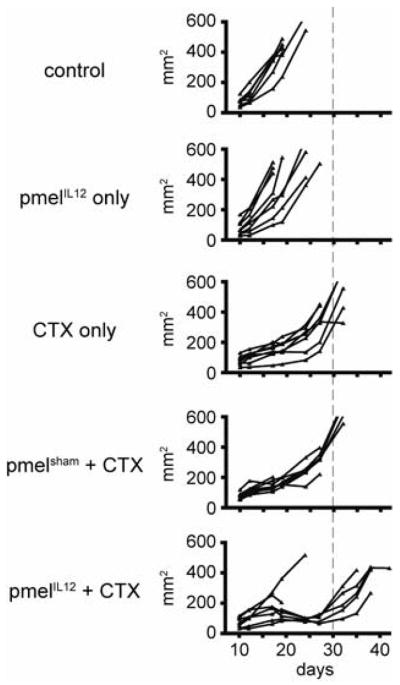
IL-12-priming during CD8+ T cell activation leads to significantly enhanced anti-tumor activity. Mice with s.c. B16 tumors were treated on day 12 as indicated with CTX. On day 13, mice were adoptively transferred with pmelIL-12 (3×106) or pmelsham (3×106) T cells. Tumors were measured blindly twice weekly. Each line represents one mouse.
Discussion
The development of improved methodologies for ACT is of critical importance to many patients with incurable cancer. This study provides novel insights into the relationship between appropriate T cell priming and host conditioning necessary for effective ACT. Specifically, we address the impact of priming tumor-specific CD8+ T cells in the context of signal 3 using the cytokine, IL-12. In addition to characterizing the modulation of phenotypic and functional molecules associated with IL-12 priming, we assessed the ability of these cells to migrate, survive, and function in mice. Importantly, we demonstrate that T cells primed in the context of IL-12 acquire a unique ability to migrate to and survive within the tumor. However, the presence of these T cells within the tumor was not sufficient to mediate significant anti-tumor immunity of established tumors. We find that host lymphodepletion with cyclophosphamide is necessary in combination with ex vivo IL-12 priming to achieve optimal tumor regression.
To assess the changes that occur during T cell activation with or without IL-12 priming in the context of pmel-1 TCR transgenic CD8+ T cells, we compared pmelIL-12 and pmelsham T cells for the expression of phenotypic markers as well as molecules associated with improved effector function. As previously reported (26,33,35), we found that IL-12 priming enhances expression of the IL-2 receptor subunits CD25, while maintaining CD122 (IL-2Rβ) and CD132 (IL-2Rγ) expression, suggesting that these cells may be more responsive to IL-2. IL-12-primed cells also had elevated levels of ICOS and OX40; perhaps indicating enhanced sensitivity to the corresponding ligands. Finally, IL-12-primed cells showed elevated expression of the transcription factor, T-bet, as previously reported (24), but reduced expression GATA-3. Upregulation of T-bet is consistent with the ability of IL-12 to direct CD8+ T cells to a Tc1 phenotype (34), while loss of GATA-3 is consistent with a loss of Th2 or Tc2 phenotype (36). As has been previously demonstrated (37), we found that IL-12 priming also enhances the expression of granzyme B. It is notable though that we find a cell concentration dependency on the magnitude of IL-12-mediated granzyme B upregulation. At higher cell concentrations, the impact of IL-12 was reduced, suggesting that other molecules, perhaps IL-2, are relevant for granzyme B expression. In addition to the effector molecule granzyme B, we also assessed the ability of T cells primed with IL-12 to produce IFNγ, another important effector molecule. Consistent with prior reports (23,24,38), we found that regardless of IL-12 priming, activated tumor-specific T cells retain the ability to produce IFNγ as measured by intracellular flow cytometry. The absolute level of IFNγ trended higher when T cells were primed with IL-12. Furthermore, T cells primed with IL-12 produced a higher basal level of IFNγ without antigen stimulation. As granzyme B and IFNγ are important molecules in mediating effector function, these data suggest that priming of CD8+ pmel-1 TCR transgenic T cells in the context of IL-12 will lead to the generation of more effective anti-tumor lymphocytes.
To determine whether T cells primed with IL-12 could localize to the tumor, we adoptively transferred lymphocytes primed with or without IL-12 into tumor-bearing mice. As others have reported previously (23,25), with ex vivo IL-12 priming we found a significant increase in the frequency of T cells in the peripheral organs such as the spleen. Strikingly, with ex vivo IL-12 priming we also found a dramatic increase in the ability of T cells to localize and survive in the tumor (Figure 3). Even the transfer of a 5-fold excess of T cells primed without IL-12 was not comparable to the tumor localization and survival of T cells primed ex vivo with IL-12. Our results are consistent with findings by Lisiero and coworkers who found that T cells primed with IL-12 were preferentially detectable in the tumor when adoptively transferred into tumor bearing mice treated with vaccination and IL-2 (26). However, our results are distinct in that mice had not received adjuvant therapy such as lymphodepletion, vaccination, or cytokine therapy. Thus, priming with IL-12 significantly augments the inherent ability of tumor-specific CD8+ T cells to migrate to and survive in the tumor.
The most important endpoint of our experiments was the evaluation of tumor immunity. Given the ability of tumor-specific CD8+ T cells to migrate to and survive within the tumor, we first determined whether the transfer of these cells alone would impact tumor growth. However, we observed no reduction in tumor growth with only the transfer of tumor-specific CD8+ T cells primed with IL-12. We hypothesized that donor cells may be functionally impaired due to competition with host lymphocytes or the impact of regulatory cells. Therefore, we evaluated whether induction of host lymphodepletion with cyclophosphamide pretreatment one day prior to adoptive transfer would improve anti-tumor immunity. Remarkably, we found that in mice pretreated with cyclophosphamide, the transfer of tumor-specific CD8+ T cells primed with IL-12 mediated dramatic anti-tumor immunity. Importantly, the transfer of T cells primed without IL-12 in mice pretreated with cyclophosphamide was not therapeutically effective. These results elegantly demonstrate a situation where combinatorial therapy is surprisingly more effective than either singular therapy by itself.
It will be important to assay whether priming other lymphocyte populations or with the use of alternative cytokines will also prove more effective as combinatorial therapy. For example, Dutton and colleagues have shown that Tc1 cells, CD8+ T cells primed with IL-12 and then cultured briefly with IL-2, have potent anti-tumor efficacy in mice (39). Furthermore, Tc17 and Th17 cells have also demonstrated potent efficacy against established tumors in mice (40,41). Whether these lymphocytes would also prove efficacious in combination with cyclophosphamide or other lymphodepleting therapies will be important questions to address.
Summary.
The inability to generate adequate numbers of non-tolerized, functionally active tumor-specific T cells, which can persist in vivo, is a significant therapeutic obstacle. Our findings demonstrate that a simple modification of the initial culture conditions, combined with host preconditioning, can significantly improve a T cell’s ability to persist and mediate anti-tumor immunity in the mouse. Consideration of these findings may aid in the development of improved culture techniques for human T cells in the context of adoptive therapy.
Acknowledgments
Supported in part by: the National Institutes of Health, 1R01CA083672-09 (DJC) Presented at the Southern Surgical Association 123rd Annual Meeting, Hot Springs, VA, December 2011.
Footnotes
Disclosure Information: Nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rosenberg SA. Karnofsky Memorial Lecture. The immunotherapy and gene therapy of cancer. J Clin Oncol. 1992;10:180–99. doi: 10.1200/JCO.1992.10.2.180. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319:1676–80. doi: 10.1056/NEJM198812223192527. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Yannelli JR, Yang JC, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–66. doi: 10.1093/jnci/86.15.1159. [DOI] [PubMed] [Google Scholar]
- 4.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heslop HE, Slobod KS, Pule MA, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood. 2010;115:925–35. doi: 10.1182/blood-2009-08-239186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson LA, Morgan RA, Dudley ME, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood. 2009;114:535–46. doi: 10.1182/blood-2009-03-211714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kalos M, Levine BL, Porter DL, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced Leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKee MD, Fichera A, Nishimura MI. T cell immunotherapy. Front Biosci. 2007;12:919–32. doi: 10.2741/2114. [DOI] [PubMed] [Google Scholar]
- 10.Dudley ME, Rosenberg SA. Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev Cancer. 2003;3:666–75. doi: 10.1038/nrc1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudley ME, Wunderlich JR, Shelton TE, et al. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–42. doi: 10.1097/00002371-200307000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–12. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosenberg SA, Restifo NP, Yang JC, et al. Adoptive cell transfer: a clinical path to effective cancer immunotherapy. Nat Rev Cancer. 2008;8:299–308. doi: 10.1038/nrc2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117:1466–76. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klebanoff CA, Gattinoni L, Torabi-Parizi P, et al. Central memory self/tumor-reactive CD8+ T cells confer superior antitumor immunity compared with effector memory T cells. Proc Natl Acad Sci U S A. 2005;102:9571–6. doi: 10.1073/pnas.0503726102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perret R, Ronchese F. Memory T cells in cancer immunotherapy: which CD8 T-cell population provides the best protection against tumours? Tissue Antigens. 2008;72:187–94. doi: 10.1111/j.1399-0039.2008.01088.x. [DOI] [PubMed] [Google Scholar]
- 17.Speiser DE, Romero P. Toward improved immunocompetence of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1467–9. doi: 10.1172/JCI25427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gattinoni L, Klebanoff CA, Palmer DC, et al. Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest. 2005;115:1616–26. doi: 10.1172/JCI24480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berger C, Jensen MC, Lansdorp PM, et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhou J, Shen X, Huang J, et al. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–52. doi: 10.4049/jimmunol.175.10.7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maus MV, Kovacs B, Kwok WW, et al. Extensive replicative capacity of human central memory T cells. J Immunol. 2004;172:6675–83. doi: 10.4049/jimmunol.172.11.6675. [DOI] [PubMed] [Google Scholar]
- 22.Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med. 2003;197:1141–51. doi: 10.1084/jem.20021910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang J, Cho JH, Lee SW, et al. IL-12 priming during in vitro antigenic stimulation changes properties of CD8 T cells and increases generation of effector and memory cells. J Immunol. 2004;172:2818–26. doi: 10.4049/jimmunol.172.5.2818. [DOI] [PubMed] [Google Scholar]
- 24.Rao RR, Li Q, Odunsi K, Shrikant PA. The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity. 2010;32:67–78. doi: 10.1016/j.immuni.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Diaz-Montero CM, El Naggar S, Al Khami A, et al. Priming of naive CD8+ T cells in the presence of IL-12 selectively enhances the survival of CD8(+)CD62L (hi) cells and results in superior anti-tumor activity in a tolerogenic murine model. Cancer Immunol Immunother. 2008;57:563–72. doi: 10.1007/s00262-007-0394-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lisiero DN, Soto H, Liau LM, Prins RM. Enhanced sensitivity to IL-2 signaling regulates the clinical responsiveness of IL-12-primed CD8(+) T cells in a melanoma model. J Immunol. 2011;186:5068–77. doi: 10.4049/jimmunol.1003317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salem ML, Kadima AN, El-Naggar SA, et al. Defining the ability of cyclophosphamide preconditioning to enhance the antigen-specific CD8+ T-cell response to peptide vaccination: creation of a beneficial host microenvironment involving type I IFNs and myeloid cells. J Immunother (1997) 2007;30:40–53. doi: 10.1097/01.cji.0000211311.28739.e3. [DOI] [PubMed] [Google Scholar]
- 28.Salem ML, Diaz-Montero CM, Al-Khami AA, et al. Recovery from cyclophosphamide-induced lymphopenia results in expansion of immature dendritic cells which can mediate enhanced prime-boost vaccination antitumor responses in vivo when stimulated with the TLR3 agonist poly(I:C) J Immunol. 2009;182:2030–40. doi: 10.4049/jimmunol.0801829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salem ML, Cole DJ. Dendritic cell recovery post-lymphodepletion: a potential mechanism for anti-cancer adoptive T cell therapy and vaccination. Cancer Immunol Immunother. 2010;59:341–53. doi: 10.1007/s00262-009-0792-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubinstein MP, Kadima AN, Salem ML, et al. Systemic administration of IL-15 augments the antigen-specific primary CD8+ T cell response following vaccination with peptide-pulsed dendritic cells. J Immunol. 2002;169:4928–35. doi: 10.4049/jimmunol.169.9.4928. [DOI] [PubMed] [Google Scholar]
- 32.Rubinstein MP, Lind NA, Purton JF, et al. IL-7 and IL-15 differentially regulate CD8+ T-cell subsets during contraction of the immune response. Blood. 2008;112:3704–12. doi: 10.1182/blood-2008-06-160945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Valenzuela J, Schmidt C, Mescher M. The roles of IL-12 in providing a third signal for clonal expansion of naive CD8 T cells. J Immunol. 2002;169:6842–9. doi: 10.4049/jimmunol.169.12.6842. [DOI] [PubMed] [Google Scholar]
- 34.Szabo SJ, Kim ST, Costa GL, et al. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–69. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 35.Li Q, Eppolito C, Odunsi K, Shrikant PA. IL-12-programmed long-term CD8+ T cell responses require STAT4. J Immunol. 2006;177:7618–25. doi: 10.4049/jimmunol.177.11.7618. [DOI] [PubMed] [Google Scholar]
- 36.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–96. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 37.Curtsinger JM, Lins DC, Johnson CM, Mescher MF. Signal 3 tolerant CD8 T cells degranulate in response to antigen but lack granzyme B to mediate cytolysis. J Immunol. 2005;175:4392–9. doi: 10.4049/jimmunol.175.7.4392. [DOI] [PubMed] [Google Scholar]
- 38.Agarwal P, Raghavan A, Nandiwada SL, et al. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J Immunol. 2009;183:1695–704. doi: 10.4049/jimmunol.0900592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dobrzanski MJ, Reome JB, Dutton RW. Therapeutic effects of tumor-reactive type 1 and type 2 CD8+ T cell subpopulations in established pulmonary metastases. J Immunol. 1999;162:6671–80. [PubMed] [Google Scholar]
- 40.Hinrichs CS, Kaiser A, Paulos CM, et al. Type 17 CD8+ T cells display enhanced antitumor immunity. Blood. 2009;114:596–9. doi: 10.1182/blood-2009-02-203935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muranski P, Boni A, Antony PA, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008;112:362–73. doi: 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]