

Abstract
The ubiquitously expressed Abl protein is a non-receptor tyrosine kinase that undergoes nuclear-cytoplasmic shuttling and is involved in many signalling pathways in the cell. Nuclear Abl is activated by DNA damage to regulate DNA repair, cell cycle checkpoints and apoptosis. Previous studies have established that the ataxia telangiectasia mutated (ATM) activates nuclear Abl via phosphorylation at serine 465 (S465) in the kinase domain in response to ionizing radiation (IR). Using a peptide biosensor that specifically reports on the Abl kinase activity, we found that an Abl-S456A mutant, which is not capable of being activated by ATM through the canonical site, was still activated rapidly after IR. We established that DNA-dependent protein kinase (DNAPK) is likely to be responsible for a second pathway to activate Abl early on in the response to IR through phosphorylation at a site other than S465. Our findings show that nuclear and cytoplasmic Abl kinase is activated early on (within 5 min) in response to IR by both ATM and DNAPK, and that while one or the other of these kinases is required, either one is sufficient to activate Abl. These results support the concept of early Abl recruitment by both the ATM and the DNAPK pathways to regulate nuclear events triggered by DNA damage and potentially communicate them to proteins in the cytoplasm.
Keywords: peptide biosensor, kinase activation, DNA damage response, c-Abl kinase
Introduction
Most techniques for monitoring kinase activity depend on isolation of the kinase from its intracellular context and/or detection of specific phosphorylation sites in endogenous proteins that aren’t always previously known. Since almost all cellular signaling events occurring in cells are affected by kinase and substrate interaction partners, this presents challenges for performing analyses of detailed signaling processes that require intracellular scaffolding and localization, or for which important endogenous sites have not yet been described and targeted with antibodies. Furthermore, since many important signaling proteins are present at low abundance in the cell and their modifications are often stoichiometrically low and/or in regions of the protein that are not amenable to protease digestion, even mass spectrometry-based proteomics cannot detect all the relevant signaling events that might be occuring in response to biological stimuli. Therefore, we have developed a complementary approach using a peptide biosensor tool that is tuned to a particular kinase and delivered into the cell to achieve cell-based functional detection of kinase activity, and applied this tool to elucidate an early event in the DNA damage response.
DNA lesions from endogenously-generated and exogenous agents are a normal consequence of cellular processes. Cells have developed sophisticated response mechanisms to recognize and repair the resulting damage.[1–3] In particular, exposure to ionizing radiation (IR) results in base modifications as well as both single and double strand breaks (SSBs and DSBs), with DSBs being the most lethal.[4, 5] When DSBs occur, the DNA damage signaling proteins ataxia telangiectasia mutatated (ATM) protein kinase and DNA-dependent protein kinase (DNAPK) are recruited to DSBs and activated to coordinate DNA repair with cell cycle checkpoints.[1, 6, 7] ATM is a member of the PIKK-family of lipid and protein kinases that phosphorylate protein targets at the S/Q and T/Q motifs.[8, 9] ATM itself also is phosphorylated at multiple residues (serine 367, 1893, and 1981 in human cells) in response to IR. These phosphorylations have different functions in vivo[10] and can be monitored to track ATM activation by IR. DNAPK, another member of the PIKK family, is composed of a catalytic subunit (DNAPKcs) and a heterodimeric DNA binding subunit (Ku70/80). Like ATM, its activity is stimulated by recruitment to DSBs by the Ku heterodimer and through autophosphorylation at multiple residues in response to IR in vivo.[7, 11, 12] DNAPK is essential for DNA repair through non-homologous end-joining (NHEJ) in mammalian cells, a pathway that promotes cell survival in response to IR.[13–16]
Among the many substrates of ATM is the Abl tyrosine kinase encoded by the ABL1 gene[17]. Cytoplasmic Abl is activated by growth factors, cytokines, cell-matrix adhesion, microbial infection and many other extracellular signals.[18, 19] One of the key functions of cytoplasmic Abl is to regulate F-actin dynamics, which underlie a wide variety of cellular processes that involve actin-polymerization and depolymerization.[20] Nuclear Abl is activated by genotoxins, in particular, those that cause DSBs in the genomic DNA[18, 21, 22] (including IR and other DNA-damaging agents) to phosphorylate diverse target proteins including Rad51, Rad52, WRN, Mdm2, and p73 to regulate DNA repair, cell cycle checkpoints and apoptosis.[3, 18, 23] Previous studies have identified interactions between Abl, ATM and DNAPK, and have focused on later events in the DNA damage response (on the scale of 1–24 hours post-IR) that control return to the cell cycle after repair and/or initiation of apoptosis. For example, when cells are exposed to IR, ATM phosphorylates Abl at S465 and activates Abl kinase activity.[17] DNAPK and Abl have been shown to phosphorylate each other in vitro,[24] although this has not yet been observed in vivo. It is not known whether ATM and DNAPK act redundantly to phosphorylate Abl at S465 and/or other residues, or if DNAPK phosphorylates a different site or sites on Abl to regulate its function in a pathway separate from that governed by ATM. This is partly due to the challenges of detecting specific phosphorylation sites in a protein like Abl, and more so to ascribing functional significance to those sites and/or the downstream phosphorylation of Abl target proteins.
To address these issues, we applied chemical biology and pharmacological tools to examine the individual roles of ATM and DNAPK in activating Abl after DNA damage via IR. To follow the intracellular activation of Abl in response to IR-induced DNA damage, we used our previously reported peptide-based biosensor substrate.[25] This biosensor (Figure 1) has the following functional modules: a known peptide substrate for Abl,[26] a photocleavable linker, an Abl SH3 domain-binding ligand peptide,[27] a biotin tag and a cell-penetrating peptide, TAT,[28] to aid delivery of the biosensor across the plasma membrane. To distinguish between the contributions of ATM and DNAPK to Abl activation in the DNA damage pathway, we generated HEK293 cell lines with stable, Tet-ON inducible expression of Abl-WT-EGFP and Abl(S465A)-EGFP, which lacks the known ATM phosphorylation site. We confirmed that the biosensor peptide is distributed and phosphorylated in relevant intracellular compartments by using subcellular fractionation. We also used the biosensor substrate to monitor Abl activity in ATM-deficient pre-B cells in the presence and absence of a DNAPK inhibitor. We found that Abl is activated very early (within 10 minutes) when cells are exposed to IR even if S465 is replaced by alanine and that a DNAPK inhibitor blocks that activation, providing support for the idea that DNAPK and ATM play unique roles in rapidly activating Abl after ionizing radiation.
Figure 1. Overall schematic for the strategy used in this work.
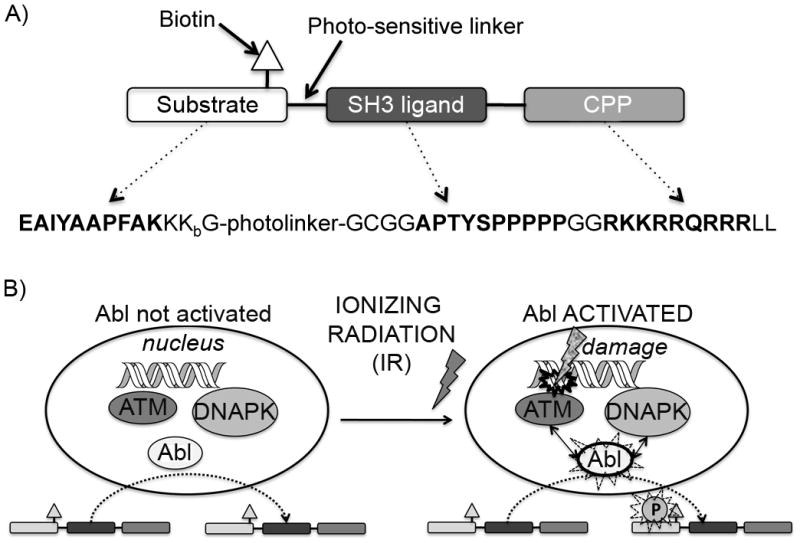
A) Shows the design of the biosensor peptide. Amino acid sequences given in bold represent the functional modules Abltide, Abl SH3 ligand and the cell penetrating peptide (CPP) TAT, respectively. The Abltide sequence is thought to be relatively specific for the Abl kinase. The Abl SH3 ligand sequence acts to further promote this specificity by providing interaction with the Abl SH3 domain, making the biosensor peptide a better mimic of native substrates, which include such protein-protein interaction modules. Kbiotin = lysine biotinylated on the γ-amino group (side chain) is included for detecting the total amount of biosensor via streptavidin blotting; The photolinker (a 3-(2-nitrobenzyl)-3-aminopropionyl residue) is incorporated in case detection with mass spectrometry is used (as we have previously reported[25]) however this is not important for the work described in this manuscript. B) Shows a cartoon representation of the interactions between Abl, ATM and DNAPK after DNA damage. The Abl kinase normally shuttles in and out of the nucleus. Upon DNA damage, ATM and DNAPK participate in phosphorylating the nuclear fraction of Abl and thus activating its kinase function, which, among other signals, initiates cascades that turn on cell cycle arrest, DNA repair machinery and eventually the decision between re-entry into the cell cycle or apoptosis. We can detect this activation in intact cells by delivering and subsequently harvesting the biosensor peptide, followed by immunoblotting to detect whether it was phosphorylated during its time in the cell.
Results
Abl biosensor phosphorylation increases after exposure to IR
Evidence suggests that Abl is activated after exposure to IR, however the current understanding of this process focuses on timeframes (>1 h) that are fairly far downstream of the initial DDR.[17, 24, 29] We first confirmed that the biosensor substrate was phosphorylated by Abl in our Abl-WT-EGFP model by inducing Abl-WT-EGFP expression via overnight treatment with doxycycline and incubation with either the biosensor peptide alone (25 μM) or the biosensor in combination with the Abl inhibitor imatinib (20 μM). In the absence of doxycycline induced overexpression of the kinase, little to no biosensor phosphory-lation was observed—however, in the presence of overexpressed Abl-WT-EGFP the peptide was phosphorylated, and this phosphorylation was inhibited by imatinib, suggesting that Abl-WT-EGFP was responsible for biosensor phosphorylation (see supporting information Figure S1 for details).
To test whether our Abl biosensor was able to detect Abl activation at earlier points in the DDR process, we induced kinase expression in our stable cell lines via incubation with doxycycline prior to addition of the peptide biosensor, followed by IR treatment and harvesting of the cells over a 10–30 min timecourse post-irradiation. As shown in Figure 2A, row 1, lanes 1 and 2, very little appreciable phosphorylation signal (measured using the antiphosphotyrosine antibody 4G10) was seen for cells where Abl-WT-EGFP was not induced. Quantification of the relative band densities for lanes corresponding to uninduced samples showed a slight increase in the ratio of 4G10/streptavidin signal, however the difference was not significant (see supporting information Figure S2). In lanes 3 and 4, within 10 minutes after irradiation, the phosphorylation signal from the biosensor peptide rapidly increased approximately four-fold (p=0.002) when compared to the phosphorylation signal from our biosensor in non-irradiated cells harvested at the same timepoint. In contrast, phosphorylation of the autophosphorylation-related Y245 in the Abl-WT-EGFP construct was only slightly increased (Figure 2A, row 5) and did not significantly change (p>0.45) during the timeframe of this experiment. Two-way ANOVA demonstrated that trends in the responses observed via these two markers were different, with nearly statistical significance (p=0.09, where p<0.05 would indicate significance).
Figure 2. Phosphorylation of the Abl kinase biosensor after exposure to IR.

The Abl-WT-EGFP and Abl-S465A-EGFP cells were prepared as described in the experimental section. The peptide substrate (25μM) was added and cells were incubated at 37°C for 5 min, then treated with IR (5 Gy). Cells were harvested at different times post IR treatment as indicated. A) Western blot analysis using quantitative two-color Licor IR-dye imaging showed that the phosphorylation of the biosensor peptide (detected via 4G10 signal just below the 6 kDa molecular weight marker, confirmed via overlaid signal from streptavidin visualized using the second channel of the Licor Odyssey scanner) was dramatically increased after IR treatment in cells overexpressing the Abl-WT-EGFP construct (row 1, lanes 4, 8 and 12). B) Unexpectedly, biosensor phosphorylation was still increased in cells expressing the Abl-S465A-EGFP mutant protein (row 1, lanes 2, 4 and 6). Immunoblotting for other proteins served as controls for IR-induced pathway and Abl activation (pATM, pDNAPK, pCrkL), construct expression/autophosphorylation (GFP, Abl(pY245)) and total protein loading (Tubulin or GAPDH). Total ATM and DNAPK blots showed consistent levels of the unphosphorylated forms of these proteins (data not shown). C), D) and E) Integrated band intensity data for the 4G10, streptavidin, phospho-c-Abl(Y245) and anti-GFP signals over independent replicate experiments were calculated and the phosphorylated signals (4G10 and phospho-c-Abl(Y245)) were divided by their respective ‘total’ marker signals (streptavidin, labelled as strept, and anti-GFP, respectively) and plotted together. Axes are labelled as 4G10/strept or norm increase (+IR), meaning the increase in the 4G10/strept signal for samples treated with IR vs non-IR controls. Error bars represent SEM, and statistical significance (p<0.05) of the change vs. non-irradiated cells is indicated using *. For the Abl-WT-EGFP biosensor data, N = 5 and for the phospho-Y245 data N = 3. For the Abl-S465A-EGFP biosensor data, N = 4 and for the phospho-Y245 data, N = 2.
Since the phosphorylation of Abl at S465 by ATM is thought to be required for IR-induced Abl activation, [17] we expected that Abl(S465A)-EGFP would not show increased activity after DNA damage. However the biosensor was still phosphorylated in this mutant (Figure 2B and C), and even showed slightly a slightly higher degree of basal phosphorylation (without IR), and a more sustained level of post-IR phosphorylation signal than the Abl-WT-EGFP (Figure 2C). A modest but significant increase (p = 0.0005 after 10 min.) in relative Abl activity after IR was still observed (albeit fewer-fold than for Abl-WT-EGFP) (Figure 2B, row 1 lanes 2, 4 and 6), which was also significantly different than the response observed via Y245 phosphorylation (2-way ANOVA p=0.037) (Figure 2B, row 5, lanes 2, 4 and 6, and Figure 2E). These data indicated that S465 might not be the only residue responsible for IR-induced Abl activation in our system. One possibility was that ATM also phosphorylated other sites that contributed to the activation of Abl. Another was that ATM was able to activate endogenous Abl in the S465 mutant cells to promote phosphorylation of the biosensor peptide. A third possibility was that another kinase besides ATM also phosphorylated Abl at a different site or sites to activate Abl to participate in the DNA damage response pathway. DNAPK is a serine/threonine kinase known to be involved in the DNA damage response that has been shown to physically interact with Abl.[24] A connection between DNAPK and Abl in regulating the DNA damage response has been implicated through in vitro and intracellular studies.[3, 30] Therefore, we examined the possibility that DNAPK is involved in activating Abl in our system after IR treatment, and addressed the potential for ATM to be acting through other sites on endogenous or expressed Abl protein.
ATM and DNAPK inhibitors indicate that both kinases function to activate Abl in the DNA damage pathway
To test the hypothesis that DNAPK participates in early Abl activation after IR, we used the ATM and DNAPK kinase inhibitors (KU55933 and NU7026, respectively) in the Abl-WT-EGFP and Abl-S465A-EGFP cell lines to dissect the potential contributions of each kinase to IR-induced Abl activation. Cells were pre-treated with an ATM inhibitor, a DNAPK inhibitor, or both for 20 min followed by addition of the biosensor peptide (5 min incubation) and irradiation at 5 Gy. As shown in Figure 3, neither single kinase inhibitor abolished the phosphorylation signal (4G10) from the Abl kinase biosensor in the Abl-WT-EGFP cell line (Figure 3A). Using our Abl(S465A)-EGFP cell line, we found that the ATM and DNAPK kinase inhibitors both attenuated Abl activation after IR—suggesting that some other phosphorylation site might be involved in ATM-dependent activation of our Abl mutant in this system and/or that off-target effects of the inhibitors were impacting the results. However, since the level of DNAPK autophosphorylation at S2056 was not substantially affected by the ATM inhibitor nor was the phosphorylation of ATM at its autophosphorylation site S1981 affected by the DNAPK inhibitor, it does not appear that off-target effects (e.g. inhibition of DNAPK by the ATM inhibitor and vice versa) were responsible.
Figure 3. Inhibition of DNAPK decreases activation of the S465A mutant.

A) In cells expressing Abl-WT-EGFP, phosphorylation of the biosensor peptide after IR (lane 2) was not significantly diminished using either inhibitor alone (lanes 3 and 4). B) However, in cells expressing Abl-S465A-EGFP, the DNAPK inhibitor NU7206 was sufficient to decrease biosensor phosphorylation levels (lane 4) to pre-IR background control levels (lane 1). B) and D) Over several replicates, cells expressing the Abl-S465A-EGFP and treated with the ATM inhibitor KU55933 also showed some decrease in Abl activation (lane 3) relative to cells with no inhibitor (lane 2)—however this decrease was not statistically significant. Differential ATM and DNAPK inhibition is shown using antiphospho-antibodies to their respective autophosphorylation/activation sites (S1981 for ATM and S2056 for DNAPK). C) and D) Data over three independent experiments were processed as described in Figure 2, averaged, tested for statistical significance relative to non-IR treated controls and plotted (p<0.05 indicated via *). Y-axes represent increase in 4G10/streptavidin signal in samples treated with IR vs. non-IR controls, and are labelled as +IR/-IR. Error bars represent SEM.
The results from performing our assay in the presence of ATM and DNAPK inhibitors in the inducible overexpression cell lines were somewhat ambiguous, showing a decrease in Abl-S465A-EGFP activation in response to the ATM inhibitor. Therefore, to better address the involvement of ATM in the activation of Abl that stimulates biosensor phosphorylation, we used an ATM-null mutant pre-B cell line that overexpresses v-Abl, which contains potentially relevant serine sites (including a residue homologous to S465 and others in the C-terminal region of the kinase) and compared IR-responsive biosensor phosphorylation in these with that observed in the cells that were wild-type for ATM expression.[31, 32] As shown in Figure 4A, in pre-B cells in which ATM expression is normal (Abl pre-B ATM2A), neither ATM nor DNAPK inhibitors alone significantly decreased the phosphorylation of the biosensor peptide after IR treatment (p>0.74 and >0.45 for ATM and DNAPK inhibitors, respectively). Likewise, in the ATM-null mutant pre-B cells (Abl pre-B ATMnull), no significant decrease in Abl activation after IR was seen following treatment with the ATM inhibitor (Figure 4B) (p>0.85), indicating that this version of v-Abl is not sensitive to some other mechanism of upstream inhibition caused by this ATM inhibitor. On the other hand, treatment of the ATM-WT cells (Abl pre-B ATM2A) with inhibitors of both ATM and DNAPK (Figure 4A, lane 5) and treatment of ATM-null cells (Abl pre-B ATMnull) with the single DNAPK inhibitor (Figure 4B, lane 4) decreased biosensor phosphorylation to a level comparable to that seen using the Abl inhibitor imatinib (Figure 4A and 4B, lane 6). Taken together, these data support the hypothesis that ATM and DNAPK both participate in early Abl activation after IR, but in a non-redundant manner, and that activation of Abl by DNAPK is not dependent on ATM.
Figure 4. IR-dependent activation of Abl kinase in murine v-Abl-expressing pre-B ATM (−/−) null cells is sensitive to a DNAPK inhibitor.

The biosensor assay was performed with v-Abl transfected preB cells that were A) WT or B) deficient for ATM expression. C) Data over two independent experiments were averaged and plotted. Statistically significant differences (p<0.05) between ATM (WT) and ATM (null) are indicated via *. Y-axis is labelled as norm increase (+IR), meaning the increase in the 4G10/strept signal for samples treated with IR vs non-IR controls. Error bars represent SEM.
Examining subcellular distribution and initial timing of biosensor phosphorylation
In principle, due to its small size our biosensor should be able to access various intracellular compartments—allowing us to detect and potentially dissect the contribution of nuclear Abl activation in response to IR. In order to examine whether the biosensor was distributed sufficiently to detect Abl activation in relevant subcellular contexts, we performed subcellular fractionation experiments using the Abl-WT-EGFP cells exposed to IR. Cells were incubated with the biosensor peptide added either concurrently with IR or for 5 min prior to IR, irradiated at 5 Gy and harvested at 5 or 10 min post-IR. The cytoplasmic and nuclear fractions were isolated as described in the materials and methods (characterized in the supporting information Figure S3) and analyzed by two-color Licor dye Western blot using streptavidin and the anti-phosphotyrosine antibody 4G10. As shown via streptavidin blotting in Figure 5A–B, the biosensor was present in both the nuclear and cytoplasmic fractions. Phosphorylation of the biosensor was increased as a function of incubation time even in cells not treated with IR (Figure 5C and D, columns 1–4), however an approximately 3-fold, statistically significant (p <0.05, both with respect to the difference from the no IR control and the difference between the 5 and 10 min time points) increase in the relative level of phosphorylation was observed by 10 min in the nuclear fraction for the cells treated concurrently with biosensor and IR (Figure 5A lane 8 and 5E column 4). These data indicated that the biosensor was being taken up and distributed very rapidly—within 5–10 min, which is consistent with observations from Stephen Dowdy’s group on the timing of uptake and nuclear activity for a TAT-tagged CRE recombinase.[33]
Figure 5. The Abl biosensor peptide is present and phosphorylated in both the cytoplasmic and nuclear fractions of Abl-WT-EGFP cells.
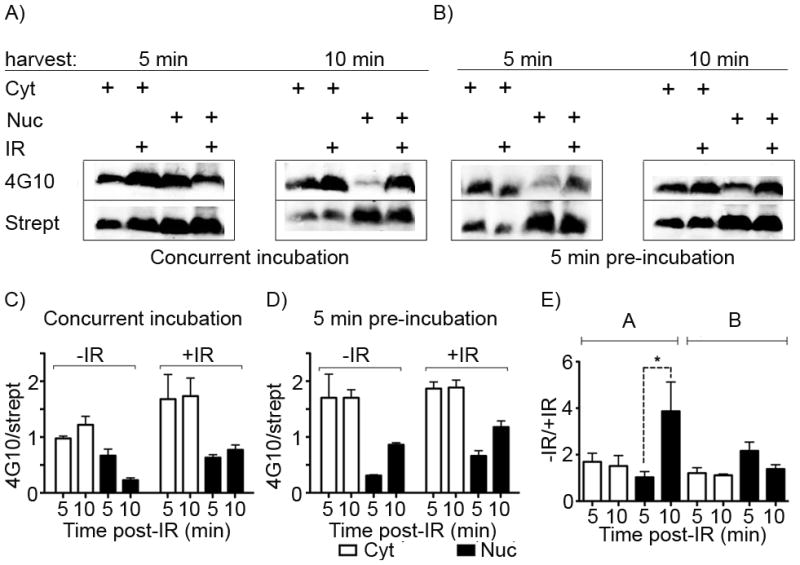
By isolating the cytoplasmic and nuclear material from cells treated with the Abl biosensor within 5 min after IR, we simultaneously examined subcellular localization and the earliest practical timepoint for post-IR sample processing. Western blotting was performed for biotinylated (Strept) and phosphorylated (4G10) peptide from 50 μg/lane of each subcellular fraction from cells incubated with the biosensor peptide A) concurrently with IR treatment or B) pre-treated for 5 min prior to IR. A) In cells expressing Abl-WT-EGFP, the biosensor peptide is observed in both the cytoplasmic (e.g. lanes 1 and 2) and nuclear (e.g. lanes 3 and 4) fractions within 5 min of incubation with and without IR. C) and D) Integrated band intensity data for 4G10 and streptavidin signals over three independent replicate experiments were calculated and the phosphorylated signal (4G10) was divided by the total’ marker signal (streptavidin) and plotted. E) These were further normalized to their change in relative intensity following IR). Y-axes are labelled as 4G10/strept or +IR/−IR, meaning the increase in the 4G10/strept signal for samples treated with IR vs non-IR controls. Error bars represent SEM, and statistical significance (p<0.05) is indicated using *.
The longer pre-IR incubation time (5 min) led to a higher level of basal biosensor phosphorylation (particularly in the cytoplasm, comparing Figure 5C columns 1–2 and 5D columns 1–2) and thus lower relative increase upon IR, however the increase in phosphorylation in the nuclear fraction by 5 min was still clearly observed (albeit not quite statistically significant, p = 0.09). Intriguingly, the data for the concurrently-treated cells (Figure 5C, columns 1 and 5) show an increase in the 4G10/streptavidin ratio for the cytoplasmic fraction by 5 min even in the absence of an increase in biosensor phosphorylation the nuclear fraction at that timepoint (Figure 5C, columns 3 and 7), suggesting the possibility that IR-activated Abl-WT-EGFP might be localizing in the cytoplasm very rapidly after IR exposure, or that the subcellular localization of the peptide itself is highly dynamic. Nuclear-cytoplasmic shuttling of Abl has been shown to be important for mediating its role in the DNA damage response,[21, 34] but has not previously been observed to occur so rapidly after damage. This trend was not statistically significant, however, and may have been confounded by the high levels of basal Abl activity in the cytoplasm resulting from the high levels of overexpression of the kinase in this system. Therefore we are currently focused on the development of optimized read-out techniques that can replace Western blot and provide both greater sensitivity in order to achieve sufficient signal to noise for detecting endogenous levels of Abl kinase activation, as well as imaging of subcellular localization, in systems that do not require overexpression.
Discussion
In this study we use cell lines engineered to express various version of Abl kinase, kinase inhibitors and an Abl kinase biosensor as chemical biology and pharmacological tools to provide evidence that both ATM and DNAPK kinases mediate the rapid activation of Abl in response to IR-induced DNA damage, and that our biosensor can detect this activation for both the nuclear and cytoplasmic fractions of Abl. We initially observed that Abl activity still increased, albeit modestly, in response to IR in cells expressing a mutant that cannot be phosphorylated at a key ATM phosphorylation site (S465 mutated to alanine). This indicates a role for other phosphorylation sites and/or kinases in stimulating IR-induced Abl activity. Using ATM and DNAPK kinase inhibitors in a v-Abl-transduced, ATM-deficient pre-B cell line, we showed that DNAPK activity was an important contributor to the rapid increase in Abl activity after IR in the absence of ATM. In fact, inhibition of both ATM and DNAPK activities abolished Abl activation in our assay, suggesting that one or the other is required, but that either kinase alone is sufficient to activate Abl during the early stages of the DDR. While no kinase inhibitor is completely specific, the concentrations used in our study were consistent with those reported by others to achieve reasonable specificity for DNAPK.[35] These results are consistent with and complementary to previous data indicating that, even though ATM typically activates Abl through the phosphorylation of S465 in response to IR,[17] there also is a functional and likely direct interaction between DNAPK and Abl that occurs in response to DNA damage.[24] Using the Tet-ON inducible Abl overexpression system, we also obtained evidence that the biosensor is reaching the nucleus, the relevant subcellular compartment for detecting DDR-related Abl activation. In addition, these experiments suggested that the cytoplasmic fraction of Abl is also activated within 5 min after IR. This provides the first evidence that Abl is activated very early on in the IR-induced DDR, and that it is either also activated in the cytoplasm by IR or may function to rapidly communicate information about DNA damage to cytoplasmic proteins.
This intracellular assay strategy therefore provides a unique insight into Abl activation that, so far, has not been apparent using traditional kinase assay methods and that we did not observe by monitoring common markers for Abl activation. For example, previous interpretations of the roles for DNAPK, ATM and Abl in the activation of one another after IR were made using in vitro kinase assays with immunoprecipitated Abl and either the C-terminal domain (CTD) of RNAPol II or a Crk construct as substrates.3,[17] These experiments provided key preliminary evidence that DNAPK and Abl interact physically and functionally, but could not address the potential for differences between interactions in vitro versus in the cell. Experiments using the RNAPol II CTD as a substrate showed no relationship between DNAPK and Abl activation as a function of IR, suggesting that phosphorylation of the RNAPol II CTD by Abl depends on other protein-protein interactions or scaffolds not provided by an in vitro experiment. However other experiments using a Crk-based substrate in vitro did show a dependence on DNAPK for Abl activation. Additionally, previous work has focused on the role of c-Abl in the later stages of DNA repair, implicating Abl as an inhibitor or deactivator of DSB repair foci[36, 37]. It has recently been suggested that Abl might function just as critically in the early, upstream stages of DSB repair, but that more comprehensive analysis of these early kinetics would be required to elucidate this process[37]. Using our intracellular biosensor substrate, we have been able to demonstrate this functional interaction in living cells to address that gap and provide a path to clarifying the respective roles of DNAPK, ATM and Abl in the IR-induced DNA damage response.
Another advantage of this biosensor substrate is that unlike native, endogenous protein substrates (which respond specifically to different exogenous stimuli), its phosphorylation seems to be relatively general. Since IR-induced endogenous substrate phosphoryla-tion is likely to be transient, localized and dependent on specific protein-protein interactions, detecting kinase activation via these endogenous proteins can be problematic. First, detection is highly dependent on either development of phosphorylation site-specific antibodies or favorable physio-chemical properties of the tryptic (or other protease-produced) peptides generated from the phosphorylation sites. For example, even though an excellent phosphospecific antibody exists for CrkL, a well-studied substrate of cAbl known to be a nuclear receptor, analysis of CrkL phosphorylation at Y207 did not provide any insight into Abl activation after IR. [38] Second, the low stoichiometry of modification can make it even more difficult to detect phosphorylation on these already low-level proteins. Working with these overexpression model systems and leveraging the increased enzymatic turnover obtained upon functional activation of the kinase, we are able to access a reasonable dynamic range for Abl kinase activity utilizing our sensor substrate to a point where it is easily detectable with a general, well-characterized antibody (4G10). It is important to point out, however, that the difference in detection sensitivity between uninduced and induced samples suggests that Abl kinase natively expressed in HEK293 cells exists at levels below the dynamic range of the Western blot-based detection method used for this assay. This is likely to be cell-line dependent and thus should be characterized in each new system used by others.
Context is important for understanding the roles of substrates and phosphorylation sites, however, there may also be value in simply knowing whether a kinase has been functionally activated or not, without relying on detailed foreknowledge of the direct, downstream consequences. Normally for the Abl kinase, phosphorylation of Y245 in the polyproline loop region thought to be involved in autoregulation, is used as a general marker for activation.[39] However, in the overexpression system used here, the extent and temporal trends of Y245 phosphorylation were distinct from the activity observed using our sensor peptide (Figure 2). Certainly some of this effect is due to the relative differences in antibody sensitivities for 4G10 vs. the anti-phospho-Abl(Y245) antibodies. However, the differences in the trends observed might be because even this ‘general’ autoactivation is context-dependent and not necessarily required for regulation of c-Abl through alternative, allosteric interactions and/or phosphorylation sites, and therefore is not the ultimate, ideal marker for intrinsic kinase activation. Given that these changes in intrinsic kinase domain activity observed using the biosensor peptide were different than those detected via the autophosphorylation site, this technique seems to provide complementary information about early Abl kinase activation after IR. FRET-based biosensors for Abl and other kinase activities also have the potential to provide this type of information. The primary advantage of such FRET-based sensors is that subcellular kinase activities can be imaged in real time—however, these techniques require sophisticated quantitative imaging and can suffer from low dynamic range in the fold-response that can be detected, since changes in FRET signal are sometimes small even upon dramatic changes in the level of phosphorylation of the sensor. Because the phosphorylation of the peptide biosensor described here can be monitored in a standard, simple manner using either the generic phosphotyrosine antibody 4G10 or label-free mass spectrometry (as we have previously demonstrated[25]), this strategy could be advantageous for molecular biology laboratories that want a simple, straightforward assay for exploring the initial timecourse of Abl’s role in the DNA damage response.
Conclusion
These results contribute to the understanding of Abl kinase activity early in the DNA damage response,[20, 40, 41] as well as provide a starting point and potential tool for further examination on the specific roles of different upstream kinases in activating Abl during this process. We are currently undertaking further studies to examine DNAPK’s role in activating Abl after IR. ATM, recruited by the MRE-11 complex, is thought to activate homologous recombination repair. Abl is known to phosphorylate Rad51 and Rad52[42] in an ATM-dependent manner [43], which are also involved in homologous recombination. DNAPK, on the other hand, is recruited by the Ku complex and stimulates other methods of repair including non-homologous end joining, typically thought of as more error-prone processes. Some preliminary evidence has shown that Abl phosphorylates WRN,[44] a helicase that is mutated in Werner’s syndrome patients and which has been linked to non-homologous end joining. Furthermore, NHEJ is known to be upregulated in chronic myelogenous leukemia cells, which overexpress the constitutively-active Abl mutant Bcr-Abl—however it is not yet clear whether this proceeds through DNAPK or alternative mechanisms.[45] This biosensor strategy could provide a tool for understanding how these two different DNA repair pathways are regulated and activated, both after exogenous damage and as a result of endogenous processes such as VDJ recombination.[32]
Additionally, ATM, DNAPK and c-Abl have each been linked to radioresistance in some cancer cell lines and tumor samples,[46–49] however specific radioresistance mechanisms related to their kinase activities have not been well characterized. There is some evidence that the Abl inhibitor imatinib can either protect against IR-induced cell death[50] or induce radio-sensitivity[51, 52] depending on the cell type. Additionally, since c-Abl inhibition could potentially have undesirable effects in some cancer contexts, such as disruption of EphB4 tumor suppressor function[53] and loss of mismatch repair-related apoptosis[54, 55], a careful analysis of the details of these signaling pathways will be necessary to design kinase inhibitor radiosensitizer treatment strategies.
Experimental Section
Kinase inhibitors
The inhibitors used in this work were: KU55933 (10 μM) (in vitro ATM kinase inhibition IC50 ~13 nM, DNAPK kinase inhibition >2 μM)(Tocris Bioscience); NU7026 (10 μM) (in vitro DNAPK kinase inhibition IC50 ~200 nM, ATM kinase inhibition >50 μM) (Tocris Bioscience) and imatinib (20 μM) (Abl kinase inhibitor, in vitro IC50 ~200 nM, LC Laboratories). Autophosphorylation of each kinase was analyzed by Western blot as a control for relative inhibitor specificity.
Plasmid constructs
The Abl-WT-EGFP coding region was amplified by PCR from the Abl-WT-EGFP-N3 vector kindly provided by Dr. Naoki Watanabe (Kyoto University, Japan), using the primers 5′-GGGGCGGCCGCATGGGGCAGCAGCCTGGAAAAGTT-3′ and 5′-GGG ACGCGTTTACTTGTACAGCTCGTCCATGCCGAGAGTGA-3′, and subcloned into the pLVX-Tight-Puro vector (Clontech Laboratories) to generate an Abl-WT-EGFP TET-inducible expression construct (pLVX-Tight-Puro-Abl-WT-EGFP). The S465A point mutation was made using the QuikChange site-directed mutagenesis kit (Stratagene) using primer (5′-GGGAATTGACCTGgCTCAGGTTT ATGAGC-3′ and 5′-GCTCATAAACCTGAGcCAGGTCAATTCCC-3′). The mutagenesis was confirmed by sequencing.
Generation of cells with TET-ON inducible Abl-WT-EGFP expression
The Abl-WT-EGFP inducible cell lines were generated by lentiviral infection of the HEK 293 Tet-On Advanced Cell Line, which expresses one of the tetracycline-controlled transactivators and the Geneticin (G418) resistance gene (Clontech Laboratories). The lentivirus was produced using the Lenti-X HT packaging system according to the manufacturer’s instructions (Clontech Laboratories), using the pLVX-Tight-Puro-Abl-WT-EGFP or Abl(S465A)-EGFP, which expresses the puromycin resistance gene. Infected cells were selected in puromycin (0.5 μg/ml) and G418 (100 μg/ml). Inducible expression of Abl-WT-EGFP or Abl(S465A)-EGFP was assessed by treatment with doxycycline and anti-GFP Western blotting. Colonies positive for expression were selected.
Cell culture
HEK293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with fetal bovine serum (10% v/v), penicillin/streptomycin (1% v/v), G418 (100 μg/ml) and puromycin (0.25 μg/ml) (all from Sigma Aldrich, Inc.) at 37°C in CO2 (5%). For the v-Abl pre-B cells [32], cells were cultured in DMEM supplemented with fetal bovine serum (10%), sodium pyruvate (1 mM), L-glutamine (2 mM), nonessential amino acids (NEAA) (0.1 mM) and 2-mercaptoethanol (0.00004% v/v) at 37°C in CO2 (5%).
Peptide synthesis and purification
Amino acids and other peptide synthesis reagents were obtained from Peptides International (Louisville, KY, USA) except for the Fmoc–biocytin (Kbiotin, obtained from Akaal Organics, Long Beach, CA, USA) and the photocleavable residue (3-(2-nitrobenzyl)-3-aminopropionic acid (purchased from Lancaster Synthesis and Fmoc-protected by J. Thomas Ippoliti’s lab at the University of St. Thomas, St. Paul, MN, USA). Peptides were synthesized at a 50-μmol scale on CLEAR-Amide resin (mixed with glass beads to avoid clumping) using solid-phase Fmoc chemistry (Fmoc-protected amino acid monomers at 100 mM final concentration) with a Prelude Parallel Peptide Synthesizer (Protein Technologies, Tucson, AZ, USA) and using “fast” protocols (coupling time: 2 × 10 min; deprotection time: 2 × 2 min) with HCTU/NMM (2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate/N-methylmorpholine, 95 mM/200 mM final concentrations) as coupling reagent. Peptides were analyzed with high-performance liquid chromatography (HPLC)/MS (Accela/LTQ, Thermo Finnigan) and MALDI TOF/TOF (Voyager 4800, Applied Biosystems, Foster City, CA, USA) and were purified using a C18 reverse-phase column on an Agilent Technologies 1200 Series preparative HPLC system (Santa Clara, CA, USA).
Biosensor assay
Abl-WT-EGFP HEK293T cells
Cells were seeded at 0.5–1.5 × 106 cells/well in 6-well plate and allowed to recover for 24 h. Induction of Abl-WT-EGFP construct expression was then achieved by treating the cells for 18–24h with doxycycline (2 μM). When desired, cells were pre-treated with kinase inhibitors (10 μM ATM and/or DNAPK inhibitors, 20 μM imatinib) in fresh medium for 20 min before removing the medium and adding the biosensor in fresh medium. The peptide biosensor (25 μM) was added and the cells incubated for 0 or 5 min before IR treatment (5 Gy). For non-fractionated samples, cells were harvested at different time-points post-IR treatment, collected, washed once with PBS and flash-frozen in PhosphoSafe Extraction Reagent (Novagen, San Diego, CA, USA) containing freshly dissolved complete protease inhibitor (Roche, Nutley, NJ, USA). For subcellular fractionation experiments, lysates of the cytoplasmic and nuclear fractions were prepared using the Nuclear Extract Kit from ActiveMotif according to the manufacturers instructions. Representative blots of fractionated material are given in the supporting information (Figure S2) to show that the biosensor was present in each fraction for the Abl-WT-EGFP and Abl-S465A-EGFP cells.
v-Abl pre-B cells
Cells were seeded at 20× 106 cells per well and pre-treated with different kinase inhibitors (10 μM ATM and/or DNAPK inhibitors, 20 μM imatinib) for 30 min. Peptide biosensor (25 μM) was added for 5 min before 5 Gy IR treatment. Cells were harvested post-IR treatment after incubating for 10 min at 37 °C, prepared and lysed as described above.
Western blot analysis
Lysate (100 μg protein/non-fractionated samples or 50 μg protein/each fraction for fractionated samples) was denatured with NuPAGE Laemmli protein gel loading buffer (Invitrogen) and run on a 4–12% Bis–Tris NuPAGE gel (Invitrogen). Proteins were transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) for 1 h at 100V and 4 °C. Membranes were blocked in 5% milk/TBS-T (Tris-buffered saline containing Tween 20) for 1 h at room temperature and cut into two segments: >15 kDa (containing most endogenous proteins) and <15 kDa (containing biosensor peptide). The >15-kDa segment was analyzed with immunoblotting using different antibodies: GFP (1:1000, rabbit, Invitrogen)), pATM (S1981, 1:1000, mouse, Abcam), pDNAPK (S2056, 1:1000, mouse, Abcam), and pCrkl (Y207, 1:1000, rabbit, Abcam). The antibody cocktail included either anti-tubulin (1:100,000, rat, Millipore) or GAPDH (1:1000, mouse, Abcam) to ensure equal loading. Membranes were incubated with the cocktail overnight at 4 °C. The <15-kDa segment was analyzed with immunoblotting using a cocktail of anti-phosphotyrosine antibody 4G10 (1:1000, mouse, Millipore) and streptavidin labeled with DyLight 680 (1:1000, Rockland Immunochemical) and incubated overnight at 4 °C. Membranes were washed (3 × 5 min, TBS-T), visualized by incubating for 1 h at room temperature with goat-derived secondary antibodies (1:10,000 for anti-mouse and anti-rabbit and 1:20,000 for anti-rat in 5% milk/TBS-T) tagged with IRDye 680 or 800 (LI-COR Biosciences) and scanned for both dyes simultaneously using a LI-COR Odyssey infrared scanner. Band densities from scanned images were determined using QuantityOne software (Bio-Rad). Data were plotted in GraphPad Prism software from biological replicate analyses (n ≥ 2 or 3).
Supplementary Material
Acknowledgments
We are very grateful to Profs. Naoki Watanabe (Kyoto University) and Barry Sleckman (Washington University St. Louis) for their kind gifts of the Abl-WT-EGFP constructs and v-Abl transduced/ATM-deficient pre-B cell lines, respectively. We also thank Prof. J. Thomas Ippoliti and his laboratory for synthesizing the Fmoc-protected photocleavable linker residue. Profs. Robert Geahlen, Tony Hazbun, Albert Bowers and Val Watts (Purdue University) provided helpful critique of the manuscript during preparation. This work was supported by an NIH/NCI K99/R00 Pathway to Independence award to LLP (CA127161).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Wang JY, Cho SK. Adv Protein Chem. 2004;69:101. doi: 10.1016/S0065-3233(04)69004-8. [DOI] [PubMed] [Google Scholar]
- 2.Davis AJ, So S, Chen DJ. Cell Cycle. 2010;9 doi: 10.4161/cc.9.16.12814. [DOI] [PubMed] [Google Scholar]
- 3.Kharbanda S, Yuan ZM, Weichselbaum R, Kufe D. Oncogene. 1998;17:3309. doi: 10.1038/sj.onc.1202571. [DOI] [PubMed] [Google Scholar]
- 4.Khanna KK, Jackson SP. Nat Genet. 2001;27:247. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 5.Shangary S, Brown KD, Adamson AW, Edmonson S, Ng B, Pandita TK, Yalowich J, Taccioli GE, Baskaran R. J Biol Chem. 2000;275:30163. doi: 10.1074/jbc.M004302200. [DOI] [PubMed] [Google Scholar]
- 6.Durocher D, Jackson SP. Curr Opin Cell Biol. 2001;13:225. doi: 10.1016/s0955-0674(00)00201-5. [DOI] [PubMed] [Google Scholar]
- 7.Yang J, Yu Y, Hamrick HE, Duerksen-Hughes PJ. Carcinogenesis. 2003;24:1571. doi: 10.1093/carcin/bgg137. [DOI] [PubMed] [Google Scholar]
- 8.Shiloh Y. Trends Biochem Sci. 2006;31:402. doi: 10.1016/j.tibs.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 9.Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER, 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ. Science. 2007;316:1160. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 10.So S, Davis AJ, Chen DJ. J Cell Biol. 2009;187:977. doi: 10.1083/jcb.200906064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottlieb TM, Jackson SP. Cell. 1993;72:131. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 12.Smith GC, Divecha N, Lakin ND, Jackson SP. Biochem Soc Symp. 1999;64:91. [PubMed] [Google Scholar]
- 13.Callen E, Jankovic M, Wong N, Zha S, Chen HT, Difilippantonio S, Di Virgilio M, Heidkamp G, Alt FW, Nussenzweig A, Nussenzweig M. Mol Cell. 2009;34:285. doi: 10.1016/j.molcel.2009.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. Oncogene. 2005;24:949. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 15.Meek K, Dang V, Lees-Miller SP. Adv Immunol. 2008;99:33. doi: 10.1016/S0065-2776(08)00602-0. [DOI] [PubMed] [Google Scholar]
- 16.Hennequin C, Giocanti N, Averbeck D, Favaudon V. Cancer Radiother. 1999;3:289. doi: 10.1016/s1278-3218(99)80070-5. [DOI] [PubMed] [Google Scholar]
- 17.Baskaran R, Wood LD, Whitaker LL, Canman CE, Morgan SE, Xu Y, Barlow C, Baltimore D, Wynshaw-Boris A, Kastan MB, Wang JY. Nature. 1997;387:516. doi: 10.1038/387516a0. [DOI] [PubMed] [Google Scholar]
- 18.Wang J. In: Handbook of Cell Signaling. 2. Bradshaw RA, Dennis EA, editors. Academic Press; Elsevier, San Diego, CA: 2010. [Google Scholar]
- 19.Pendergast AM. Adv Cancer Res. 2002;85:51. doi: 10.1016/s0065-230x(02)85003-5. [DOI] [PubMed] [Google Scholar]
- 20.Woodring PJ, Hunter T, Wang JY. J Cell Sci. 2003;116:2613. doi: 10.1242/jcs.00622. [DOI] [PubMed] [Google Scholar]
- 21.Wang JY. Cell Res. 2005;15:43. doi: 10.1038/sj.cr.7290263. [DOI] [PubMed] [Google Scholar]
- 22.Colicelli J. Sci Signal. 2010;3:re6. doi: 10.1126/scisignal.3139re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshida K, Miki Y. Cell Cycle. 2005;4:777. doi: 10.4161/cc.4.6.1746. [DOI] [PubMed] [Google Scholar]
- 24.Kharbanda S, Pandey P, Jin S, Inoue S, Bharti A, Yuan ZM, Weichselbaum R, Weaver D, Kufe D. Nature. 1997;386:732. doi: 10.1038/386732a0. [DOI] [PubMed] [Google Scholar]
- 25.Placzek EA, Plebanek MP, Lipchik AM, Kidd SR, Parker LL. Anal Biochem. 2010;397:73. doi: 10.1016/j.ab.2009.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou S, Carraway KL, 3rd, Eck MJ, Harrison SC, Feldman RA, Mohammadi M, Schlessinger J, Hubbard SR, Smith DP, Eng C, et al. Nature. 1995;373:536. doi: 10.1038/373536a0. [DOI] [PubMed] [Google Scholar]
- 27.Pisabarro MT, Serrano L. Biochemistry. 1996;35:10634. doi: 10.1021/bi960203t. [DOI] [PubMed] [Google Scholar]
- 28.Wadia JS, Dowdy SF. Advanced Drug Delivery Reviews. 2005;57:579. doi: 10.1016/j.addr.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Shafman T, Khanna KK, Kedar P, Spring K, Kozlov S, Yen T, Hobson K, Gatei M, Zhang N, Watters D, Egerton M, Shiloh Y, Kharbanda S, Kufe D, Lavin MF. Nature. 1997;387:520. doi: 10.1038/387520a0. [DOI] [PubMed] [Google Scholar]
- 30.Amrein L, Davidson D, Shawi M, Petruccelli LA, Miller WH, Jr, Aloyz R, Panasci L. Leuk Res. 2011 doi: 10.1016/j.leukres.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Mahowald GK, Baron JM, Mahowald MA, Kulkarni S, Bredemeyer AL, Bassing CH, Sleckman BP. Proc Natl Acad Sci U S A. 2009;106:18339. doi: 10.1073/pnas.0902545106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bredemeyer AL, Helmink BA, Innes CL, Calderon B, McGinnis LM, Mahowald GK, Gapud EJ, Walker LM, Collins JB, Weaver BK, Mandik-Nayak L, Schreiber RD, Allen PM, May MJ, Paules RS, Bassing CH, Sleckman BP. Nature. 2008;456:819. doi: 10.1038/nature07392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wadia JS, Stan RV, Dowdy SF. Nat Med. 2004;10:310. doi: 10.1038/nm996. [DOI] [PubMed] [Google Scholar]
- 34.Preyer M, Shu CW, Wang JY. Cell Death Differ. 2007;14:1139. doi: 10.1038/sj.cdd.4402119. [DOI] [PubMed] [Google Scholar]
- 35.Amrein L, Loignon M, Goulet AC, Dunn M, Jean-Claude B, Aloyz R, Panasci L. J Pharmacol Exp Ther. 2007;321:848. doi: 10.1124/jpet.106.118356. [DOI] [PubMed] [Google Scholar]
- 36.Meltser V, Ben-Yehoyada M, Reuven N, Shaul Y. Cell Death Dis. 2010;1:e20. doi: 10.1038/cddis.2009.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meltser V, Ben-Yehoyada M, Shaul Y. Cell Death Differ. 2011;18:2. doi: 10.1038/cdd.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rhodes J, York RD, Tara D, Tajinda K, Druker BJ. Exp Hematol. 2000;28:305. doi: 10.1016/s0301-472x(99)00148-4. [DOI] [PubMed] [Google Scholar]
- 39.Brasher BB, Van Etten RA. J Biol Chem. 2000;275:35631. doi: 10.1074/jbc.M005401200. [DOI] [PubMed] [Google Scholar]
- 40.Woodring PJ, Hunter T, Wang JY. J Biol Chem. 2001;276:27104. doi: 10.1074/jbc.M100559200. [DOI] [PubMed] [Google Scholar]
- 41.Wang JY. Nat Cell Biol. 2004;6:3. doi: 10.1038/ncb0104-3. [DOI] [PubMed] [Google Scholar]
- 42.Popova M, Shimizu H, Yamamoto K, Lebechec M, Takahashi M, Fleury F. FEBS Lett. 2009;583:1867. doi: 10.1016/j.febslet.2009.04.044. [DOI] [PubMed] [Google Scholar]
- 43.Chen G, Yuan SS, Liu W, Xu Y, Trujillo K, Song B, Cong F, Goff SP, Wu Y, Arlinghaus R, Baltimore D, Gasser PJ, Park MS, Sung P, Lee EY. J Biol Chem. 1999;274:12748. doi: 10.1074/jbc.274.18.12748. [DOI] [PubMed] [Google Scholar]
- 44.Cheng WH, von Kobbe C, Opresko PL, Fields KM, Ren J, Kufe D, Bohr VA. Mol Cell Biol. 2003;23:6385. doi: 10.1128/MCB.23.18.6385-6395.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sallmyr A, Tomkinson AE, Rassool FV. Blood. 2008;112:1413. doi: 10.1182/blood-2007-07-104257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, Tofilon PJ. Cancer Res. 2003;63:7377. [PubMed] [Google Scholar]
- 47.Rainey MD, Charlton ME, Stanton RV, Kastan MB. Cancer Res. 2008;68:7466. doi: 10.1158/0008-5472.CAN-08-0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Beskow C, Skikuniene J, Holgersson A, Nilsson B, Lewensohn R, Kanter L, Viktorsson K. Br J Cancer. 2009;101:816. doi: 10.1038/sj.bjc.6605201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eschrich S, Zhang H, Zhao H, Boulware D, Lee JH, Bloom G, Torres-Roca JF. Int J Radiat Oncol Biol Phys. 2009;75:497. doi: 10.1016/j.ijrobp.2009.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maiani E, Diederich M, Gonfloni S. Biochem Pharmacol. 2011 doi: 10.1016/j.bcp.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 51.Chung HW, Wen J, Lim JB, Bang S, Park SW, Song SY. Int J Radiat Oncol Biol Phys. 2009;75:862. doi: 10.1016/j.ijrobp.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 52.Weigel MT, Dahmke L, Schem C, Bauerschlag DO, Weber K, Niehoff P, Bauer M, Strauss A, Jonat W, Maass N, Mundhenke C. BMC Cancer. 2010;10:412. doi: 10.1186/1471-2407-10-412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang JY. Nat Cell Biol. 2006;8:785. doi: 10.1038/ncb0806-785. [DOI] [PubMed] [Google Scholar]
- 54.Li LS, Morales JC, Hwang A, Wagner MW, Boothman DA. J Biol Chem. 2008;283:21394. doi: 10.1074/jbc.M709954200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wagner MW, Li LS, Morales JC, Galindo CL, Garner HR, Bornmann WG, Boothman DA. J Biol Chem. 2008;283:21382. doi: 10.1074/jbc.M709953200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.