

Abstract
The anticoagulant serpin, protein Z-dependent protease inhibitor (ZPI), is catalytically activated by its cofactor, protein Z (PZ), to regulate the function of blood coagulation factor Xa on membrane surfaces. The X-ray structure of the ZPI-PZ complex has shown that PZ binds to a unique site on ZPI centered on helix G. In the present study, we show by Ala-scanning mutagenesis of the ZPI-binding interface, together with native PAGE and kinetic analyses of PZ binding to ZPI, that Tyr240 and Asp293 of ZPI are crucial hot spots for PZ binding. Complementary studies with protein Z–protein C chimeras show the importance of both pseudocatalytic and EGF2 domains of PZ for the critical ZPI interactions. To understand how PZ acts catalytically, we analyzed the interaction of reactive loop–cleaved ZPI (cZPI) with PZ and determined the cZPI X-ray structure. The cZPI structure revealed changes in helices A and G of the PZ-binding site relative to native ZPI that rationalized an observed 6-fold loss in PZ affinity and PZ catalytic action. These findings identify the key determinants of catalytic activation of ZPI by PZ and suggest novel strategies for ameliorating hemophilic states through drugs that disrupt the ZPI-PZ interaction.
Introduction
Protein Z (PZ)–dependent protease inhibitor (ZPI), an anticoagulant protein of the serpin superfamily, is activated by its cofactor, PZ, to rapidly and specifically inhibit membrane-associated coagulation factor Xa. ZPI also rapidly inhibits factor XIa, but PZ activation is not required for this inhibition and cleaved ZPI is the predominant product.1–3 The importance of ZPI for regulation of factor Xa activity is evident from the observation that ZPI or PZ deficiency results in an increased risk of thrombosis, especially when combined with the factor V Leiden mutation or other risk factors.4–6 ZPI circulates in blood plasma as a tight complex with PZ,7 a vitamin K–dependent protein with a domain structure similar to that of factors VII, IX, and X and protein C, but with a nonfunctional protease domain.8 Biochemical and structural studies have suggested that PZ promotes ZPI inhibition of membrane-associated factor Xa primarily though a bridging mechanism in which the binding of PZ to ZPI enables the ZPI-PZ complex to bind to a membrane surface and encounter membrane-bound factor Xa.9,10 PZ acts catalytically in promoting the ZPI–factor Xa reaction, as evidenced by the observation that once ZPI forms an inhibited complex with factor Xa, it dissociates from PZ.11 This catalytic action may be important for sparing PZ, because normal plasma concentrations of PZ are limiting relative to ZPI.2,12
The X-ray structure of the ZPI-PZ complex was determined recently by 2 different groups of investigators.9,10 The structure shows that ZPI interacts with PZ through 3 clusters of salt bridges involving residues D74, D238, K239, and D293 and through hydrophobic interactions involving residues M71 and Y240 at a unique site centered on helix G. In the present study, we performed Ala-scanning mutagenesis of these 6 ZPI contact residues in the binding interface to determine their relative contributions to PZ binding. Kinetic analysis of the effect of these mutations on the cofactor-dependent ZPI–factor Xa reaction, together with native PAGE analysis of PZ binding to the ZPI mutants, revealed large differential contributions of these residues to PZ binding, with only 2 residues accounting for the bulk of the binding energy. Complementary studies with PZ chimeras confirmed that the key ZPI residues interacted with both pseudocatalytic and EGF2 domains of PZ. Kinetic competition studies showed that reactive loop–cleaved ZPI (cZPI) bound PZ with a 6-fold lower affinity than the native serpin. Solution of the crystal structure of cZPI and its alignment with native ZPI in the ZPI-PZ complex revealed that changes in helices A and G of the PZ-binding site accounted for the loss in ZPI affinity for PZ. These findings provide new insights into the mechanism of catalytic activation of ZPI by PZ and have implications for the design of small molecules that could disrupt the ZPI-PZ interface and potentially ameliorate hemophilic states.
Methods
Proteins
Plasma-derived human factor Xa, ZPI, PZ, and factor XIa were obtained from Enzyme Research Laboratories or were purified as described previously.3 Recombinant human ZPI and ZPI mutants were expressed in baculovirus-infected insect cells and purified as described previously.9,11 A human ZPI Y387R variant and wild-type mouse ZPI were expressed in Escherichia coli and purified as described previously.10 The reactive center loop cleaved form (cZPI) was prepared from a Y387R ZPI variant with enhanced substrate reactivity1,9 by reacting with catalytic factor Xa followed by purification.10 DNA mutagenesis was carried out by PCR using the Quick Exchange kit (Stratagene). Mutations were confirmed by DNA sequencing. Molar concentrations of ZPIs were determined from the absorbance at 280 nm using a calculated extinction coefficient of 31 525M−1s−1.13 Protease concentrations were determined by standard activity assays that were calibrated based on active-site titrations.11
Recombinant PZ and protein Z/protein C (PZ/PC) chimeric proteins were produced by manipulating PZ cDNA to encode the sequence for PZ/PC chimeric proteins using standard techniques. The pre-pro-leader sequence of PZ was replaced with that of prothrombin to improve protein expression levels, and the γ-carboxyglutamic acid domain (Gla), epidermal growth factor 1 domain (EGF1), epidermal growth factor 2 domain (EGF2), or Gla through EGF2 domains (Gla-EGF2) of PZ were replaced with the corresponding domains of PC. cDNAs encoding the PZ/PC chimeric proteins and wild-type PZ were engineered into the pcDNA4/TO (Invitrogen) vector with placement of a 6-His tag at their C-termini and transfected (lipofectamine; Invitrogen) into T-Rex-293 cells (Invitrogen). Cells were cultured in DMEM with tetracycline-reduced FBS (10%, Invitrogen), l-glutamine (2mM), vitamin K (10 μg/mL), basticidin (5 μg/mL), and Zeocin (300 μg/mL). At confluence, cells were washed and the media replaced with FreeStyle 293 expression medium (Invitrogen) with tetracycline (1 μg/mL) supplemented with the same levels of glutamine, vitamin K, basticidin, and Zeocin. After 24 hours, conditioned medium was collected, benzamidine added (5mM), and the expressed proteins were isolated by metal chelate (Talon; Clontech), Resource Q, and Superose 12 (GE Health Care) chromatography. The chimeric proteins were expressed at levels similar to PZ wild-type and reacted with the following available mAbs in a fashion equivalent to that of native PZ or PC. The mAbs tested included: anti-PZ EGF1 domain (mAb 2048.ED9), anti-PZ pseudocatalytic domain (mAb 2306.AC5, mAb 2306.BF12, mAb 4260.2B9, and mAb 4260.5F5), anti-PC Gla domain (mAb CaC-11), and anti-PC EGF1 domain (mAb 1518.EG9).
Phospholipids
Small, unilamellar phospholipid vesicles were prepared from a 7:3 mixture (by weight) of dioleyl phosphatidylcholine and dioleoyl phosphatidylserine (Avanti Polar Lipids) as described previously.9,11 The phospholipid concentration was determined by colorimetric assay.14
Kinetics of ZPI-FXa reactions
The rate of factor Xa inactivation by wild-type and mutant ZPIs in the absence and presence of cofactors was measured under pseudo-first-order conditions in 50mM HEPES, 0.1M NaCl, 0.1% PEG 8000 buffer, pH 7.4, at 25°C by discontinuous or continuous assays as described previously.9,11 In the discontinuous assay, reactions were quenched at various times by 10- to 20-fold dilution into substrate (100μM Spectrozyme FXa [American Diagnostica] or 50μM Pefafluor FXa [Centerchem]) and the residual factor Xa activity was measured from the initial rate of absorbance change (405 nm) or fluorescence change (λex 380 nm, λem 440 nm). Quench solutions contained 10mM EDTA for reactions with calcium. Because of the slow rate of dissociation of the ZPI–factor Xa complex during the assay, initial velocities of substrate hydrolysis by residual factor Xa were determined from computer fits by a second-order polynomial function.11 Observed pseudo-first-order rate constants (kobs) were obtained by fitting the loss of protease activity by an exponential decay function with a nonzero end point that reflected < 10% degraded protease more resistant to inhibition. For continuous assays, reactions included 50μM Pefafluor FXa substrate and progress curves of protease inhibition were obtained by continuously monitoring the decrease in rate of substrate hydrolysis to a steady-state level of inhibited enzyme activity under conditions in which substrate consumption was < 10% and the rate of hydrolysis was linear in the absence of inhibitor. Reaction progress curves were fit over 5-10 half-lives by an exponential plus steady-state equation to obtain kobs.11
Binding of wild-type and mutant ZPIs to PZ was quantitated from kinetic titrations of the accelerating effect of PZ on kobs for the ZPI–factor Xa reaction in the presence of 25μM lipid and 1mM calcium. To ensure that kobs was linearly dependent on the ZPI-PZ complex concentration, reactions were done at ZPI-PZ complex concentrations below the KM of approximately 50nM for PZ-ZPI-FXa ternary complex formation.11 To avoid lipid oxidation and achieve reproducible reaction rates, frozen aliquots of phospholipid vesicles prepared by sonication were thawed and used without additional sonication. Second-order association rate constants for reactions in the absence (kass,uncat) or presence of PZ, lipid, and calcium cofactors (kass,cat) and the KD for the ZPI-PZ complex interaction were obtained by fitting the dependence of kobs on the ZPI or PZ concentration by the equation:
kobs = kdiss + (kass,cat [ZPI-PZ] + kass,uncat [ZPI]f)/(1 + [S]o/KM,S)
where kdiss is the first-order rate constant for ZPI-FXa complex dissociation, [ZPI-PZ] and [ZPI]f (free ZPI) are functions of total ZPI and PZ concentrations and the KD for the ZPI-PZ interaction as given by the quadratic equilibrium binding equation,11 [S]o is the fluorogenic substrate concentration, and KM,S is the Michaelis constant for substrate hydrolysis by factor Xa. A KM,S of 119 ± 3μM was determined under the conditions of the kinetic experiments. For ZPI variants with high PZ affinity, the uncatalyzed reaction term and kdiss could be neglected. For ZPI variants with low PZ affinity, kass,uncat and kdiss were fixed at values determined independently for the ZPI–factor Xa reaction in the absence of PZ but in the presence of 25μM lipid and 1mM CaCl2, and kass,cat was assumed to be equal to the wild-type value to allow fitting of KD.
Experiments examining the competitive effect of cZPI on the PZ-dependent reactions of native ZPI or M71A ZPI with factor Xa were done at 5.5nM ZPI, 5.5nM PZ, variable cZPI (0-500nM), 0.1nM factor Xa, 25μM lipid, and 5mM CaCl2. Reactions were initiated by adding a mixture of ZPI and cZPI to a preincubated solution of PZ, factor Xa, lipid, and calcium, and progress curves were measured by the discontinuous assay to obtain kobs. The decrease in kobs as a function of cZPI concentration was fit by the kinetic equation above except that the quadratic equation for the [ZPI-PZ] term was replaced by the cubic equation that defines the ZPI-PZ complex concentration in the presence of a competitor.15 M71A ZPI–factor Xa reaction data were fit by fixing the KD for the M71A ZPI-PZ interaction at the measured value and fitting the KD for the cZPI-PZ interaction as a parameter. Wild-type ZPI–factor Xa reaction data were fit by fixing the cZPI-PZ interaction KD and fitting the wild-type ZPI-PZ interaction KD as a parameter. Stoichiometric factors for ZPI and cZPI interactions with PZ were assumed to be 1. Simulations of ZPI-PZ complex reactions with factor Xa were done using Explorer Version 2.5 software (KinTech).
Stoichiometry of ZPI-protease reactions
The stoichiometry of inhibition of factor Xa or factor XIa by ZPI was determined by end point titrations of approximately 100nM protease with increasing molar ratios of ZPI to protease from 3:1 to 8:1 in the absence or presence of PZ (equimolar with ZPI), phospholipid (25 μm), and CaCl2 (5mM) under the conditions of kinetic studies.9,11
PAGE
Nondenaturing PAGE was performed at 4°C using the Laemmli buffer system16 with 5.5% gels and running times of 2-5 hours at 100-150 V. SDS-PAGE used the Laemmli buffer system and 7.5%-10% polyacrylamide gels. Protein bands were detected by Coomassie blue staining.
Characterization of PZ/PC chimeric proteins
Plasma PZ, recombinant wild-type PZ (rPZ), and the rPZ/PC chimeric proteins were tested for their ability to accelerate the inhibition of factor Xa (0.5nM) by plasma ZPI (40nM) in the presence of phospholipid (15μM rabbit brain cephalin; Pentapharm) and Ca2+ (4mM) in 0.1M NaCl, 0.02M HEPES, pH 7.4, with 1 mg/mL of BSA, similar to previous studies.2 PZ, factor Xa, phospholipid, and Ca2+ were incubated for 3 minutes at 37°C and then ZPI was added to start the reaction. Alternatively, ZPI, PZ, phospholipid, and Ca2+ were incubated for 3 minutes at 37° and then factor Xa was added. After 60 seconds, a sample (50 μL) was added to prewarmed buffer (100 μL) and CaCl2 (25mM, 50 μL) and the clotting time was measured after the addition of factor X–deficient plasma (George King Biomedical) in a fibrometer (BBL). Remaining factor Xa activity was determined based on a factor Xa standard curve.
The rPZ/PC chimeric proteins were tested for their ability to bind ZPI with a microtiter-plate assay. rPZ/PC proteins (2 μg/mL in 0.1M NaCl, 0.02M HEPES, pH 7.4) were allowed to bind to the microtiter wells (100 μL, 2 hours) followed by washing and blocking with PBS containing Tween 20 (0.05%; PBST). ZPI in PBST was then added (0-2 μg/mL, 100 μL) and incubated for 30 minutes. After washing with PBST, bound ZPI was detected using biotin-conjugated mAb 4249.3 anti-ZPI (2 μg/mL), avidin-HRP (Pierce), and 3,3′,5,5′-tetramethylbenzidine (Sigma-Aldrich) with PBST washes between steps. The same method and a mAb against the pseudocatalytic domain of PZ (mAb 2306.AC5) showed comparable binding of the PZ/PC chimeric proteins in replicate microtiter wells.
Crystallization and X-ray structure determination of ZPIs
Crystals of cZPI were grown in 1.8M sodium/potassium phosphate, pH 5, or 10% 2-propanol, 0.2M Li2SO4, 0.1M Na acetate, pH 4.5, using the sitting drop method. Crystals of mouse ZPI were grown in 4M NaCl, 50mM MES buffer, pH 6. cZPI crystals were quickly soaked in cryo-solution (the precipitant plus 15% ethylene glycol) and snap-frozen in liquid nitrogen. Mouse ZPI crystals were snap-frozen without cryo-solution. Diffraction data for cZPI and mouse ZPI were collected from single crystals at the Diamond Synchrotron stations I02 and I04-1 and processed with Mosflm Version 7.0.5 software.17 The cZPI and mouse structures were solved by molecular replacement with Phaser Version 2.3.0 software18 using the reactive loop cleaved antitrypsin structure (pdb 7API) and native human ZPI (pdb 3F1S) as search models. cZPI crystals with space groups I2 and P41 were observed, each with one copy of cZPI in the corresponding asymmetric unit. Structures were built in COOT Version 0.6.2,19 refined using Refmac Version 5.6.0117,20 and validated by MolProbity Version 3.1921 (supplemental Table 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). All residues of the I2 cZPI structure apart from 1-40 and 387 were built. In the P41 cZPI structure, residues 1-34, 115-116, 288, and 387-389 were not built due to poor density. Mouse ZPI was built with residues 47-425 (except for the 7 reactive loop residues 380-386 due to poor density).
Results
Engineering and functional activity of ZPI variants
ZPI variants were engineered in which the 3 clusters of salt bridges and hydrophobic interactions in the ZPI-PZ interface involving Met71 and Asp74 in helix A and its C-terminal extension; Asp238, Lys239, and Tyr240 in the loop connecting strands 3 and 4 of sheet C; and Asp293 in helix G were each mutated to Ala (Figure 1). A double Asp74/Asp293 mutant was also engineered. The mutants were expressed and purified as in previous studies and yielded amounts of protein comparable to wild-type.9 All mutants inhibited factor Xa or factor XIa in the absence of cofactors at a rate similar to wild-type (Figure 2A and supplemental Figure 1). Moreover, the inhibition stoichiometries for reaction of the variant ZPIs with these proteases were similar to or only modestly elevated relative to wild-type reactions (Table 1 and supplemental Table 2). These results indicated that the mutations did not perturb the cofactor-independent inhibitory function of ZPI.
Figure 1.

ZPI contact residues in the ZPI-PZ complex interface. ZPI-PZ complex structure (pdb 3F1S) with ZPI in green and PZ pseudocatalytic and EGF2 domains in cyan. The reactive loop (RCL, red), sheet A (blue), helix A (orange), and helix G (purple) of ZPI are highlighted. The 6 ZPI residues of the contact interface in helix A, helix G, and sheet C are represented in stick.
Figure 2.

Kinetic analysis of mutant ZPI reactions with factor Xa in the absence and presence of cofactors. (A) Progress curves for reactions of 480nM wild-type and mutant ZPIs as indicated with 3nM factor Xa in the absence of cofactors. (B) Progress curves for reactions of 16nM wild-type and mutant ZPIs as indicated with 0.25nM factor Xa in the presence of 13nM PZ, 25μM phospholipid, and 5mM Ca2+. Solid lines are fits by a single exponential function.
Table 1.
Kinetic constants and stoichiometries of inhibition for ZPI-factor Xa reactions in the absence and presence of cofactors and KDs for ZPI-PZ interactions
| ZPI | No cofactors |
Cofactors |
|||||
|---|---|---|---|---|---|---|---|
| kass,uncat, M−1s−1 | SI, mol I/mol E | kass,uncat × SI, M−1s−1 | kass,cat, M−1s−1 | SI, mol I/mol E | kass,cat × SI, M−1s−1* | KD ZPI-PZ, M | |
| Wild-type | 1.0 ± 0.1 × 104 | 3.6 ± 0.4 | 3.6 ± 0.8 × 104 | 3.1 ± 0.1 × 106 | 2.8 ± 0.2 | 8.7 ± 0.9 × 106 | 1.2 ± 0.1 × 10−9 |
| M71A | 8.6 ± 0.7 × 103 | 3.7 ± 0.1 | 3.2 ± 0.3 × 104 | 2.8 ± 0.3 × 106 | 2.9 ± 0.1 | 8.1 ± 1.1 × 106 | 1.7 ± 0.5 × 10−8 |
| D74A | 9.5 ± 0.1 × 103 | 4.1 ± 0.1 | 3.9 ± 0.1 × 104 | 3.3 ± 0.2 × 106 | 2.6 ± 0.1 | 8.6 ± 0.9 × 106 | 3.4 ± 0.5 × 10−8 |
| D238A | 6.0 ± 0.4 × 103 | 5.5 ± 0.4 | 3.3 ± 0.5 × 104 | 3.1 ± 0.1 × 106 | 2.9 ± 0.1 | 9.0 ± 0.9 × 106 | ≤ 1 × 10−9 |
| K239A | 9.3 ± 0.2 × 103 | 4.0 ± 0.1 | 3.7 ± 0.2 × 104 | 2.4 ± 0.1 × 106 | 4.2 ± 0.1 | 10 ± 1 × 106 | ≤ 1 × 10−9 |
| Y240A | 5.8 ± 0.1 × 103 | 4.7 ± 0.1 | 2.7 ± 0.1 × 104 | ND | ND | 1-1.5 × 10−6 | |
| D293A | 7.6 ± 0.6 × 103 | 3.8 ± 0.1 | 2.9 ± 0.3 × 104 | ND | ND | 4.0 ± 1.4 × 10−5 | |
| D74A/D293A | 1.1 ± 0.1 × 104 | 4.7 ± 0.1 | 5.2 ± 0.6 × 104 | ND | ND | 2.4 ± 1.6 × 10−4 | |
Rate constants in the absence of cofactors (kass,uncat) were obtained from the slope of linear plots of pseudo-first-order rate constants (kobs) on ZPI concentration in the range of 100-800nM. Rate constants for reactions in the presence of cofactors (kass,cat) were obtained from the data shown in Figure 4. kass,cat could not be determined for ZPI variants with low PZ affinity (Y240A, D293A, and D74A/D293A). Stoichiometry of inhibition (SI) values were determined from stoichiometric titrations of factor Xa with ZPI as described in “Stoichiometry of ZPI-protease reactions.” The product of kass,uncat or kass,cat and SI represents the corrected association rate constant for reaction through the inhibitory pathway. KDs for ZPI-PZ interactions were determined from fits of the kinetic titrations shown in Figures 4 and 6 using the equation provided in “Kinetics of ZPI-FXa reactions.”
ND indicates not determined.
Native PAGE analysis of ZPI-PZ interactions
To determine whether the ZPI mutations impaired the ability to bind PZ, we analyzed binding by nondenaturing PAGE.9,11 Binding of PZ to ZPI induces a large shift in ZPI electrophoretic mobility, resulting in a complex band migrating intermediate between that of ZPI and PZ (Figure 3). A minor complex band with slower mobility than the major complex band likely reflects the binding of a minor thrombin-cleaved Gla-domainless PZ species to ZPI.9 When mixed with a slight molar excess of PZ, bands for D238A and K239A ZPI variants were shifted completely to positions corresponding to complex, similar to the behavior of wild-type ZPI. Slight variations in complex mobility reflected the altered charge of ZPI variants (supplemental Figure 2). In contrast, PZ caused partial band shifts of M71A, D74A, and Y240A variants and no band shift for the D293A and D74A/D293A ZPI variants. These results indicated that mutation of ZPI binding-interface residues produces variable impairments in PZ binding.
Figure 3.
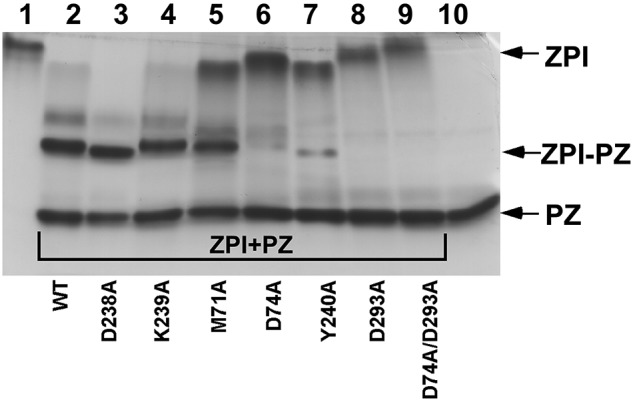
Native PAGE analysis of mutant ZPI binding to PZ. Lane 1 is wild-type ZPI; lanes 2-9, wild-type or mutant ZPIs (approximately 3μM), as indicated, mixed with a molar excess of plasma PZ; and lane 10, PZ.
Cofactor-dependent activity of ZPI variants
To confirm the PZ-binding defects of the ZPI variants, the kinetics of the ZPI–factor Xa reaction were analyzed in the presence of PZ equimolar with ZPI and with lipid and calcium cofactors. Contrasting the similar rates at which the mutant ZPIs inhibited factor Xa in the absence of cofactors, the mutants showed strikingly different cofactor-dependent rates of factor Xa inhibition (Figure 2B). Whereas D238A and K239A mutants showed rapid rates of factor Xa inhibition similar to wild-type ZPI, the M71A, D74A, Y240A, D293A, and D74A/D293A mutants showed progressively greater reductions in the cofactor-dependent rate of factor Xa inhibition. Relative to the wild-type ZPI reaction, the pseudo-first-order inhibition rate constant (kobs) was decreased for mutant ZPI reactions by 2-fold for M71A, 5-fold for D74A, 50-fold for Y240A, 400-fold for D293A, and 1300-fold for D74A/D293A. Stoichiometries of inhibition for the mutant ZPI reactions in the presence of cofactors showed small or no changes from wild-type (Table 1). These results suggested markedly different contributions of ZPI contact residues in the PZ-binding interface to bind PZ and accelerate the reaction of ZPI with membrane-associated factor Xa. These contributions increased in the order D293 > Y240 > D74 > M71, with D238 and K239 residues making no detectable contributions. Such findings were in agreement with the native PAGE analysis.
Quantification of ZPI-PZ interactions
To quantify the PZ-binding defects of the mutant ZPIs, we performed kinetic titrations of the accelerating effect of PZ on the ZPI–factor Xa reaction in the presence of lipid and calcium. For wild-type, D238A, and K239A ZPIs, kobs increased with increasing ZPI concentration to an end point reached at a ZPI concentration equimolar with PZ, indicating tight stoichiometric binding (Figure 4A). The end point for the K239A variant was somewhat reduced from wild-type and D238A variants because of an increased stoichiometry of inhibition (Table 1). Fitting of the data by the equilibrium-binding equation indicated a poorly determined subnanomolar affinity in all cases. For M71A and D74A variants, kobs showed a more gradual increase to an end point similar to wild-type, consistent with a significant weakening of PZ-binding affinity. Fitting of these titrations provided KDs of 17 ± 5nM and 34 ± 5nM, respectively, indicating minimal 20- to 30-fold decreases in affinity for these mutants. Titrations with Y240A, D293A, and D74A/D293A ZPI variants showed barely detectable increases in kobs over the same ZPI concentration range, indicating weak binding. Extending the ZPI concentration range revealed a clear saturable increase in kobs for the Y240 variant, from which a KD of 1000 ± 100nM was determined (Figure 4B). However, D293A and D74A/D293A variants still showed no detectable increases in kobs over this range of ZPI concentrations relative to wild-type ZPI in the absence of PZ. By titrating the PZ-dependent rate enhancement at a fixed level of ZPI and increasing PZ concentrations, significant linear increases in kobs were detectable for these mutants, verifying a weak, PZ-dependent enhancement of the inhibition rate constant (Figure 4B). Assuming that these mutations only affected PZ binding to ZPI and that the maximal rate constant at saturating PZ was equivalent to wild-type, estimates of KD for the binding of Y240A, D293A, and D74A/D293A ZPI variants to PZ of 1500 ± 100nM, 40 000 ± 14 000nM, and 240 000 ± 160 000nM, respectively, were made. These results confirmed that massive binding defects result from mutations of Y240 and D293.
Figure 4.

Kinetic titrations of mutant ZPI binding to PZ. (A) Titrations of the PZ-dependent increase in kobs for wild-type and mutant ZPI reactions with factor Xa (0.04nM) in the presence of 8.8nM PZ, variable ZPI, 25μM phospholipid, and 1mM Ca2+. (B) Titrations of the low-PZ-affinity variants of ZPI from panel A over an extended range of ZPI concentrations, along with control wild-type ZPI in the absence of PZ. Inset shows titrations of the PZ-dependent increase in kobs for reactions of low-PZ-affinity ZPI variants with factor Xa at 55nM ZPI, variable PZ, 25μM phospholipid, and 1mM Ca2+. kobs values represent averages of 2-3 independent measurements. Solid lines are fits of data by the equation given in “Kinetics of ZPI-FXa reactions” from which values of KD for the ZPI-PZ interaction and kass,cat were determined (Table 1). For ZPI variants with low PZ affinity, kass,cat was fixed at the wild-type value.
ZPI binding to PZ/PC chimeras
Of the 4 ZPI contact residues in which mutations resulted in impaired binding to PZ, 3 contacted the pseudocatalytic domain of PZ, whereas one, Y240, bound in a hydrophobic pocket formed by the pseudocatalytic domain and the EGF2 domain of PZ (Figure 1). To assess the importance of the EGF2 domain in this interaction, PZ chimeras in which the Gla, EGF1, and EGF2 domains of PZ were replaced with the homologous domains of protein C individually or together were tested for their ability to promote ZPI inhibition of factor Xa in the presence of lipid and calcium. The purity and appropriate size of the chimeras was confirmed by SDS-PAGE (supplemental Figure 3). The chimeric proteins were expressed at the same level as PZ wild-type and reacted in a fashion equivalent to native PZ with a bank of mAbs, suggesting that they were folded appropriately. The EGF1 chimera enhanced the rate of ZPI inhibition of factor Xa to the same extent as recombinant or plasma forms of wild-type PZ, indicating that this domain was not important for PZ rate enhancement (Figure 5A). The Gla domain chimera showed a reduced rate-enhancing effect, which is consistent with previous studies showing that a PZ–Gla domain interaction with the factor Xa Gla domain made an important contribution to the PZ-dependent acceleration of the ZPI–factor Xa reaction on a membrane surface.3,11,22 The EGF2 chimera or combined Gla, EGF1, and EGF2 chimeras showed no ability to enhance the rate of the ZPI reaction with membrane-associated factor Xa, which is consistent with an important role for the EGF2 domain in the PZ-dependent rate enhancement. Solid-phase binding assays of the PZ chimeras to ZPI showed similar strong interactions of ZPI with wild-type and EGF1 or Gla-domain chimeras and no detectable binding of the EGF2 or combined Gla, EGF1, and EGF2 chimeras (Figure 5B). These results are consistent with an important role for the PZ EGF2 domain, but not the EGF1 or Gla domains, for binding ZPI in addition to the well-established role of the pseudocatalytic domain for binding.22
Figure 5.

Characterization of cofactor activities of PZ-PC chimeric proteins. (A) Comparison of the ability of PZ and PZ-PC chimeras to accelerate the inhibition of 0.5nM factor Xa by 40nM plasma ZPI in the presence of phospholipid (15μM) and Ca2+ (4mM). Reactions were initiated by adding ZPI last and were allowed to proceed for 60 seconds before measuring residual factor Xa activity by coagulation assay. Reactions initiated with factor Xa showed similar relative ZPI reactivities (not shown). (B) Analysis of the binding of ZPI to the recombinant forms of PZ as assessed in a microtiter-plate assay described in “Characterization of PZ/PC chimeric proteins.”
Binding of cZPI to PZ
PZ dissociates from ZPI after ZPI forms an inhibited complex with factor Xa, allowing PZ to recycle as a catalyst.11 To determine the extent of the PZ affinity loss for ZPI in the ZPI–factor Xa complex, we analyzed PZ binding to cZPI. This was based on findings that the serpin moiety of the serpin-protease complex is structurally indistinguishable from cleaved serpin23,24 and that both cZPI and ZPI–factor Xa complex products of the ZPI–factor Xa reaction similarly fail to bind PZ at nM levels at which native ZPI binds.11 cZPI reduced kobs for the reaction of 5nM native ZPI-PZ complex with factor Xa in the presence of lipid and calcium in a dose-dependent manner (Figure 6A), with 50% reduction occurring at approximately 50nM cZPI (Figure 6B). This was consistent with cZPI competing with native ZPI for binding PZ, thereby reducing the level of ZPI-PZ complex competent to inhibit membrane-associated factor Xa. Because the KD for the wild-type ZPI-PZ interaction was not well determined, we also analyzed the effect of cZPI on the membrane-dependent reaction of M71A ZPI-PZ complex with factor Xa. As expected, cZPI competed more effectively with M71A ZPI for PZ, as evidenced by the 50% reduction in kobs at approximately 10nM cZPI (Figure 6B). Fitting of both datasets by the cubic equation for competitive binding15 by fixing the KD for the M71A ZPI-PZ interaction at 17nM (Figure 4) indicated KDs for wild-type ZPI-PZ and cZPI-PZ interactions of 1.2 ± 0.1nM and 6.8 ± 0.5nM, respectively. Reactive loop cleavage of ZPI thus reduces PZ affinity by approximately 6-fold. The ability of cZPI to bind PZ was confirmed by native PAGE analysis (supplemental Figure 4). Simulations of the reaction of the ZPI-PZ complex with factor Xa at physiologic concentrations of ZPI and PZ, assuming the measured KDs for native ZPI and the cZPI–factor Xa complex, showed that the decrease in PZ affinity is sufficient to account for the catalytic effect of PZ (supplemental Figure 5).
Figure 6.

Competitive effect of cZPI on the PZ-dependent ZPI–factor Xa reaction. (A) Progress curves for the reaction of 5.5nM ZPI with 0.1nM factor Xa in the presence of 5.5nM PZ, 25μM lipid, 5mM Ca2+, and increasing concentrations of cZPI. Solid lines are fits to a single exponential function from which kobs was determined. (B) Percentage decrease in kobs for the PZ-accelerated reactions of wild-type ZPI (●) and M71A ZPI (▴) with factor Xa plotted as a function of the competitor cZPI concentration. Solid lines are fits by the cubic equation for competitive binding of cZPI and wild-type or M71A ZPI to PZ after fixing the KD for the M71A ZPI-PZ interaction at the value determined from the data shown in Figure 4.
Structure of cZPI
Two crystal forms of cZPI were obtained from different crystallization conditions, one from the I2 space group diffracting up to 2.09 Å and a second from the P41 space group diffracting up to 2.65 Å. The statistics of data collection and refinements are shown in supplemental Table 1. Overall, the 2 structures share similar features except for the position of helix D and confirm that ZPI undergoes the typical stressed to relaxed conformational transition of serpins on reactive loop cleavage (Figure 7A and supplemental Figure 6A). Compared with the structure of the ZPI-PZ complex (Figure 1), the cZPI structures show that there are marked changes in both helix A and helix G apart from the expansion of sheet A that accompanies reactive loop insertion into sheet A. Helix A, which is considerably longer in ZPI than in other serpins, is bent in the ZPI-PZ complex, presumably to allow hydrophobic interactions between W46 and neighboring leucines. These interactions are broken in cZPI by the repositioning of helix A when sheet A expands, allowing helix A to straighten (supplemental Figure 6B). Helix G is partially unwound in both cZPI structures relative to the ZPI-PZ complex, indicating that this structural change is not an artifact of crystal packing. To determine whether uncomplexed ZPI adopts a similar conformation, we solved the structure of mouse ZPI. Mouse ZPI has a typical native serpin structure that resembles human ZPI in the ZPI-PZ complex, including helix G, and differs only in the reactive loop and position of helix A. Helix G unwinding thus appears to be induced in cZPI by the stressed to relaxed transition (supplemental Figure 7). When the cZPI structures are overlaid with ZPI in the ZPI-PZ complex, movements of the relative positions of the 4 key residues involved in binding PZ are observed (Figure 7B). Specifically, Y240 in sheet C is slightly shifted away from D74 in the loop extending from helix A and D293 of helix G is shifted closer to M71 of helix A, with the latter change being the most significant (supplemental Figure 8).
Figure 7.

Comparison of the structures of cZPI and ZPI. (A) Ribbon structure of cZPI (I2 space group) in gray with sheet A in blue, the cleaved reactive loop in red, helix A in orange, and helix G in purple. (B) Superposition of the PZ-binding sites of native (cyan) and cleaved (green) ZPI by aligning strand 6 of sheet A. The 4 key PZ-interacting residues are represented in stick.
Discussion
In the present study, we have elucidated the relative contributions of 6 ZPI residues that form prominent salt-bridge and hydrophobic contacts with PZ in the X-ray structure of the ZPI-PZ complex to binding PZ and to reducing PZ affinity when ZPI undergoes cleavage in the ZPI–factor Xa complex. These residues were chosen for mutagenesis based on their complementary interactions with PZ residues in the binding interface and their conservation in mouse, rat, and chicken ZPIs. Surprisingly, only 2 of the 6 contact residues on ZPI, D293 on helix G and Y240 in the gate region loop of sheet C (Figure 1), were found to be critical for binding PZ. Unitary binding energies25 of −8.4 kcal/mol for Y240 and −10.4 kcal/mol for D293 can be calculated for these residues based on estimated KDs for Y240A and D293A interactions, which together account for essentially all of the binding energy of the interaction. Such “hot spot” binding residues are common in protein-protein interactions and indicate that binding energy is not distributed additively over the binding interface, but instead is realized through cooperative interactions of key residues.26 The importance of these residues was not predictable from the X-ray structures of the ZPI-PZ complex based on the energetics of burial of contact residues (supplemental Table 3). Salt bridges of D293 in ZPI with R298 and H210 of PZ and the hydrophobic interaction of Y240 of ZPI in the hydrophobic cavity formed by the EGF2 and pseudocatalytic domains of PZ thus appear to be crucial determinants of the ZPI-PZ interaction.
We found that replacement of the EGF2 domain of PZ with the homologous domain of protein C completely abrogates the ability of PZ to bind ZPI or to enhance the rate of the ZPI reaction with membrane-associated factor Xa. This finding reinforces the critical importance of the ZPI Y240 interaction with the PZ pseudocatalytic and EGF2 domains for the ZPI-PZ interaction. Surprisingly, a previous study of a PZ-factor Xa chimera in which the Gla, EGF1, and EGF2 domains of PZ were replaced with those of factor Xa showed normal binding to ZPI.22 In contrast to the work presented herein, the solid phase-binding assay used in that study detected substantially weaker interactions of adsorbed ZPI with solution-phase PZ and reported that the binding could be detected in the presence of calcium ions, but not EDTA, implying a divalent cation dependence of the PZ-ZPI interaction. The reason for these discrepancies is not clear. Whereas D293 and Y240 residues of ZPI are of overriding importance for binding PZ, interactions of M71 and D74 in helix A of ZPI with PZ also were found to make significant, albeit much smaller, contributions to binding. Notably, our results demonstrate that D238 and K239 contact residues of ZPI do not contribute to PZ binding.
Our determination of the structure of cleaved ZPI and studies of its interactions with PZ have provided new insight into how PZ acts as a catalyst in promoting ZPI inhibition of membrane-associated factor Xa. This catalysis results from a loss in PZ affinity for ZPI after it is cleaved in the ZPI–factor Xa complex, causing PZ to dissociate and bind another ZPI molecule.11 Kinetic competition studies revealed that cZPI binds PZ with a 6-fold lower affinity than native ZPI. Simulations showed that such an affinity loss accounts for PZ catalytic action and the sparing of PZ under physiologic conditions in which PZ levels are limiting relative to ZPI.2 Whereas the reduction in ZPI affinity is not sufficient to cause 100% of bound PZ to dissociate from the ZPI–factor Xa complex, this may not be necessary given that ZPI levels only modestly exceed those of PZ, so the fraction of PZ that does dissociate is sufficient to ensure that all ZPI is activatable by PZ. Indeed, catalytic levels of PZ are sufficient to accelerate the reaction of much higher levels of ZPI and factor Xa in the presence of lipid and calcium.11
Alignment of the structures of cleaved and native ZPI suggested how structural changes in ZPI induced by factor Xa cleavage cause the observed 6-fold loss in PZ affinity. The structure of the ZPI-binding site was largely maintained in cZPI, which is consistent with the secondary structural elements of the binding site moving as a rigid unit in the conformational transition accompanying cleavage, as occurs in other cleaved serpins.27 Nevertheless, structural changes unique to cZPI, including the straightening of helix A and the unwinding of helix G, caused the relative positions of the key binding residue side chains of these helices to be similarly altered in both cleaved structures, suggesting that these changes were induced by the native to cleaved conformational transition rather than by an inherent flexibility of the side chains (supplemental Figure 8). The modestly reduced affinity of PZ for cleaved ZPI may thus arise from a reduced complementarity of the secondary Met71 and Asp74 residues in the binding site once the critical Tyr240 and Asp293 residues have bound PZ.
The results of the present study have additional implications for designing small molecules that could disrupt the ZPI-PZ interaction and thereby reduce the anticoagulant regulation of factor Xa at sites of factor X activation. Such molecules might be targeted to disrupt either of the 2 “hot spot” interactions that mediate the ZPI-PZ interaction. This could possibly restore a balance between anticoagulant and procoagulant factors in certain hemophilia disorders to support normal hemostasis. The feasibility of such an approach has been demonstrated with several other protein-protein interactions.28
Supplementary Material
Acknowledgments
The authors thank S. Paul Bajaj (University of California-Los Angeles) for providing the mAb against the PC Gla domain (CaC-11), and Peter Gettins (University of Illinois-Chicago) for providing critical comments on the manuscript.
This study was supported by the National Institutes of Health (grants R37 HL39888 to S.T.O. and HL60782 to G.J.B.), the American Heart Association (Scientist Development Grant SDG4880022 to X.H.), and the British Heart Foundation (grant PG/09/072 to A.Z.). A.Z. was supported by The Program for Professor of Special Appointment (Eastern Scholar) at Shanghai Institutions of Higher Learning and by Innovation Program of Shanghai Municipal Education Commission (no. 12ZZ113).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: X.H., G.J.B., A.Z., and S.T.O. designed the experiments; X.H., Y.Y., Y.T., and J.G. performed the experiments; and X.H., Y.Y., G.J.B., A.Z., and S.T.O. analyzed the data and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for Y.T. is Astellas Research Institute of America, Skokie, IL.
Correspondence: Steven Olson, Center for Molecular Biology of Oral Diseases, University of Illinois at Chicago, 801 S Paulina St, Chicago, IL 60612; e-mail: stolson@uic.edu; or Aiwu Zhou, Key Laboratory of Cell Differentiation and Apoptosis of Chinese Ministry of Education, and Shanghai Key Laboratory of Tumor Microenvironment and Inflammation, Shanghai Jiao-Tong University School of Medicine, Chongqing South Road 280, Shanghai 200025, China; e-mail: aiwuzhou@googlemail.com; or George J. Broze Jr, Division of Hematology, Washington University, 660 S Euclid Ave, St Louis, MO 63110; e-mail: gbroze@dom.wustl.edu.
References
- 1.Han X, Huang ZF, Fiehler R, Broze GJ., Jr The protein Z-dependent protease inhibitor is a serpin. Biochemistry. 1999;38(34):11073–11078. doi: 10.1021/bi990641a. [DOI] [PubMed] [Google Scholar]
- 2.Han X, Fiehler R, Broze GJ., Jr Characterization of the protein Z-dependent protease inhibitor. Blood. 2000;96(9):3049–3055. [PubMed] [Google Scholar]
- 3.Han X, Fiehler R, Broze GJ., Jr Isolation of a protein Z-dependent plasma protease inhibitor. Proc Natl Acad Sci U S A. 1998;95(16):9250–9255. doi: 10.1073/pnas.95.16.9250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang J, Tu Y, Lu L, Lasky N, Broze GJ., Jr Protein Z-dependent protease inhibitor deficiency produces a more severe murine phenotype than protein Z deficiency. Blood. 2008;111(10):4973–4978. doi: 10.1182/blood-2007-12-126391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sofi F, Cesari F, Tu Y, et al. Protein Z-dependent protease inhibitor and protein Z in peripheral arterial disease patients. J Thromb Haemost. 2009;7(8):731–735. doi: 10.1111/j.1538-7836.2009.03325.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kemkes-Matthes B, Nees M, Kuhnel G, Matzdorff A, Matthes KJ. Protein Z influences the prothrombotic phenotype in Factor V Leiden patients. Thromb Res. 2002;106(4-5):183–185. doi: 10.1016/s0049-3848(02)00181-0. [DOI] [PubMed] [Google Scholar]
- 7.Tabatabai A, Fiehler R, Broze GJ., Jr Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thromb Haemost. 2001;85(4):655–660. [PubMed] [Google Scholar]
- 8.Sejima H, Hayashi T, Deyashili Y, Nishioka J, Suzuki K. Primary structure of vitamin K-dependent human protein Z. Biochem Biophys Res Com. 1990;171(2):661–668. doi: 10.1016/0006-291x(90)91197-z. [DOI] [PubMed] [Google Scholar]
- 9.Huang X, Dementiev A, Olson ST, Gettins PG. Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. J Biol Chem. 2010;285(26):20399–20409. doi: 10.1074/jbc.M110.112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wei Z, Yan Y, Carrell RW, Zhou A. Crystal structure of protein Z-dependent inhibitor complex shows how protein Z functions as a cofactor in the membrane inhibition of factor X. Blood. 2009;114(17):3662–3667. doi: 10.1182/blood-2009-04-210021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang X, Swanson R, Broze GJ, Jr, Olson ST. Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. J Biol Chem. 2008;283(44):29770–29783. doi: 10.1074/jbc.M805214200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miletich JP, Broze GJ., Jr Human plasma protein Z antigen: range in normal subjects and the effect of warfarin therapy. Blood. 1987;69(6):1580–1586. [PubMed] [Google Scholar]
- 13.Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989;182(2):319–326. doi: 10.1016/0003-2697(89)90602-7. [DOI] [PubMed] [Google Scholar]
- 14.Stewart JC. Colorimetric determination of phospholipids with ammonium ferrothiocyanate. Anal Biochem. 1980;104(1):10–14. doi: 10.1016/0003-2697(80)90269-9. [DOI] [PubMed] [Google Scholar]
- 15.Lindahl P, Raub-Segall E, Olson ST, Björk I. Papain labelled with fluorescent thiol-specific reagents as a probe for characterization of interactions between cysteine proteinases and their protein inhibitors by competitive titrations. Biochem J. 1991;276(Pt 2):387–394. doi: 10.1042/bj2760387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227(5259):680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 17.Evans PR. In: Sawyer L, Isaacs N, Baily S, editors. Proceedings of the CCP4 Study Weekend Data Collection and Processing; Darsbury, United Kingdom: Darsbury Laboratory; 1993. [Google Scholar]
- 18.McCoy AJ, Grosse-Kunstleve RW, Storoni LC, Read RJ. Likelihood-enhanced fast translation functions. Acta Crystallogr D Biol Crystallogr. 2005;61(4):458–464. doi: 10.1107/S0907444905001617. [DOI] [PubMed] [Google Scholar]
- 19.Emsley P, Cowtan K. Coot: model-buildfing tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 20.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D Biol Crystallogr. 2001;57(Pt 1):122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 21.Davis IW, Leaver-Fay A, Chen VB, et al. Molprobity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. Web Server issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rezaie AR, Bae J-S, Manithody C, Qureshi SH, Yang L. Protein Z-dependent protease inhibitor binds to the C-terminal domain of protein Z. J Biol Chem. 2008;283(29):19922–19926. doi: 10.1074/jbc.M802639200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huntington JA, Read RJ, Carrell RW. Structure of a serpin-protease complex shows inhibition by deformation. Nature. 2000;407(6806):923–926. doi: 10.1038/35038119. [DOI] [PubMed] [Google Scholar]
- 24.Peterson FC, Gettins PGW. Insight into the mechanism of serpin-proteinase inhibition from 2D (1H-15N) NMR studies of the 69 kDa alpha1-proteinase inhibitor Pittsburgh-trypsin covalent complex. Biochemistry. 2001;40(21):6284–6892. doi: 10.1021/bi010100x. [DOI] [PubMed] [Google Scholar]
- 25.Lewis SD, Shields PP, Shafer JA. Characterization of the kinetic pathway for liberation of fibrinopeptides during assembly of fibrin. J Biol Chem. 1985;260(18):10192–10199. [PubMed] [Google Scholar]
- 26.Clackson T, Wells JA. A hot spot of binding energy in a hormone-receptor interface. Science. 1995;267(5196):383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 27.Whisstock JC, Skinner R, Carrell RW, Lesk AM. Conformational changes in serpins: I. The native and cleaved conformations of alpha1-antitrypsin. J Mol Biol. 2000;296(2):685–699. doi: 10.1006/jmbi.1999.3520. [DOI] [PubMed] [Google Scholar]
- 28.Shahian T, Lee GM, Lazic A, et al. Inhibition of a viral enzyme by a small-molecule dimer disruptor. Nat Chem Biol. 2009;5(9):640–646. doi: 10.1038/nchembio.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.