

Abstract
Thalidomide and lenalidomide constitute an important part of effective myeloma therapy. Recent data from the Intergroup Francophone du Myélome, Cancer and Leukemia Group B, and Gruppo Italiano Malattie Ematologiche dell Adulto MM-015 trials suggest that lenalidomide maintenance therapy is associated with a higher incidence of second primary malignancies (SPMs), including both hematologic and solid malignancies. In the present study, we analyzed data from the Total Therapy 2 (TT2) trial, along with the 2 Total Therapy 3 (TT3) trials. TT2 patients were assigned randomly to either a control group (no thalidomide) or to the experimental group (thalidomide during induction, between transplantations, and during consolidation and maintenance). The 2 TT3 trials used thalidomide and bortezomib during induction, before and in consolidation after tandem melphalan-based transplantation; TT3A applied VTD (bortezomib, thalidomide, dexamethasone) in the first year of maintenance and TD for 2 more years, whereas TT3B used VRD (bortezomib, lenalidomide, dexamethasone) maintenance for 3 years. The cumulative incidence of SPMs did not differ significantly among the TT trial components when measured from enrollment (P = .78) or from initiation of maintenance (P = .82). However, a pairwise comparison of the TT2 arms suggested a lower incidence of hematologic SPMs in the thalidomide maintenance arm (hazard ratio = 0.38; P = .09). These trials are registered at www.clinicaltrials.gov as NCT00573391 (TT2), NCT00081939 (TT3A), and NCT00572169 (TT3B).
Introduction
The past decade has seen a remarkable increase in overall survival for patients with multiple myeloma (MM), which is attributable to the introduction of the proteasome inhibitor bortezomib (V) and the immunomodulatory drugs thalidomide (T) and lenalidomide (R) in combination with dexamethasone (D) in the frontline setting.1 Several clinical trials have investigated whether it is safe and efficacious to use V, T, and R as maintenance therapy in both transplantation-eligible2–4 and transplantation-ineligible5,6 patients. This question was answered for T showing significant benefit initially in extending progression-free survival and, with longer follow-up, overall survival (ref). Three large randomized phase 3 trials (IFM 0502, Cancer and Leukemia Group B [CALGB] 100104, and GEMEMA MM-015) demonstrated improved progression-free survival in patients receiving R compared with the control arms.5,7,8 However, recent studies have also suggested that R maintenance may be associated with a higher frequency of myeloid and lymphoid malignancies, although the overall benefit from R may outweigh this risk (Table 1). Because we had used T maintenance in one arm of the Total Therapy 2 (TT2) trial9 and in the first Total Therapy 3 (TT3A) trial,10 and R maintenance with TT3B,11 a comparison of these successive, otherwise similar trials enrolling 668, 303, and 177 patients, respectively, offered the unique opportunity to address the issue of second primary malignancies (SPMs) relative to the use of T, VTD, and VRD in maintenance.
Table 1.
Summary of published data on SPMs in MM patients
| Study (median follow-up) | Treatment schedule | Overall SPMs | % SPMs |
|---|---|---|---|
| IFM 2005-027 (24 mo) | R | 17/299 | 5.5% |
| Placebo | 3/292 | 1% | |
| CALGB 1001048 (28 mo) | R | 15/231 | 6.5% |
| Placebo | 6/229 | 2.6% | |
| MM-015 9 (30 mo) | MPR-R/MPR | 11/335 | 3.1% |
| Placebo | 2/154 | 1.3% |
M indicates melphalan; and P, prednisone.
Methods
The details of the TT2, TT3A, and TT3B trials and patient outcomes have been reported previously.9–11 Briefly, trial TT2 included 668 patients enrolled between October 1998 and February 2004 with a median follow-up of 112 months. Patients were randomized to a control (T−) or thalidomide (T+) arm. Both arms called for 4 cycles of induction with D(T)–PACE (dexamethasone and thalidomide plus 4-day continuous infusions of cisplatin, doxorubicin, cyclophosphamide, and etoposide), followed by a melphalan (200 mg/m2)–based tandem autologous transplantation and 4 cycles of dose-reduced D(T)–PACE consolidation. Maintenance was with IFN-α-2b in both arms until progression, and dexamethasone pulsing was applied in year 1. T+ patients continued on T until progression or unacceptable toxicity.
TT3A included 303 patients and TT3B 177 patients enrolled between January 2004 and July 2006 and November 2006 and August 2009, respectively, with median follow-up times of 75 and 46 months. Both used identical induction therapy with 2 cycles of VTD-PACE (bortezomib, thalidomide, dexamethasone plus 4-day continuous infusions of cisplatin, doxorubicin, cyclophosphamide, and etoposide), followed by a melphalan 200 mg/m2–based tandem autologous transplantation and 2 cycles of dose-reduced VTD-PACE consolidation. Maintenance consisted of VTD in the first year and TD for years 2 and 3 in TT3A, and TT3B used VRD for all 3 years.
All patients signed an informed consent in keeping with institutional and federal guidelines. The protocols were approved by the institutional review board and monitored by a data safety and monitoring board, and had outcome data audited by an independent team of reviewers.
For the purposes of this analysis, our myeloma database was interrogated for the development of SPM after enrollment in these 3 TT trials. This was based on frequently scheduled positron emission tomography scanning, performed at baseline and on average every 3 months in the first year and semiannually thereafter for majority of TT2 patients and all TT3A/B patients. For the early detection of potential myelodysplasia or acute myeloid leukemia, BM examinations were performed at least semiannually during maintenance and follow-up beyond maintenance, along with metaphase karyotyping and interphase FISH for the detection of MM, myelodysplasia, and acute myeloid leukemia, and acute myeloid leukemia–associated abnormalities.12,13 This uniform stringency in therapy and subsequent patient follow-up is a hallmark of the TT trials program and has allowed the investigators to make many clinically meaningful and novel observations.
χ2 and Fisher exact tests were used to compare baseline characteristics among protocols.14 Cox proportional hazard regression was used to model associations between baseline covariates and onset of SPM.15 Cumulative incidence of SPMs in the presence of death as a competing risk was estimated using the method described by Gooley et al.16
Results
As reported previously, baseline characteristics were similar across the 3 trials except for differences in the prevalence of decreased albumin (< 3.5g/dL), elevated β-2-microglobulin (≥ 3.5mg/L), and gene-expression profiling 70- and 80-gene risk status (Table 2). To account for differences in follow-up time among the 3 trials, we implemented cumulative incidence analyses with death as a competed risk to compare SPM incidence. No difference in the incidence of any SPM was observed among the 4 trial components, between the 2 TT2 trial arms, or between the 2 TT3 trials whether examined from induction of therapy or from initiation of maintenance therapy (Figure 1).
Table 2.
Patient characteristics for the TT2/TT3 trials
| Factor | Overall | TT2 − T | TT2 + T | TT3A | TT3B | P |
|---|---|---|---|---|---|---|
| Female sex | 451/1148 (39%) | 135/345 (39%) | 137/323 (42%) | 110/303 (36%) | 69/177 (39%) | .481 |
| Age ≥ 65 y | 266/1148 (23%) | 72/345 (21%) | 64/323 (20%) | 84/303 (28%) | 46/177 (26%) | .061 |
| Albumin < 3.5 g/dL | 275/1143 (24%) | 59/343 (17%) | 60/321 (19%) | 78/303 (26%) | 78/176 (44%) | < .001 |
| B2M ≥ 3.5 mg/L | 478/1145 (42%) | 126/345 (37%) | 117/323 (36%) | 136/303 (45%) | 99/174 (57%) | < .001 |
| LDH ≥ 190 U/L | 328/1145 (29%) | 98/344 (28%) | 106/322 (33%) | 81/303 (27%) | 43/176 (24%) | .175 |
| Cytogenetic abnormalities | 367/1136 (32%) | 104/339 (31%) | 93/322 (29%) | 100/302 (33%) | 70/173 (40%) | .058 |
| GEP70 high-risk | 123/792 (16%) | 20/176 (11%) | 26/175 (15%) | 40/275 (15%) | 37/166 (22%) | .039 |
| GEP80 high-risk | 56/792 (7%) | 6/176 (3%) | 9/175 (5%) | 20/275 (7%) | 21/166 (13%) | .006 |
| Transplantation 1 | 1020/1148 (89%) | 292/345 (85%) | 278/323 (86%) | 285/303 (94%) | 165/177 (93%) | < .001 |
| Transplantation 2 | 834/1148 (73%) | 233/345 (68%) | 213/323 (66%) | 252/303 (83%) | 136/177 (77%) | < .001 |
Data are shown as n/N, where n indicates the number of patients with the factor and N the number of patients with valid data for the factor.
B2M indicates β-2-microglobulin; LDH, lactate dehydrogenase; and GEP, gene-expression profiling.
Figure 1.
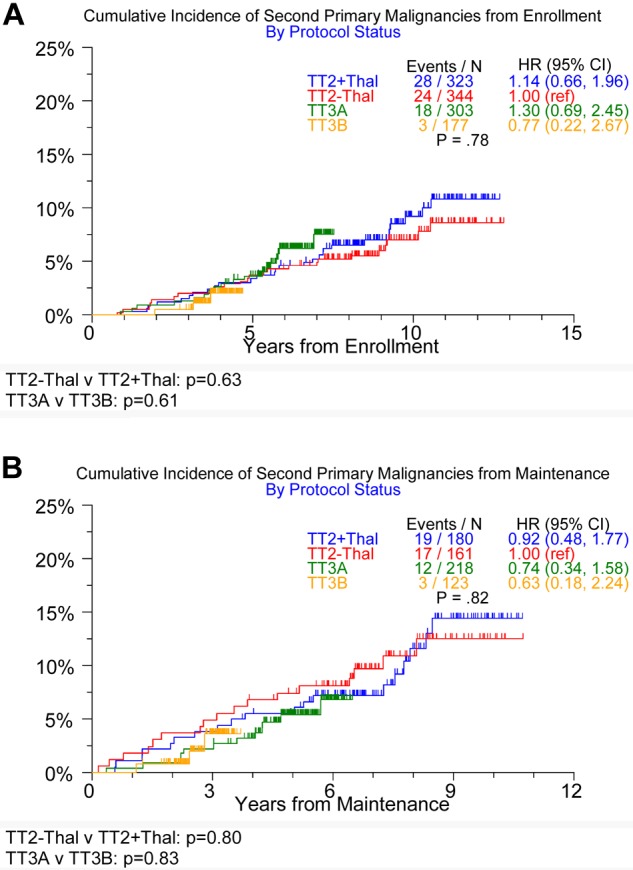
Timing of onset of all SPMs in TT2, TT3A, and TT3B. From enrollment (A) and from maintenance (B).
Examination of the incidence of SPMs within a subtype revealed no differences in the development of hematologic or solid-tissue SPMs between the 2 TT3 trials or in the development of solid-tissue SPMs between the 2 TT2 arms (Table 3). There was, however, a trend toward an increased risk of solid SPMs (Figure 2) from initiation of maintenance therapy for the TT2 + T arm compared with the TT2 − T arm (P = .23 from enrollment; P = .31 from maintenance). Interestingly, although no differences were observed in the cumulative incidence of hematologic malignancies between the TT2 arms and the 2 TT3 trials from enrollment, a trend for lower incidence of hematologic malignancies was observed in TT2 + T compared with TT2 − T from initiation of maintenance (Figure 3). Cox regression analysis identified age ≥ 65 years and the presence of cytogenetic abnormalities as being associated with an increased incidence of SPMs (hazard ratio = 2.06, P = .004, and hazard ratio = 1.64, P = .05, respectively; Table 4) when modeled from trial enrollment; no variables were linked to SPMs when the analysis was performed from the start of maintenance.
Table 3.
Types of SPMs observed
| TT2 − T | TT2 + T | TT3A | TT3B | Total | |
|---|---|---|---|---|---|
| Total patients enrolled | 345 | 323 | 303 | 177 | 1148 |
| Total person-years | 2164 | 2135 | 1521 | 577 | 6397 |
| Solid-tissue SPMs | 13 | 10 | 11 | 2 | 36 |
| ALL | 0 | 0 | 1 | 0 | 1 |
| AML | 3 | 0 | 3 | 0 | 6 |
| NHL | 0 | 1 | 2 | 0 | 3 |
| MDS | 10 | 9 | 5 | 2 | 26 |
| Hematological SPMs | 11 | 18 | 7 | 1 | 37 |
| Bladder cancer | 1 | 1 | 1 | 0 | 3 |
| Breast cancer | 2 | 3 | 1 | 0 | 6 |
| Colon cancer | 0 | 2 | 0 | 1 | 3 |
| Lung cancer | 0 | 1 | 0 | 0 | 1 |
| Nonmelanoma cancer | 4 | 2 | 2 | 0 | 8 |
| Prostate cancer | 2 | 5 | 2 | 0 | 9 |
| Renal cancer | 0 | 2 | 1 | 0 | 3 |
| Thyroid cancer | 2 | 2 | 0 | 0 | 4 |
| Total | 24 | 28 | 18 | 3 | 73 |
Note that these frequencies should not be used as a means to assess by-protocol differences due to large differences in follow-up. For a by-protocol comparison, see Figures 2 through 4.
ALL indicates acute lymphoblastic leukemia; AML, acute myeloid leukemia; NHL, non-Hodgkin lymphoma; and MDS, myelodysplastic syndrome.
Figure 2.
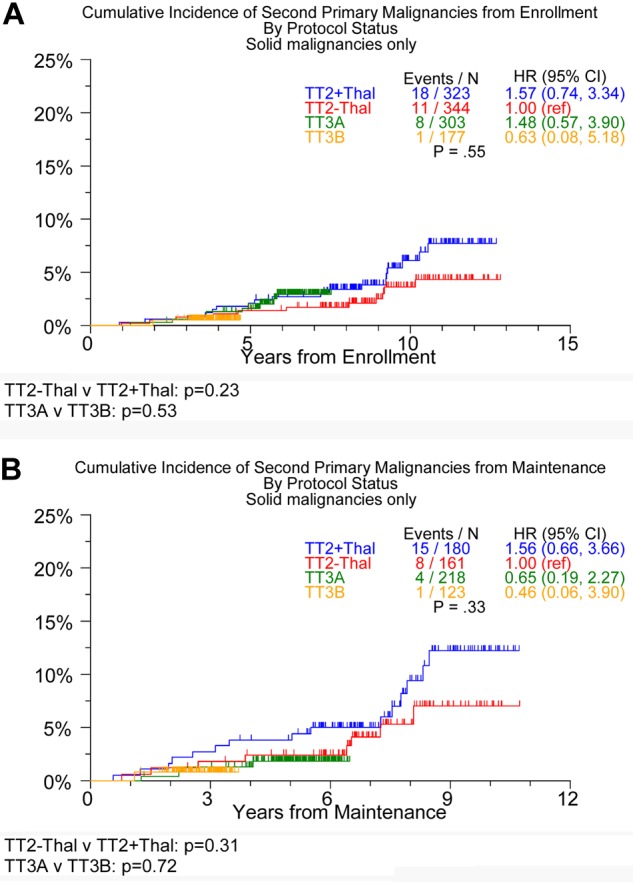
Timing of onset of solid-tissue SPMs in TT2, TT3A, and TT3B. From enrollment (A) and from maintenance (B).
Figure 3.
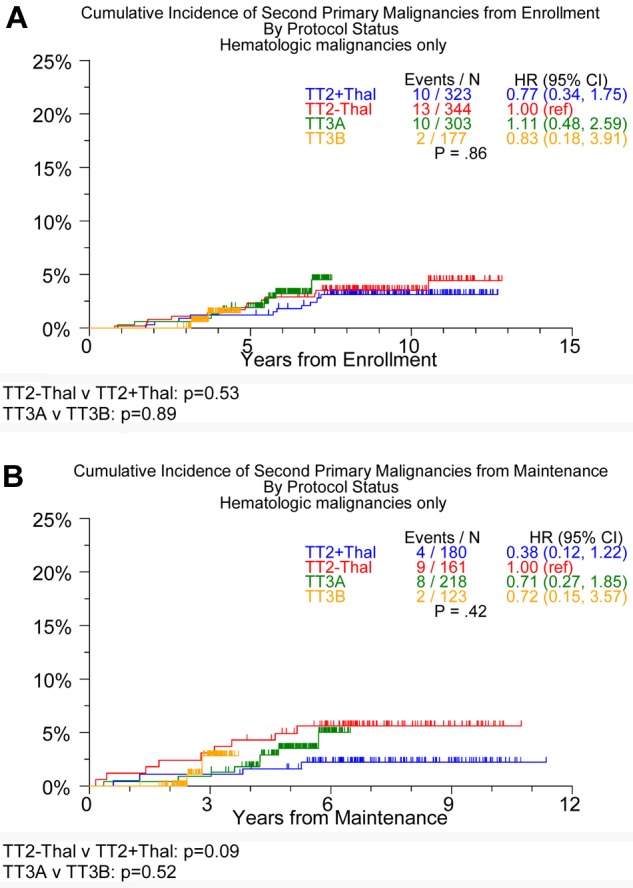
Timing of onset of hematologic SPMs in TT2, TT3A, and TT3B. From enrollment (A) and from maintenance (B).
Table 4.
Univariate Cox proportional hazards regression modeling time from enrollment to SPMs
| Variable | n/N (%) | Time to SPMs from enrollment |
|
|---|---|---|---|
| HR (95% CI) | P* | ||
| Female sex | 451/1148 (39%) | 0.77 (0.47-1.25) | .288 |
| White race | 1008/1148 (88%) | 2.65 (0.97-7.25) | .058 |
| Age ≥ 65 y | 266/1148 (23%) | 2.06 (1.25-3.39) | .004 |
| IgA isotype | 270/1143 (24%) | 0.83 (0.46-1.48) | .524 |
| IgG isotype | 624/1143 (55%) | 0.85 (0.54-1.34) | .487 |
| Albumin < 3.5 g/dL | 275/1143 (24%) | 0.66 (0.33-1.33) | .249 |
| B2M ≥ 3.5 mg/L | 478/1145 (42%) | 1.03 (0.64-1.68) | .895 |
| B2M > 5.5 mg/L | 238/1145 (21%) | 1.43 (0.80-2.56) | .233 |
| IPSS Stage I | 565/1141 (50%) | 1.24 (0.76-2.00) | .392 |
| IPSS Stage II | 339/1141 (30%) | 0.60 (0.33-1.10) | .098 |
| IPSS Stage III | 238/1145 (21%) | 1.43 (0.80-2.56) | .233 |
| CRP ≥ 8 mg/L | 423/1136 (37%) | 0.85 (0.51-1.39) | .512 |
| Creatinine ≥ 2.0 mg/dL | 96/1133 (8%) | 1.81 (0.87-3.79) | .113 |
| Hb < 10 g/dL | 312/1146 (27%) | 0.90 (0.52-1.57) | .716 |
| LDH ≥ 190 U/L | 328/1145 (29%) | 1.30 (0.78-2.16) | .309 |
| Platelet count < 150 × 109/L | 171/1146 (15%) | 0.53 (0.22-1.33) | .177 |
| Cytogenetic abnormalities | 367/1136 (32%) | 1.64 (1.01-2.66) | .045 |
| GEP70 high-risk | 123/792 (16%) | 0.71 (0.22-2.30) | .571 |
| GEP80 high-risk | 56/792 (7%) | 0.68 (0.09-4.92) | .701 |
| K/L FLC ratio | N = 478 | 1.00 (1.00-1.00) | .515 |
| Kappa FLC | N = 614 | 1.00 (1.00-1.00) | .464 |
| Kappa FLC > 168 | 70/614 (11%) | 1.23 (0.43-3.51) | .697 |
| Lambda FLC | N = 615 | 1.00 (1.00-1.00) | .456 |
| Thalidomide | 803/1148 (70%) | 1.16 (0.71-1.89) | .561 |
HR indicates hazard ratio; 95% CI, 95% confidence interval; B2M, β-2-microglobulin; IPSS, International Prognostic Scoring System; CRP, C-reactive protein; Hb, hemoglobin; LDH, lactate dehydrogenase; GEP, gene-expression profiling; and FLC, free light chain.
P value from Wald χ2 test in cox regression. All univariate P values are reported regardless of significance. The multivariate model uses stepwise selection with an entry level of P = .1 and the variable remains if it meets the P = .05 level. A multivariate P value > .05 indicates a variable forced into the model with significant variables chosen using stepwise selection.
Discussion
In December 2010, the IFM for the first time raised the question of whether SPMs are linked to L therapy. Such SPM excess in MM patients was also reported by others subsequently and has been reviewed comprehensively by Thomas et al across the decades since the 1960s.17 Significant differences between the quoted trials in the incidence of solid versus hematologic malignancies, the use of induction treatment, and application or not of melphalan-based autotransplantations prevent definitive conclusions, especially because SPMs had not been observed in earlier R trials for advanced myeloma or in the newly diagnosed setting. Furthermore differences in length or intensity of follow-up between treatment and control arms, as well as reporting bias from different centers in multicenter trials, may have led to underreporting of certain malignancies. However, in light of the recent observation of improved overall survival in the CALGB trial, a consensus has emerged that the benefits of R maintenance outweighs its detriments regarding SPM potential. Palumbo et al, who calculated the risk of death or progression from myeloma at 2 years as being 45% on the R arm compared with 84% on the control arm, found that the risk of developing SPMs was 3% and < 1%, respectively. Whereas it is not randomized, our successive TT3 trials enrolled sizable patient cohorts and their main difference pertained to the maintenance phase. The prospectively planned use of positron emission tomography scanning as part of myeloma staging and assessment of response and relapse represented an ideal circumstance for detecting SPMs without bias and with a high level of sensitivity in patients followed at our center at intervals of 3-6 months during the 3-year maintenance phase and then at 6-12 months thereafter. The overall incidence of SPMs in our TT trials (6.4%) was comparable with those reported for IFM 2005-02 (5.5%) and CALGB 100104 (6.5%), both of which also included melphalan-based transplantations. The 2 randomized phase 3 trials (CALGB 100104 and IFM 2005-02) showed clearly the increased risk of SPMs associated with R maintenance. Our present data, albeit nonrandomized and with variable follow-up times, do not indicate significant differences in SPMs when combining T or R with VD during maintenance. However, it will be important to monitor closely for any emerging differences as patient follow-up times increase.
Footnotes
There is an Inside Blood commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: S.Z.U. designed the study and wrote the manuscript; S.Z.U., R.S., A.H., N.C., N.P., J.C., and B.B. analyzed the data; S.Z.U., C.J.H., B.N., S.W., Y.A.S., N.A., S.A., F.v.R., and B.B. contributed patients; and all authors reviewed the manuscript.
Conflict-of-interest disclosure: S.Z.U. is a consultant for Celgene, Millennium, and Onyx and has received research funding from Onyx and Celgene and speaking honoraria from Celgene. B.B. has received research funding from Celgene and Novartis, is a consultant for Celgene and Genzyme, has received speaking honoraria from Celgene and Millennium, and is a coinventor on patents and patent applications related to use of gene-expression profiling in cancer treatment. The remaining authors declare no competing financial interests.
Correspondence: Saad Z. Usmani, MD, FACP, Assistant Professor of Medicine, Myeloma Institute for Research & Therapy, University of Arkansas for Medical Sciences, 4301 W Markham St, Little Rock, AR 72205-7199; e-mail: susmani@uams.edu.
References
- 1.Kyle RA, Rajkumar SV. Multiple Myeloma. N Engl J Med. 2004;351(18):1860–73. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 2.Attal M, Harousseau JL, Leyvraz S, et al. Maintenance therapy with thalidomide improves survival in patients with multiple myeloma. Blood. 2006;108(10):3289–3294. doi: 10.1182/blood-2006-05-022962. [DOI] [PubMed] [Google Scholar]
- 3.Abdelkefi A, Ladeb S, Torjman L, et al. Single autologous stem-cell transplantation followed by maintenance therapy with thalidomide is superior to double autologous transplantation in multiple myeloma: results of a multicenter randomized clinical trial. Blood. 2008;111(4):1805–1810. doi: 10.1182/blood-2007-07-101212. [DOI] [PubMed] [Google Scholar]
- 4.Palumbo A, Bringhen S, Rossi D, et al. A prospective, randomized phase III study of bortezomib, melphalan, prednisone and thalidomide (VMPT) versus bortezomib, melphalan and prednisone (VMP) in elderly newly diagnosed myeloma patients [abstract]. Blood (ASH Annual Meeting Abstracts) 2008;114:652. [Google Scholar]
- 5.Palumbo A, Hajek R, Delforge M, et al. Continuous lenalidomide treatment for newly diagnosed multiple myeloma. N Engl J Med. 2012;366(19):1759–1769. doi: 10.1056/NEJMoa1112704. [DOI] [PubMed] [Google Scholar]
- 6.Mateos MV, Oriol A, Martinez J, et al. A prospective, multicenter, randomized, trial of bortezomib/melphalan/prednisone (VMP) versus bortezomib/thalidomide/prednisone (VTP) as induction therapy followed by maintenance treatment with bortezomib/thalidomide (VT) versus bortezomib/prednisone (VP) in elderly untreated patients with multiple myeloma older than 65 years [abstract]. Blood (ASH Annual Meeting Abstracts) 2009;114:3. [Google Scholar]
- 7.Attal M, Lauwers VC, Marit G, et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1782–1791. doi: 10.1056/NEJMoa1114138. [DOI] [PubMed] [Google Scholar]
- 8.McCarthy PL, Owzar K, Hofmeister CC, et al. Lenalidomide after stem-cell transplantation for multiple myeloma. N Engl J Med. 2012;366(19):1770–1781. doi: 10.1056/NEJMoa1114083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barlogie B, Tricot G, Anaissie E, et al. Thalidomide and hematopoietic-cell transplantation for multiple myeloma. N Engl J Med. 2006;354(10):1021–30. doi: 10.1056/NEJMoa053583. [DOI] [PubMed] [Google Scholar]
- 10.Barlogie B, Anaissie E, van Rhee F, et al. Incorporating bortezomib into upfront treatment for multiple myeloma: early results of total therapy 3. Incorporating bortezomib into upfront treatment for multiple myeloma: early results of total therapy 3. Br J Haematol. 2007;138(2):176–85. doi: 10.1111/j.1365-2141.2007.06639.x. [DOI] [PubMed] [Google Scholar]
- 11.Nair B, van Rhee F, Shaughnessy JD, Jr, et al. Superior results of Total Therapy 3 (2003-33) in gene expression profiling-defined low-risk multiple myeloma confirmed in subsequent trial 2006-66 with VRD maintenance. Blood. 2010;115(21):4168–4173. doi: 10.1182/blood-2009-11-255620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barlogie B, Tricot G, Haessler J, et al. Cytogenetically defined myelodysplasia after melphalan-based autotransplantation for multiple myeloma linked to poor hematopoietic stem-cell mobilization: the Arkansas experience in more than 3000 patients treated since 1989. Blood. 2008;111(1):94–100. doi: 10.1182/blood-2007-06-097444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papanikolaou X, Barlogie B, Usmani SZ. Therapy-related myeloid malignancies in myeloma. Mediterr J Hematol Infect Dis. 2011;3(1):e2011047. doi: 10.4084/MJHID.2011.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agresti A. A survey of exact inference for contingency tables. Stat Sci. 1992;7(1):131–153. [Google Scholar]
- 15.Cox DR. Regression tables and life tables. J R Stat Soc. 1972;34(2):187–202. [Google Scholar]
- 16.Gooley T, Leisenring W, Crowley J, Storer B. Estimation of failure probabilities in the presence of competing risks: new representations of old estimators. Stat Med. 1999;18(6):695–706. doi: 10.1002/(sici)1097-0258(19990330)18:6<695::aid-sim60>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 17.Thomas A, Mailankody S, Korde N, et al. Second malignancies after multiple myeloma: from 1960s to 2010s. Blood. 2012;119(12):2731–2737. doi: 10.1182/blood-2011-12-381426. [DOI] [PMC free article] [PubMed] [Google Scholar]