

Abstract
Neutrophil recruitment and extravasation at sites of inflammation provide a mechanism for host defense. We showed previously that heparan sulfate, a type of sulfated glycosaminoglycan, facilitates neutrophil recruitment based on the reduction of neutrophil infiltration in mice in which the overall sulfation of the chains was reduced by selective inactivation of N-acetylglucosamine N-deacetylase-N-sulfotransferase (Ndst1) in endothelial cells. Here we show that inactivation of uronyl 2-O-sulfotransferase in endothelial cells (Hs2st), an enzyme that acts downstream from Ndst1, results in enhanced neutrophil recruitment in several models of acute inflammation. Enhanced neutrophil infiltration resulted in part from reduced rolling velocity under flow both in vivo and in vitro, which correlated with stronger binding of neutrophil L-selectin to mutant endothelial cells. Hs2st-deficient endothelial cells also displayed a striking increase in binding of IL-8 and macrophage inflammatory protein-2. The enhanced binding of these mediators of neutrophil recruitment resulted from a change in heparan sulfate structure caused by increased N-sulfation and 6-O-sulfation of glucosamine units in response to the decrease in 2-O-sulfation of uronic acid residues. This gain-of-function phenotype provides formidable evidence demonstrating the importance of endothelial heparan sulfate in inflammation and suggests a novel enzyme target for enhancing the innate immune response.
Introduction
The inflammatory response, initiated by injury or infection, begins by release of cytokines, such as TNF-α and IL-1, from resident macrophages. These effectors stimulate endothelial cells, resulting in the secretion of von Willebrand factor, which aids in platelet recruitment and thrombus formation, and the rapid appearance of P-selectin on the apical (lumenal) face of endothelial cells. P-selectin interacts with its primary ligand, P-selectin glycoprotein ligand-1 expressed on neutrophils, enabling rolling of the cells under blood flow. Chemokines released by resident inflammatory and stromal cells transcytose across the endothelium and bind to the apical surface. Their presentation to rolling leukocytes then activates G-protein–coupled receptors, which in turn activates integrins and causes firm adhesion of the leukocytes to the endothelium. Changes in cell shape and spreading occur followed by transcellular and paracellular extravasation. After entry into tissues, neutrophils release proteases and reactive oxygen species that kill foreign pathogens and prepare the tissue for repair.
A large body of evidence suggests the importance of heparan sulfate in inflammation. Cytokines (TNF-α, IL-1, and others), L- and P-selectins, and chemokines bind to therapeutic heparin and purified heparan sulfate. Furthermore, heparin blocks L- and P-selectin in vivo in mice,1,2 which may explain in part its anti-inflammatory activity in humans.3 Recently, we showed that mice bearing an endothelial-specific mutation of the biosynthetic enzyme, N-acetylglucosamine N-deacetylase-N-sulfotransferase-1 (Ndst1) exhibit reduced neutrophil extravasation in vivo in several acute inflammatory models.4 Decreased neutrophil infiltration was partially the result of enhanced rolling velocity in vitro, which correlated with weaker binding of L-selectin to endothelial cells. Inactivation of Ndst1 also resulted in reduced chemokine transcytosis across endothelial cells and reduced presentation on the cell surface. Ndst1-deficient mice exhibit decreased allergen-induced airway hyper-responsiveness and inflammation characterized by a significant reduction in airway recruitment of inflammatory cells (eosinophils, macrophages, neutrophils, and lymphocytes).5
Heparan sulfate has a complex structure, consisting of variably sulfated disaccharides arranged in clusters, which make up the binding sites for ligands.6 Ndst1 and other Ndst isoforms catalyze the initial sulfation of subsets of glucosamine residues, creating the substrates for 2-O-sulfation of adjacent uronic acid residues and 6-O-sulfation (and more rarely 3-O-sulfation) of N-sulfoglucosamine units.7 Thus, inactivation of Ndst1 results in the overall diminution of sulfation, leaving open the question of how sulfation at other positions might affect interactions with ligands involved in the inflammatory response. In the current study, we inactivated uronyl 2-O-sulfotransferase (Hs2st) in mouse endothelial cells and examined inflammatory responses in the mutant mice. The unique altered structure of heparan sulfate generated by this mutation unexpectedly resulted in elevated neutrophil trafficking in response to several inflammatory stimuli in vivo. Altering heparan sulfate structure in this way enhanced L-selectin–dependent rolling in vivo and in vitro and dramatically increased the binding of L-selectin, IL-8, and macrophage inflammatory protein-2 (MIP-2) to endothelial cells. These findings demonstrate that endothelial heparan sulfate has the inherent plasticity to regulate the intensity of inflammatory responses and define Hs2st as a novel target for enhancing the innate immune response.
Methods
Mouse strains
Hs2stf/fTie2Cre+ and Hs2stf/fLysMCre+ mice were generated by crossing Hs2st1f/f mice8 to Tie2Cre and LysMCre transgenic mice, respectively9,10 All animals were fully backcrossed on C57Bl/6 background. Ndst1f/f Tie2Cre+ mice were generated as described previously.4 The study was approved by the University of California, San Diego (UCSD) Animal Care and Use Committee.
Structural analysis of endothelial heparan sulfate
Primary lung microvascular endothelial cells (CD31+) were isolated from wild-type and mutant mice as described.4 Blood endothelial cells (CD105+ CD31+) were separated from lymphatic endothelial cells (CD105− CD31+) using magnetic bead selection. Blood endothelial cells were metabolically labeled with 0.1 mCi/mL of [35S] Na2SO4 for 18 hours in F12/nutrient mixture medium (Invitrogen), and radiolabeled heparan sulfate was isolated by diethylaminoethyl chromatography.11 Endothelial heparan sulfate was digested with heparin lyase III (10 mU/mL) for 18 hours at 37°C, and the resulting oligosaccharides (1.5 × 106 cpm) were resolved on a Bio-Gel P-10 column (0.7 cm × 100 cm) equilibrated in 0.5M ammonium bicarbonate (pH 7.8). For disaccharide analysis, [35S]heparan sulfate was digested with heparin lyase I, II, and III (5 mU each) for 18 hours at 37°C, and the products were analyzed by strong anion exchange HPLC with a Propac PA1 (4 × 250 mm; Dionex) column as described.12
Thioglycolate-induced peritonitis
Eight- to 12-week-old mice were injected intraperitoneally with 2 mL sterile 3% thioglycolate broth (BD Biosciences). The peritoneal cavity was lavaged with 8 mL PBS containing 3mM EDTA 4 hours or 18 hours after injection, and the total number of cells was counted on a particle counter (Beckman Coulter). After counting, a portion of the cells were stained with anti–mouse Ly6C/G (Gr-1) FITC-conjugated antibody (Invitrogen) and anti–mouse F4/80 PE-conjugated antibody (eBioscience) and analyzed by flow cytometry (FACSCalibur). Flow cytometry data were analyzed using FlowJo Version 8.87 software (TreeStar), and neutrophils and monocytes were identified based on side and forward scattering and expression profile of Gr-1, F4/80, CD11b, and CD11c.
Dorsal air pouch model
Dorsal air pouches were created by subcutaneous injection of 2.5 mL sterile air. Lipopolysaccharide (LPS), IL-8, or MIP-2 (1 μg) was injected in 0.5% solution of carboxymethyl cellulose (sodium salt; Sigma-Aldrich) in PBS into the air pouches. After 4 hours, the pouches were lavaged with PBS containing 3mM EDTA and the cells were counted using a hemacytometer. The cells were stained and analyzed as described in the previous section. In selected experiments, mice were injected intravenously with 25 μg of a monoclonal blocking antibody to L-selection (MEL-14, eBioscience) 1 hour before injection of LPS into the air pouch.
Intravital microscopy
Male mice were anesthetized using a mixture of ketamine (Ketaset), xylazine (AnaSed), and atropine (Sigma-Aldrich) and kept on a thermally controlled platform throughout the experiments. The cremaster muscle was prepared as described previously.13 Briefly, the cremaster muscle was exteriorized and superfused with bicarbonate-buffered saline (37°C) throughout the experiment. Venules with a diameter of 25-40 μm (average, ∼ 30 μm) and leukocytes were observed using a microscope (Zeiss Axioskop) fitted with a water immersion objective (20×, 0.5 W; Carl Zeiss). Video recordings were captured using a DAGE Mti. 66x SIT camera and Panasonic S-VHS recorder. The recordings were then further analyzed by digitalizing them using the Turtle Beach AD Fullcap Version 1.0 software (Voyetra Turtle Beach). Two venules per mouse (n = 8 wild-type mice and n = 24 mutant mice) were analyzed, and the velocities of 6-11 rolling leukocytes were determined in each venule. Rolling velocities were calculated by individually tracking leucocytes rolling along the endothelium using a cell tracking software based on the MATLAB 2009b software (MathWorks).13
Flow chambers
Neutrophils were freshly isolated from mouse bone marrow with more than 90% purity as described.14 Lung endothelial cells were grown to confluence on gelatin-coated glass coverslips. The coverslips were then placed in the bottom of a parallel-plate flow chamber (100 μm in thickness). In selected experiments, endothelial cells were stimulated with 50 ng/mL of TNF-α for 4 hours before analysis. In some experiments, neutrophils were treated with 10 μg/mL of rat IgG2a or MEL-14 monoclonal antibody for 20 minutes at room temperature. The flow chamber was then perfused with a suspension of mouse neutrophils (2 × 105 cells/mL) under a shear stress of 1 dyne/cm2 for a period of 5 minutes, and the interaction of neutrophils with endothelial cells was visualized with a Leitz Wetzlar inverted microscope. The images were recorded on video for subsequent analysis.
L-selectin, IL-8, and MIP-2 binding
Monolayer cultures of lung blood endothelial cells were detached using Accutase (Millipore) and incubated with recombinant mouse L-selectin/Fc chimera (10 μg/mL; R&D Systems) or biotinylated IL-815 (40 μg/mL) or mouse MIP-2 (50 μg/mL, PeproTech) for 1 hour in PBS at 4°C. The cells were then incubated at 4°C for 20 minutes with DyLight 488-conjugated rabbit anti–human IgG (10 μg/mL; Jackson ImmunoResearch Laboratories), or streptavidin-conjugated PE (1:1000; eBioscience), or biotinylated antimouse MIP-2 (1 μg/mL) followed by streptavidin-conjugated PE, respectively, and flow cytometry (FACSCalibur; BD Biosciences). Flow cytometry data were analyzed using FlowJo Version 8.87 software (TreeStar).
IL-8 binding to [35S]heparan sulfate
Wild-type or mutant endothelial [35S]heparan sulfate (3 × 104 cpm) was incubated with biotinylated IL-8 (100 ng) in 20 μL PBS for 1 hour at room temperature. Streptavidin-conjugated magnetic beads (10 μL, Dynabeads; Invitrogen) were added to the IL-8/heparan sulfate mixture for 15 minutes to capture biotinylated IL-8. The bead/IL-8 complexes were washed with PBS using a magnet, and the radioactivity retained on the beads was measured by a liquid scintillation spectrometry. The bound counts were expressed as a percentage of total input counts.
Statistical methods
Statistical analysis was performed using the Prism Version 4.0 software (GraphPad Software). All comparisons we analyzed using 1-way t test, and data are presented as mean ± SEM. P values less than .05 were considered significant.
Results
Endothelial and myeloid-specific disruption of Hs2st
Vertebrates have only one gene encoding uronyl 2-O-sulfotransferase, Hs2st, and the enzyme is ubiquitously expressed in tissues based on the universal presence of 2-O-sulfated uronic acids in tissue heparan sulfates. The enzyme installs sulfate groups at C2 of uronic acids, with preference for iduronic acid over glucuronic acid,16 consistent with the concentration of 2-O-sulfate groups in domains rich in N-sulfoglucosamine residues and iduronic acid.17 Systemic inactivation of Hs2st results in neonatal death primarily because of renal agenesis.18 To study the role of Hs2st in specific tissues, we generated a loxP-flanked conditional allele (Hs2stf/f).8 Interbreeding Hs2stf/f mice with transgenic mice expressing Cre recombinase under the control of the Tie2 promotor (Tie2Cre) created cell-specific disruption of Hs2st in endothelial and myeloid cells.19 Similarly, interbreeding with transgenic mice expressing Cre under the control of the M lysozyme promotor (LysMCre) disrupted Hs2st specifically in monocytes/macrophages and neutrophils.10 Southern blot analysis showed the loss of Hs2st in endothelial cells and macrophages in Hs2stf/fTie2Cre+ (supplemental Figure 1, available on the Blood Web site; see the Supplemental Materials link at the top of the online article). Both Hs2stf/fTie2Cre+ and Hs2stf/fLysMCre+ mice reproduced normally. No differences in blood leukocyte counts were noted (supplemental Table 1). Staining of lung and liver sections with CD31 showed no gross changes in the microvasculature (supplemental Figure 2). Peripheral node addressin, which is normally expressed in the high endothelial venules of the lymph node, was expressed as expected in Hs2stf/fTie2Cre+ mice and was absent from lung and liver endothelium (supplemental Figure 2).
To analyze the structural change of heparan sulfate induced by inactivation of Hs2st in endothelial cells, we analyzed the disaccharide composition of chains purified from pulmonary endothelial cells derived from Hs2stf/fTie2Cre+ mice. Initial attempts to characterize the chains chemically met with complications because of inclusion of heparin in the growth medium, which was necessary for growth and viability of the cells. Therefore, cells were incubated with 35SO4, a precursor of the sulfate groups on the chains. The [35S]heparan sulfate was isolated and then digested with bacterial heparin lyases, which depolymerize the chain into 35S-labeled disaccharides.20 Separation of the disaccharides by anion exchange chromatography showed that inactivation of Hs2st greatly reduced all 2-O-sulfate containing disaccharides (Figure 1A; disaccharides designated as D2A0, D2S0, D2A6, and D2S6). In addition, the N-sulfated disaccharide D0S6 was greatly increased in the mutant, resulting in enhanced overall N-sulfation (from 43% of sulfated disaccharides in wild-type to 60% in mutant) and 6-O-sulfation (from 31% of sulfated disaccharides in wild-type to 38% in mutant) (Figure 1B). Similar changes have been observed in Hs2st-deficient CHO cells21 and in various tissues and cells derived from Hs2st-deficient mice.8,22,23 Treatment of the 35S-labeled endothelial heparan sulfate with heparin lyase III, which is restricted in its action to regions devoid of 2-O-sulfation,20 showed that heparan sulfate from the mutant fragmented to a much greater extent, resulting in loss of tetrasaccharide (dp4), hexasaccharide (dp6), and octasaccharide (dp8) peaks compared with fragmentation of material from wild-type cells (Figure 1C). These findings confirm the overall reduction of 2-O-sulfation in the mutant, and the increase in N-sulfation.
Figure 1.
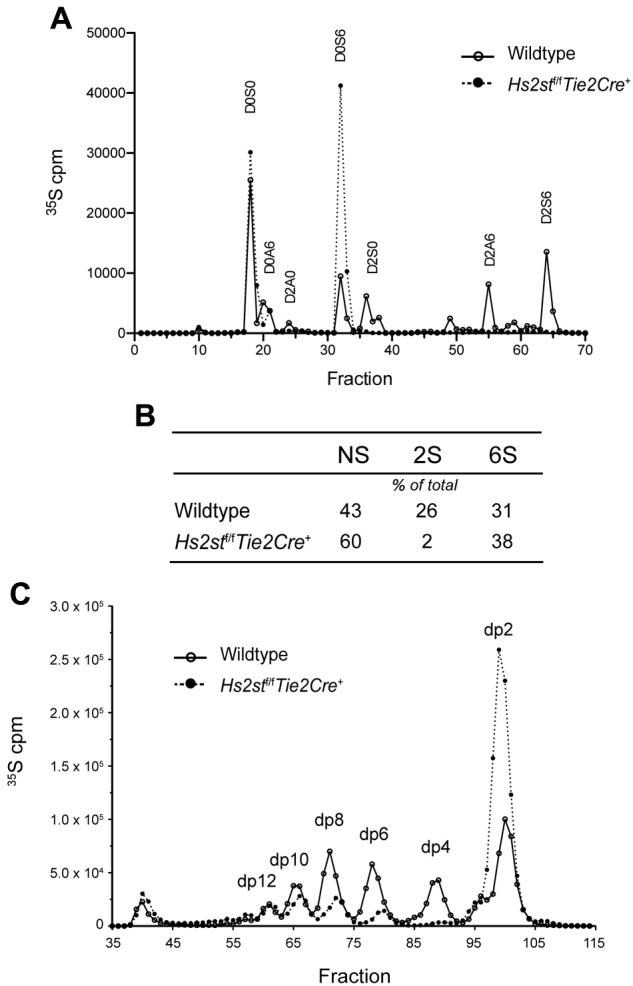
Structural analysis of endothelial heparan sulfate isolated from Hs2stf/fTie2Cre+ mice. (A) Lung endothelial cells were metabolically labeled with 35SO4, and purified [35S]heparan sulfate was digested to completion with a combination of heparin lyases I, II, and III. The resulting disaccharides were analyzed by liquid chromatography. The 4-letter disaccharide structure codes (DSC) describe the composition of each recovered disaccharide.49 In DSC, the D designates a Δ4,5-unsaturated uronic acid and the absence or presence of a 2-O-sulfate group is designated by 0 or 2, respectively. The N-substituent of the glucosamine unit is designated A or S for acetate or sulfate, respectively. The absence or presence of 6-O-sulfate groups on the glucosamine units is designated by the numerals 0 or 6, respectively. (B) The number of [35S]sulfate groups at each position was determined and expressed as a percentage of the total. (C) Endothelial cells were metabolically labeled with 35SO4, and purified [35S]heparan sulfate was digested with heparin lyase III to completion. The resulting oligosaccharide fragments was resolved by gel filtration on a Bio-Gel P-10 column. dp indicates degree of polymerization (refers to the size of the oligosaccharide in the individual peaks).
Accentuated neutrophil infiltration in thioglycolate-induced peritonitis
To characterize the effect of inactivation of Hs2st on inflammation, we injected thioglycolate intraperitoneally into Hs2stf/fTie2Cre+ mice, inducing a peritonitis characterized by infiltration of neutrophils and monocytes. Wild-type mice injected with thioglycolate showed a marked increase of peritoneal neutrophils after 4 hours compared with saline-injected mice. Unexpectedly, Hs2stf/fTie2Cre+ mice showed a 57% increase in neutrophil accumulation compared with wild-type mice (5.5 ± 0.6 × 106 vs 3.5 ± 0.3 × 106, respectively; P < .01; Figure 2A). In contrast, Ndst1f/f Tie2Cre+ mice, which exhibit reduced overall sulfation of heparan sulfate, showed a 40% reduction in neutrophil infiltration compared with wild-type (2.1 ± 0.4 × 106 vs 3.5 ± 0.3 × 106, respectively; P < .05), as described previously.4 The Tie2-Cre transgene is expressed in endothelial cells and in the myeloid and lymphoid lineages. To determine whether altering heparan sulfate on neutrophils caused the enhanced infiltration, we generated Hs2stf/fLysMCre+ mice, which express Cre only in the myeloid lineage. No significant difference in neutrophil infiltration was observed in Hs2stf/fLysMCre+ mice compared with the wild-type, indicating that the enhanced neutrophil infiltration in Hs2st1f/fTie2Cre+ mice resulted from alteration of heparan sulfate specifically in endothelial cells. The difference in leukocyte infiltration between Hs2stf/fTie2Cre+ and Hs2stf/fLysMCre+ mice showed the same trend (P = .06). The total number of monocytes in the peritoneum 18 hours after thioglycolate injection showed a similar trend as neutrophils, but the differences did not achieve significance (Figure 2B). Analysis of monocyte subpopulations suggested enhanced accumulation of Gr-1mediumF4/80medium-highCD11clow cells (supplemental Figure 3),24,25 but not other monocyte subsets. In subsequent studies, we focused only on neutrophil infiltration.
Figure 2.
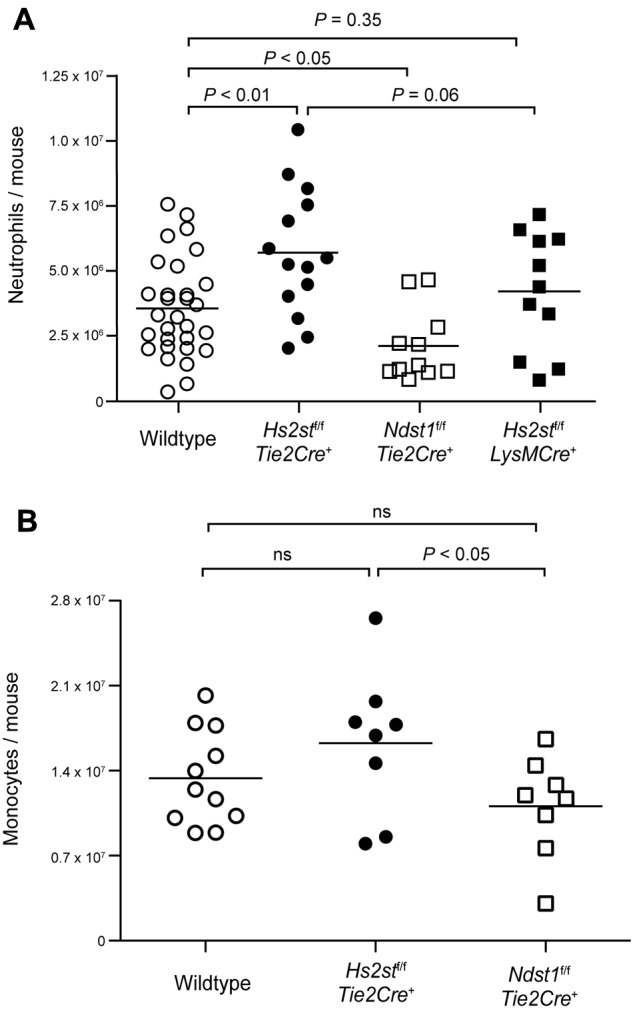
Hs2stf/fTie2Cre+ mice show increased neutrophil and monocyte infiltration in thioglycolate-induced peritonitis. (A) Mice were injected intraperitoneally with sterile 3% thioglycolate broth. After 4 hours, the peritoneum was lavaged and infiltrated cells were collected. The cells were stained with a Gr-1 specific antibody followed by flow cytometry to quantify the total number of neutrophils. (B) In another set of animals, the peritoneum was lavaged after 18 hours and infiltrated cells were harvested. The cells were stained with antibodies specific for Gr-1 and F4/80 to quantify the total number of monocytes by flow cytometry. Each symbol represents a single mouse. ns indicates not significant.
Enhanced neutrophil rolling of neutrophils on Hs2stf/fTie2Cre+ endothelial cells
Infiltration of neutrophils occurs through an orderly process in which cells initially tether to the endothelial cell surface and roll under shear flow. To determine whether Hs2st deficiency affected this initial step, we measured neutrophil rolling in cremaster muscle venules by intravital microscopy. Approximately 6-11 leukocytes were tracked in each of 2 vessels that measured 25-40 μm in diameter, and the frequency of cells rolling at a particular velocity was determined (n = 8 wild-type mice, and n = 24 mutant mice). Figure 3A shows the cumulative rolling velocity, which demonstrated that the median velocity was reduced in the mutant (11 μm/s in the mutant vs 19 μm/s in the wild-type). Similarly, the average velocity was reduced to almost half (14.2 ± 1.4 μm/s in the mutant vs 26.6 ± 3.4 μm/s in the wild-type; P < .0001; Figure 3B). In general, this difference in rolling velocity led to an increase in the number of cells tethered to the endothelial cell surface in the mutant compared with the wild-type.
Figure 3.

Hs2stf/fTie2Cre+ mice show reduced neutrophil rolling velocity. (A) Rolling velocity of neutrophils in cremaster muscle venules was assessed by intravital microscopy. Up to 10 individual leucocytes were manually tracked over time using MATLAB software, and the velocity of the leucocytes was calculated. Each dot represents the cumulative frequency (summation of the velocity frequency and all frequencies below it). (B) Quantification of the rolling velocity of neutrophils in Hs2stf/fTie2Cre+ mutant mice and wild-type littermate control (n = 6-10). (C) Neutrophil rolling was assessed in vitro using flow chambers. Endothelial cells were grown to confluence on glass slides, and bone marrow-derived wild-type neutrophils were infused through the slides under a shear stress of 1 dyne/cm2. The rolling velocity of neutrophils was quantified with or without pretreatment with TNF-α (n = 3). (D) The number of rolling neutrophils was quantified. In a separate experiment, endothelial cells were pretreated with TNF-α (n = 3 for each experimental condition). Error bar represents SEM.
Neutrophil rolling was also analyzed in vitro using flow chambers containing a monolayer of purified murine lung microvascular endothelial cells. Under a shear stress of 1 dyne/cm2, the rolling velocity of wild-type neutrophils on wild-type endothelial cells was rapid compared with the rolling velocity measured in vivo (107 ± 8 μm/s in vitro vs 19 μm/s in vivo; Figure 3C). Nevertheless, wild-type neutrophils rolled with lower average velocity on Hs2st-deficient endothelial cells (80 ± 5 μm/s, P < .01). Moreover, the number of rolling neutrophils was greater on mutant endothelial cells than on the wild-type (34 ± 2.5 cells vs 26 ± 1.8 cells, respectively; P < .05; Figure 3D). Pretreatment of the endothelial cells with the inflammatory cytokine TNF-α increased the overall number of rolling neutrophils, but the differences in velocity and number of adherent cells between the mutant and wild-type persisted (Figure 3C-D).
L-selectin-heparan sulfate interactions
Previously, we showed that neutrophil rolling velocity was increased on Ndst1f/fTie2Cre+ endothelial cells, which we attributed to decreased L-selectin binding to the undersulfated heparan sulfate chains in the mutant. To test whether the decrease in rolling velocity in Hs2st-deficient endothelial cells might reflect a common mechanism involving L-selectin, we examined L-selectin interactions in vivo using a dorsal skin air pouch model. Injection of LPS into an air pouch resulted in extensive neutrophil intravasation in wild-type animals (3.9 ± 0.3 × 106 cells; Figure 4A). More neutrophils were recovered in air pouches in Hs2stf/fTie2Cre+ mice than in wild-type animals (6.1 ± 0.6 × 106), consistent with the enhanced neutrophil infiltration observed in thioglycolate–induced peritonitis (Figure 2). When mice were injected intravenously with the L-selectin blocking antibody MEL-14 before LPS challenge, the number of neutrophils decreased significantly in both mutant and wild-type mice (Figure 4A), reducing the number of neutrophils to comparable levels (3.4 ± 0.6 × 106 in the mutant and 2.7 ± 0.6 × 106 cells in the wild-type). Injection of MEL-14 has minimal effect on the peripheral neutrophil counts (supplemental Figure 4), consistent with the idea that the reduction in neutrophil migration by MEL-14 was probably the result of blockade of L-selection–mediated rolling.
Figure 4.

L-selectin contributes to the enhanced neutrophil infiltration in Hs2stf/fTie2Cre+ mice. (A) LPS (1 μg per mouse) was injected into dorsal air pouches; and after 4 hours, infiltrated neutrophils were collected and quantified. A set of wild-type and Hs2stf/fTie2Cre+ mice were injected intravenously with a blocking antibody to L-selectin (MEL-14) 1 hour before LPS injection. Each symbol represents a single mouse. (B) Effect of MEL-14 on neutrophil rolling velocity was assessed in vitro using flow chambers. Neutrophils were pretreated with either control IgG or MEL-14 and infused through the slides (n = 6). (C) The number of rolling neutrophils was quantified after pretreatment with either control IgG or MEL-14 (n = 6). Error bar represents SEM. (D) Binding of L-selectin-Fc fusion protein to wild-type or mutant lung endothelial cells was measured by flow cytometry. Black represents binding to wild-type cells; red, binding to Hs2stf/fTie2Cre+ cells; and blue, binding to Ndst1f/fTie2Cre+. The control sample was incubated only with anti–Fc-DyLight488 and is shown as filled gray histogram. ns indicates not significant.
To show unambiguously that L-selectin–heparan sulfate interactions contribute to neutrophil rolling, we blocked L-selectin with MEL-14 and examined neutrophil rolling using flow chambers. The rolling velocity increased significantly on both wild-type and mutant endothelial cells. Furthermore, the difference of rolling velocity between wild-type and mutant endothelial cells vanished after MEL-14 treatment (Figure 4B). Blockade of L-selectin also diminished the number of rolling neutrophils on Hs2stf/fTie2Cre+ endothelial cells to a level that was indistinguishable from the number of rolling neutrophils on wild-type cells (Figure 4C). Thus, the decreased rolling velocity of neutrophils on Hs2stf/fTie2Cre+ endothelial cells was caused by enhanced L-selectin–heparan sulfate interactions.
We obtained direct evidence for enhanced L-selectin interaction with heparan sulfate on Hs2st-deficient cells by measuring the binding of recombinant soluble L-selectin to wild-type and Hs2st-deficient endothelial cells using flow cytometry. As shown in Figure 4D, binding of L-selectin to Hs2st-deficient endothelial cells was increased by 35% compared with wild-type cells (Figure 4D). In contrast, L-selectin binding to Ndst1-deficient endothelial cells was decreased by ∼ 40%.
Elevated chemokine binding
Endothelial heparan sulfate also plays an important role in chemokine presentation to rolling leukocytes, which is a prerequisite for leukocyte firm adhesion and transmigration.4 To examine whether the altered structure of endothelial heparan sulfate in Hs2stf/fTie2Cre+ mice might also affect chemokine presentation, MIP-2, one of the major neutrophil chemoattractant chemokines in mice, and its human analog IL-8 were injected into dorsal skin air pouches. Hs2stf/fTie2Cre+ mice responded more robustly to IL-8 injection, resulting in 2.2-fold more neutrophil infiltration into the pouch compared with wild-type (5.7 ± 0.5 × 106 vs 2.6 ± 0.4 × 106 cells; Figure 5A). Similarly, the response of mutant mice to MIP-2 was 1.7-fold higher than that of wild-type mice (4.3 ± 0.5 × 106 vs 2.5 ± 0.4 × 106 cells; Figure 5A). As expected, pretreatment with MEL-14 significantly reduced neutrophil infiltration induced by IL-8 in both wild-type (from 2.6 ± 0.4 × 106 to 1.7 ± 0.3 × 106 cells) and Hs2stf/fTie2Cre+ mice (from 5.7 ± 0.5 × 106 to 2.3 ± 0.3 × 106 cells; Figure 5A).
Figure 5.

Hs2stf/f Tie2Cre+ mice show increased neutrophil infiltration to IL-8 and MIP-2. (A) IL-8 or MIP-2 was injected into preformed dorsal air pouches. After 4 hours, pouches were lavaged and infiltrating neutrophils were quantified by flow cytometry. In a separate experiment, animals were treated with MEL-14 antibody by intravenous injection before injection of IL-8 into the pouch. Each symbol represents a single mouse. ns indicates not significant. (B) Binding of biotinylated IL-8 to wild-type or mutant lung endothelial cells was measured by flow cytometry. Black represents binding to wild-type cells; red, binding to Hs2stf/fTie2Cre+ cells; and blue, binding to Ndst1f/fTie2Cre+. The control sample was incubated only with streptavidin-PE and is shown as filled gray histogram. (C) Binding of MIP-2 to wild-type or mutant lung endothelial cells. (D) Binding of biotinylated FGF-2 to wild-type or mutant lung endothelial cells.
Direct binding of biotinylated IL-8 to Hs2st-deficient endothelial cells also was dramatically enhanced, increasing by 10-fold compared with the wild-type (Figure 5B; relative fluorescence units [RFU] = 93 vs 9.3). In contrast, binding of IL-8 to Ndst1-deficient endothelial cells decreased to 43% of the wild-type level (RFU = 4 vs 9.3; Figure 5B), as reported previously.4 Binding of MIP-2 to Hs2st-deficient endothelial cells also increased, but not as dramatically (2.5-fold compared with the wild-type level, Figure 5C; RFU = 55 vs 22). The enhanced binding of IL-8 was also observed in the Hs2st-deficient Chinese hamster ovary cell mutant, pgsF-17 (RFU = 350 in the mutant vs 110 in the wild-type; supplemental Figure 5).21 As a control, we measured binding of FGF2, which is known to depend on 2-O-sulfation.26,27 As expected, binding of FGF2 to Hs2st-deficient endothelial cells was reduced ∼ 40-fold compared with wild-type (Figure 5D, RFU = 5 vs 200). Furthermore, increased binding of IL-8 to mutant endothelial cells caused a 30% increase in neutrophil adhesion under static conditions compared with wild-type cells (supplemental Figure 6).
Finally, we measured binding of IL-8 to heparan sulfate in solution using purified [35S]heparan sulfate. Binding of IL-8 to heparan sulfate isolated from Hs2st-deficient endothelial cells increased 1.8-fold compared with heparan sulfate derived from wild-type endothelial cells (Figure 6). Interestingly, treatment of the [35S]heparan sulfate with Escherichia coli K5 lyase, an enzyme that cleaves only within nonsulfated regions of heparan sulfate, abolished binding of IL-8 to both wild-type and mutant-derived heparan sulfate, consistent with the prevailing “horseshoe” model for interaction of IL-8 dimers with heparan sulfate.28 These data show that binding of IL-8 to heparan sulfate does not require 2-O-sulfation and might favor an arrangement of N-sulfate and 6-O-sulfate groups generated by inactivation of Hs2st.
Figure 6.
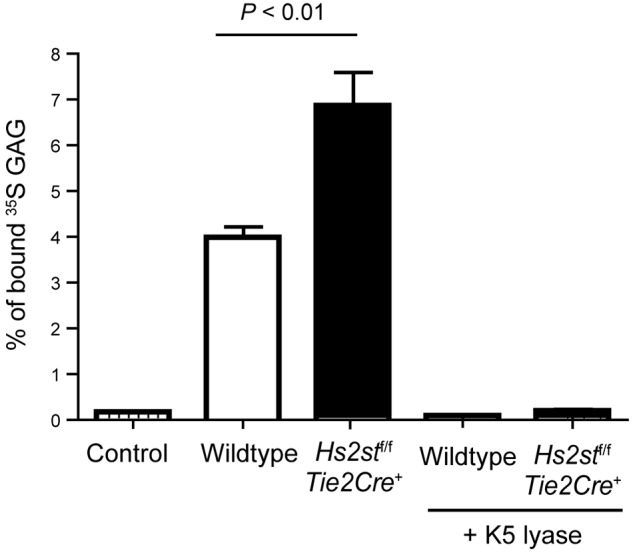
IL-8 shows enhanced binding to purified heparan sulfate derived from Hs2st-deficient endothelial cells. [35S]Heparan sulfate purified from mutant or wild-type endothelial cells was incubated with biotinylated IL-8 and collected by streptavidin-conjugated magnetic beads. One set of samples was treated with K5 lyases before incubation with IL-8. IL-8 was not added in the control sample.
Discussion
In this report, we characterized inflammatory phenotypes in mice harboring a tissue-specific inactivation of the heparan sulfate biosynthetic enzyme, Hs2st, which specifically adds sulfate to C-2 of uronic acids. Only a single Hs2st isoform exists; thus, its inactivation results in nearly complete elimination of 2-O-sulfate groups in heparan sulfate. Its systemic inactivation results in perinatal lethality because of renal agenesis,8,18 necessitating selective tissue-specific inactivation of the gene using the Cre-loxP system. Prior studies of Ndst1f/fTie2Cre+ mice, which exhibit an overall decrease in sulfation of endothelial heparan sulfate, showed reduced infiltration of neutrophils in several inflammatory models, and thus we expected a similar phenotype in Hs2stf/fTie2Cre+ mice. Unexpectedly, Hs2stf/fTie2Cre+ mice displayed a gain-of-function phenotype in which neutrophil infiltration was enhanced (Figures 2, 4, and 5). Although the Tie2Cre driver causes recombination in both endothelial cells and throughout the myeloid and lymphoid lineages, the enhanced infiltration of neutrophils results from the change in heparan sulfate structure specifically in the endothelium because inactivation of the gene in the myeloid lineage using LysMCre had no effect. We attribute the increase in neutrophil infiltration to a combination of enhanced chemokine and L-selectin binding, resulting in an increase in adhesion to the endothelium. To our knowledge, this is the first report showing that inactivation of a heparan sulfate biosynthetic enzyme (loss-of-function) results in enhanced binding of 3 heparin-binding proteins, L-selectin, IL-8, and MIP-2 (gain-of-function). Although most monocytes share L-selectin expression and responses to ELR+ CXC chemokines with neutrophils (eg, CXCL1 and CXCL2), only a subset of monocytes exhibited a significant increase in infiltration. Whether this reflects differences in L-selectin (CD62L) expression or response to chemokines will require further study.
Many studies have demonstrated participation of L-selectin in leukocyte recruitment to sites of acute inflammation.29 Rolling depends on both P-selectin and L-selectin in the mouse, but the relative contribution varies with the endothelial bed and leukocyte population under study as well as experimental conditions. The nature of the L-selectin ligands has been controversial. Potential ligands include heparan sulfate30–32 and sulfated ligands on TNF-α–stimulated microvascular and macrovascular endothelial cells,33,34 P-selectin glycoprotein ligand-1,35 and endoglycan.36 L-selectin interaction with P-selectin glycoprotein ligand-1 also facilitates neutrophil-neutrophil interactions or “secondary capture” of cells, resulting in pavementing of neutrophils.37,38 In 2005, we provided genetic evidence suggesting that endothelial heparan sulfate facilitates neutrophil rolling on different endothelial beds based on inactivation of Ndst1 in the endothelium in Ndst1f/fTie2Cre+ mice.4 Loss of Ndst1 resulted in reduced overall sulfation of heparan sulfate, reduced binding of L-selectin, enhanced rolling velocity, and decreased neutrophil infiltration in vivo. Subsequently, Bao et al inactivated a subunit of the heparan sulfate copolymerase Ext1 in endothelial cells, which presumably resulted in shortened or reduced numbers of chains.39 Although this latter study focused on lymphocyte adhesion to high endothelial venules in lymph nodes and in recruitment of resident dendritic cells through lymphatic vessels to the lymph nodes, lymphocyte rolling velocity was increased as in Ndst1f/fTie2Cre+ mice. The opposite phenotypes observed in Hs2st-deficient mice versus Ndst1 and Ext1 mice demonstrate through independent genetic models the importance of heparan sulfate as a ligand for L-selectin. The observation that intravenously injected heparin blocks L-selectin–dependent neutrophil infiltration in acute peritonitis induced by thioglycolate supports this conclusion as well.2
The gain-of-function phenotypes in Hs2stf/fTie2Cre+ mice (enhanced L-selectin and chemokine binding, increased neutrophil infiltration) are unusual. In general, reduction of sulfation or inhibition of iduronic acid formation in heparan sulfate decreases binding and/or activity of most ligands, presumably because of removal of favored arrangements of negatively charged groups required for electrostatic interactions with positively charged residues in the ligands. For example, binding of FGF2 to Hs2stf/fTie2Cre+ endothelial cells was dramatically reduced, consistent with previous observations showing that FGF2 binding depends on 2-sulfoiduronic acid units in addition to N-sulfoglucosamine residues.26,27 Although one could argue that the enhanced binding of L-selectin and chemokines might result from removal of 2-O-sulfate groups that sterically interfere with binding, competition studies with selectively desulfated forms of heparin suggest that removal of 2-O-sulfate groups partially reduces its efficacy as an inhibitor.2,28 Thus, we think that the increased capacity to bind L-selectin, IL-8, and MIP-2 is probably the result of the increase in N-sulfate and 6-O-sulfate groups on glucosamine residues, perhaps by increasing the frequency of binding sites. Indeed, L-selectin will preferentially bind to highly sulfated tetradecasaccharides derived from heparin and endothelial heparan sulfate.30,31,40
Chemokines usually exist as dimers or oligomers and in several the heparin binding sites lie on opposite faces of the dimer (eg, IL-8, MCP-1, MIP-1α, and IFN-γ).28,41–44 Changes in sulfation that facilitate the adoption of a “horseshoe” conformation, in which 2 N-sulfated domains each containing 2-3 highly sulfated disaccharide units separated by a spacer, enable interaction with 2 IL-8 monomers in an antiparallel fashion. The stretch of oligosaccharide connecting the 2 N-sulfated domains, which is not sulfated or very sparsely sulfated, has to satisfy a certain length requirement (6-7 disaccharide units) in order for the 2 N-sulfated domains to interact simultaneously with the IL-8 dimer. It seems probable that the occurrence of such unique domain structures in heparan sulfate might be increased in Hs2st-deficient cells because of the increase of N- and 6-O-sulfation. Whether L-selectin exhibits similar preference is unknown, but it too can exist as a dimer.45
An extremely important, yet poorly studied, question in inflammatory biology is whether changes in heparan sulfate structure, such as those observed in Hs2stf/fTie2Cre+ and Ndst1f/fTie2Cre+ mice, can occur naturally as part of a response to inflammatory insult. Changes in mRNAs encoding several enzymes involved in heparan sulfate biosynthesis have been noted after cytokine stimulation of endothelial cells,46 which may result in cleavage of the chains or structural changes that result in altered binding of chemokines and cell adhesion molecules.47,48 Whether these changes are beneficial to the host or merely reflect a pathophysiologic response is unknown.
In conclusion, we show sharply reduced 2-O-sulfation, enhanced N-sulfation, and moderately elevated 6-O-sulfation in endothelial cells lacking Hs2st, resulting in increased binding of ELR+ CXC chemokines and of L-selectin. These changes probably synergize to produce a net increase in neutrophil recruitment to sites of inflammation in Hs2stf/fTie2Cre+ mice.
Supplementary Material
Acknowledgments
The authors thank UCSD Glycotechnology Core Resource for assistance with disaccharide analysis, UCSD Histology core facility for assistance with immunohistology, UCSD hematology core facility for hematology analysis, and C. Lefort and Y. Kuwano for assistance with intravital microscopy.
This work was supported by the National Institutes of Health (grants P01 HL57345 and P01 HL107150, J.D.E.; grant R01 AI37113, T.M.H.; and grant R01 HL 111969, K.L.), the Swedish Research Council (postdoctoral grant, J.A.), the Tegger Foundation (postdoctoral grant, J.A.), and the American Heart Association (Postdoctoral Fellowship 0825274F, D.X.).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.A., D.X., K.L., P.S., and J.D.E. designed the research, analyzed and interpreted data, and wrote the manuscript; J.A., D.X., B.N.K., and J.K.N. performed the work and statistical analyses; and T.M.H. contributed reagents and reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeffrey D. Esko, Department of Cellular and Molecular Medicine, University of California, San Diego, 9500 Gilman Dr, La Jolla, CA 92093-0687; e-mail: jesko@ucsd.edu.
References
- 1.Ley K, Cerrito M, Arfors KE. Sulfated polysaccharides inhibit leukocyte rolling in rabbit mesentery venules. Am J Physiol. 1991;260:1667–1673. doi: 10.1152/ajpheart.1991.260.5.H1667. [DOI] [PubMed] [Google Scholar]
- 2.Wang LC, Brown JR, Varki A, Esko JD. Heparin's anti-inflammatory effects require glucosamine 6-O-sulfation and are mediated by blockade of L- and P-selectins. J Clin Invest. 2002;110(1):127–136. doi: 10.1172/JCI14996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Young E. The anti-inflammatory effects of heparin and related compounds. Thromb Res. 2008;122(6):743–752. doi: 10.1016/j.thromres.2006.10.026. [DOI] [PubMed] [Google Scholar]
- 4.Wang L, Fuster M, Sriramarao P, Esko JD. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6(9):902–910. doi: 10.1038/ni1233. [DOI] [PubMed] [Google Scholar]
- 5.Zuberi RI, Ge XN, Jiang S, et al. Deficiency of endothelial heparan sulfates attenuates allergic airway inflammation. J Immunol. 2009;183(6):3971–3979. doi: 10.4049/jimmunol.0901604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108(2):169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem. 2002;71:435–471. doi: 10.1146/annurev.biochem.71.110601.135458. [DOI] [PubMed] [Google Scholar]
- 8.Stanford KI, Wang L, Castagnola J, et al. Heparan sulfate 2-O-sulfotransferase is required for triglyceride-rich lipoprotein clearance. J Biol Chem. 2010;285(1):286–294. doi: 10.1074/jbc.M109.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Constien R, Forde A, Liliensiek B, et al. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 2001;30(1):36–44. doi: 10.1002/gene.1030. [DOI] [PubMed] [Google Scholar]
- 10.Clausen BE, Burkhardt C, Reith W, Renkawitz R, Forster I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999;8(4):265–277. doi: 10.1023/a:1008942828960. [DOI] [PubMed] [Google Scholar]
- 11.Bame KJ, Esko JD. Undersulfated heparan sulfate in a Chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J Biol Chem. 1989;264:8059–8065. [PubMed] [Google Scholar]
- 12.Skidmore MA, Guimond SE, Dumax-Vorzet AF, Yates EA, Turnbull JE. Disaccharide compositional analysis of heparan sulfate and heparin polysaccharides using UV or high-sensitivity fluorescence (BODIPY) detection. Nat Protoc. 2010;5(12):1983–1992. doi: 10.1038/nprot.2010.145. [DOI] [PubMed] [Google Scholar]
- 13.Jung U, Bullard DC, Tedder TF, Ley K. Velocity differences between L- and P-selectin-dependent neutrophil rolling in venules of mouse cremaster muscle in vivo. Am J Physiol. 1996;271(6):H2740–H2747. doi: 10.1152/ajpheart.1996.271.6.H2740. [DOI] [PubMed] [Google Scholar]
- 14.Siemsen DW, Schepetkin IA, Kirpotina LN, Lei B, Quinn MT. Neutrophil isolation from nonhuman species. Methods Mol Biol. 2007;412:21–34. doi: 10.1007/978-1-59745-467-4_3. [DOI] [PubMed] [Google Scholar]
- 15.Allen SJ, Hamel DJ, Handel TM. A rapid and efficient way to obtain modified chemokines for functional and biophysical studies. Cytokine. 2011;55(2):168–173. doi: 10.1016/j.cyto.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rong J, Habuchi H, Kimata K, Lindahl U, Kusche-Gullberg M. Substrate specificity of the heparan sulfate hexuronic acid 2-O-sulfotransferase. Biochemistry. 2001;40(18):5548–5555. doi: 10.1021/bi002926p. [DOI] [PubMed] [Google Scholar]
- 17.Maccarana M, Sakura Y, Tawada A, Yoshida K, Lindahl U. Domain structure of heparan sulfates from bovine organs. J Biol Chem. 1996;271:17804–17810. doi: 10.1074/jbc.271.30.17804. [DOI] [PubMed] [Google Scholar]
- 18.Bullock SL, Fletcher JM, Beddington RS, Wilson VA. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes Dev. 1998;12(12):1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol. 2001;230(2):230–242. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 20.Linhardt RJ. Analysis of glycosaminoglycans with polysaccharide lyases. Curr Protoc Mol Biol. 2001 doi: 10.1002/0471142727.mb1713bs48. Chapter 17:Unit17 13B. [DOI] [PubMed] [Google Scholar]
- 21.Bai XM, Esko JD. An animal cell mutant defective in heparan sulfate hexuronic acid 2-O-sulfation. J Biol Chem. 1996;271:17711–17717. doi: 10.1074/jbc.271.30.17711. [DOI] [PubMed] [Google Scholar]
- 22.Merry CLR, Bullock SL, Swan DC, et al. The molecular phenotype of heparan sulfate in the Hs2st−/− mutant mouse. J Biol Chem. 2001;276(38):35429–35434. doi: 10.1074/jbc.M100379200. [DOI] [PubMed] [Google Scholar]
- 23.Ledin J, Staatz W, Li JP, et al. Heparan sulfate structure in mice with genetically modified heparan sulfate production. J Biol Chem. 2004;279(41):42732–42741. doi: 10.1074/jbc.M405382200. [DOI] [PubMed] [Google Scholar]
- 24.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- 25.Sunderkotter C, Nikolic T, Dillon MJ, et al. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172(7):4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- 26.Jemth P, Kreuger J, Kusche-Gullberg M, Sturiale L, Giménez-Gallego G, Lindahl U. Biosynthetic oligosaccharide libraries for identification of protein-binding heparan sulfate motifs: exploring the structural diversity by screening for fibroblast growth factor (FGF) 1 and FGF2 binding. J Biol Chem. 2002;277(34):30567–30573. doi: 10.1074/jbc.M203404200. [DOI] [PubMed] [Google Scholar]
- 27.Kreuger J, Salmivirta M, Sturiale L, Giménez-Gallego G, Lindahl U. Sequence analysis of heparan sulfate epitopes with graded affinities for fibroblast growth factors 1 and 2. J Biol Chem. 2001;276(33):30744–30752. doi: 10.1074/jbc.M102628200. [DOI] [PubMed] [Google Scholar]
- 28.Spillmann D, Witt D, Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J Biol Chem. 1998;273:15487–15493. doi: 10.1074/jbc.273.25.15487. [DOI] [PubMed] [Google Scholar]
- 29.Rosen SD. Ligands for L-selectin: homing, inflammation, and beyond. Annu Rev Immunol. 2004;22:129–156. doi: 10.1146/annurev.immunol.21.090501.080131. [DOI] [PubMed] [Google Scholar]
- 30.Norgard-Sumnicht KE, Varki A. Endothelial heparan sulfate proteoglycans that bind to L-selectin have glucosamine residues with unsubstituted amino groups. J Biol Chem. 1995;270:12012–12024. doi: 10.1074/jbc.270.20.12012. [DOI] [PubMed] [Google Scholar]
- 31.Norgard-Sumnicht KE, Varki NM, Varki A. Calcium-dependent heparin-like ligands for L-selectin in nonlymphoid endothelial cells. Science. 1993;261:480–483. doi: 10.1126/science.7687382. [DOI] [PubMed] [Google Scholar]
- 32.Giuffre L, Cordey AS, Monai N, Tardy Y, Schapira M, Spertini O. Monocyte adhesion to activated aortic endothelium: role of L-selectin and heparan sulfate proteoglycans. J Cell Biol. 1997;136(4):945–956. doi: 10.1083/jcb.136.4.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zakrzewicz A, Gräfe M, Terbeek D, et al. L-selectin-dependent leukocyte adhesion to microvascular but not to macrovascular endothelial cells of the human coronary system. Blood. 1997;89:3228–3235. [PubMed] [Google Scholar]
- 34.Tu LL, Chen AJ, Delahunty MD, et al. L-selectin binds to P-selectin glycoprotein ligand-1 on leukocytes: interactions between the lectin, epidermal growth factor, and consensus repeat domains of the selectins determine ligand binding specificity. J Immunol. 1996;157:3995–4004. [PubMed] [Google Scholar]
- 35.Rivera-Nieves J, Burcin TL, Olson TS, et al. Critical role of endothelial P-selectin glycoprotein ligand 1 in chronic murine ileitis. J Exp Med. 2006;203(4):907–917. doi: 10.1084/jem.20052530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leppanen A, Parviainen V, Ahola-Iivarinen E, Kalkkinen N, Cummings RD. Human L-selectin preferentially binds synthetic glycosulfopeptides modeled after endoglycan and containing tyrosine sulfate residues and sialyl Lewis x in core 2 O-glycans. Glycobiology. 2010;20(9):1170–1185. doi: 10.1093/glycob/cwq083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alon R, Fuhlbrigge RC, Finger EB, Springer TA. Interactions through L-selectin between leukocytes and adherent leukocytes nucleate rolling adhesions on selectins and VCAM-1 in shear flow. J Cell Biol. 1996;135:849–865. doi: 10.1083/jcb.135.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fuhlbrigge RC, Alon R, Puri KD, Lowe JB, Springer TA. Sialylated, fucosylated ligands for L-selectin expressed on leukocytes mediate tethering and rolling adhesions in physiologic flow conditions. J Cell Biol. 1996;135:837–848. doi: 10.1083/jcb.135.3.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bao X, Moseman EA, Saito H, et al. Endothelial heparan sulfate controls chemokine presentation in recruitment of lymphocytes and dendritic cells to lymph nodes. Immunity. 2010;33(5):817–829. doi: 10.1016/j.immuni.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Koenig A, Norgard-Sumnicht K, Linhardt R, Varki A. Differential interactions of heparin and heparan sulfate glycosaminoglycans with the selectins: implications for the use of unfractionated and low molecular weight heparins as therapeutic agents. J Clin Invest. 1998;101(4):877–889. doi: 10.1172/JCI1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stringer SE, Forster MJ, Mulloy B, Bishop CR, Graham GJ, Gallagher JT. Characterization of the binding site on heparan sulfate for macrophage inflammatory protein 1alpha. Blood. 2002;100(5):1543–1550. [PubMed] [Google Scholar]
- 42.Lau EK, Paavola CD, Johnson Z, et al. Identification of the glycosaminoglycan binding site of the CC chemokine, MCP-1: implications for structure and function in vivo. J Biol Chem. 2004;279(21):22294–22305. doi: 10.1074/jbc.M311224200. [DOI] [PubMed] [Google Scholar]
- 43.Sarrazin S, Bonnaffe D, Lubineau A, Lortat-Jacob H. Heparan sulfate mimicry: a synthetic glycoconjugate that recognizes the heparin binding domain of interferon-gamma inhibits the cytokine activity. J Biol Chem. 2005;280(45):37558–37564. doi: 10.1074/jbc.M507729200. [DOI] [PubMed] [Google Scholar]
- 44.Lortat-Jacob H, Turnbull JE, Grimaud JA. Molecular organization of the interferon gamma-binding domain in heparan sulphate. Biochem J. 1995;310:497–505. doi: 10.1042/bj3100497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li X, Steeber DA, Tang MLK, Farrar MA, Perlmutter RM, Tedder TF. Regulation of L-selectin-mediated rolling through receptor dimerization. J Exp Med. 1998;188:1385–1390. doi: 10.1084/jem.188.7.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krenn EC, Wille I, Gesslbauer B, Poteser M, van Kuppevelt TH, Kungl AJ. Glycanogenomics: a qPCR-approach to investigate biological glycan function. Biochem Biophys Res Commun. 2008;375(3):297–302. doi: 10.1016/j.bbrc.2008.07.144. [DOI] [PubMed] [Google Scholar]
- 47.Celie JW, Rutjes NW, Keuning ED, et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. 2007;170(6):1865–1878. doi: 10.2353/ajpath.2007.070061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Celie JW, Beelen RH, van den Born J. Heparan sulfate proteoglycans in extravasation: assisting leukocyte guidance. Front Biosci. 2009;14:4932–4949. doi: 10.2741/3578. [DOI] [PubMed] [Google Scholar]
- 49.Lawrence R, Lu H, Rosenberg RD, Esko JD, Zhang L. Disaccharide structure code for the easy representation of constituent oligosaccharides from glycosaminoglycans. Nat Methods. 2008;5(4):291–292. doi: 10.1038/nmeth0408-291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.