

Abstract
The adenosine agonist [3H]CGS21680 (2-[4-[[2-carboxyethyl]phenyl]ethylamino]-5'-N-ethylcarboxamidoadenosine) bound to A2 receptors in human striatal membranes with a Kd of 17.8±1.1 nM and a Bmax of 313± 10 fmol/mg protein. The addition of 100 μM GTP diminished both the affinity of agonist radioligand for A2 adenosine binding sites and the total binding, resulting in Kd and Bmax values of 28.6±1.0 nM and 185± 22 fmol/mg of protein. Adenosine ligands competed for [3H]CGS21680 with the expected potency order. The adenosine antagonist [3H]XAC (8-[4-[[[[(2-aminoethyl)-amino]carbonyl]methyl]oxy]phenyl]-1,3-dipropylxanthine), although A1-selective in the rat, binds to human striatal A2 receptors with high affinity. 25 nM CPX (8-cyclopentyl-1,3-dipropylxanthine), an A1-selective antagonist, was added to the incubation medium and effectively eliminated 91% of [3H]XAC (1 nM) binding to human A1 receptors, yet preserved 90% of binding to A2 receptors. [3H]XAC exhibited saturable, specific binding (50% of total) to A2 sites with a Kd of 2.98±0.54 nM and a Bmax of 0.71±0.23 pmol/mg protein (25°C, non-specific binding defined with 100 μM NECA). The potency order for antagonists against 1 nM [3H]XAC was CGS15943A > XAC ≈ PD115,199 > PAPA-XAC > CPX > HTQZ ≈ XCC ≈ CP-66,713 > theophylline ≈ caffeine, indicative of an A2-type binding site. A2a-receptors were found to be present in the human cortex, albeit at a much lower density than in the striatum. Photoaffinity labeling using 125I-PAPA-APEC revealed a molecular weight of 45K, but proteolytic cleavage was observed, resulting in fragments of MW 43K and 37K. In the absence of proteolytic inhibitors the 37K fragment, which still bound 125I-PAPA-APEC, was predominant.
INTRODUCTION
A2-adenosine receptors mediate the anti-platelet-aggregatory effects (1) and vasodilatory effects (2,3) of adenosine. Two putative subtypes of A2-adenosine receptors have been distinguished: A high affinity A2a receptor and a low affinity A2b receptor (23). In the central nervous system, A2a-adenosine receptors (4), are localized mainly in the striatum and olfactory tubicle (5). Both A1 and A2-adenosine receptors mediate the depression of neuronal firing elicited by adenosine. A selective A2-adenosine agonist was found to act as a locomotor depressant, through a centrally-mediated mechanism (6).
Recently, two selective agonist radioligands with high affinity for A2-receptors, [3H]CGS21680 and 125I-PAPA-APEC (2-[4-[2-[2-(4-aminophenylacetyl)aminoethyl]-aminocarbonyl]ethyl]phenyl]ethylamino]-5'-N-ethylcarboxamidoadenosine), have been reported (7,17). The development of antagonist radioligands for A2-receptors has been impeded by the lack of truly selective agents. In this study, we have taken advantage of the unusual high affinity (but not selectivity) at human A2-receptors of the antagonist [3H]XAC. This is consistent with previously noted species differences (27,28) in affinity of xanthine derivatives for adenosine receptors.
Recently, a G-protein linked receptor for which the amino acid sequence was determined by recombinant DNA methodology using mRNA from the dog thyroid, was identified as an A2-receptor (10). When expressed in COS cells, the protein was found to resemble the high affinity A2a-receptor in radioligand binding and in stimulation of adenylate cyclase. The A2a-receptor has been characterized previously by photoaffinity labeling methods (7–9) and found to be a glycoprotein of molecular weight 45K Daltons (bovine and rat). The A2a-adenosine receptor in rabbit striatum has a molecular weight of 47K (9), and in the absence of proteolytic inhibitors undergoes proteolytic cleavage to yield a 38K fragment, still capable of binding radioligands with the appropriate pharmacology. In this work, it is shown that the human A2a-receptor also undergoes proteolytic cleavage to yield a 37K fragment. Because of the difficulty of obtaining fresh human brain samples, some proteolytic cleavage will be unavoidable, even in the presence of proteolytic inhibitors. Therefore, we chose to study the 37K proteolytic product, and protease inihbitors were deliberately left out after the membrane preparations.
MATERIALS AND METHODS
XAC, CPA (N6-cyclopentyladenosine), ADAC (N6-[4[[[4-[[[(2-aminoethyl)amino]carbonyl]methyl]anilino]carbonyl]-methyl]phenyl]adenosine), DPMA (N6-[2-(3,5-dimethoxyphenyl)-2-(2-methylphenyl)ethyl]adenosine), CGS21680, NECA (5'-N-ethylcarboxamidoadenosine), 2-chloroadenosine, 8-p-sulfophenyltheophylline and CPX were obtained from Research Biochemicals, Inc. (Natick, MA). The A2-adenosine agonists APEC (2-[4-[2-[2-aminoethyl]aminocarbonyl]ethyl]phenyl]ethylamino]-5'-N-ethylcarbox-amidoadenosine), its Nα-p-aminophenylacetyl derivative, PAPA-APEC, and other derivatives were prepared as described (12). Ac-ADAC, the N-acetyl amide of ADAC, was prepared as described (11). m-DITC (m-phenylenediisothiocyanate) derivatives of ADAC and APEC were prepared as described (12). PD115,199 (8-[4-[[(2-dimethylaminoethyl)N-methylamino]sulfonyl]phenyl]-1,3-dipropylxanthine) (13) and HTQZ (3-(3-hydroxyphenyl)-5H-thiatolo[2,3-b]quinazoline) (14) were the gift of Dr. J. Bristol, Parke-Davis Warner Lambert, Ann Arbor MI. CGS15943A (9-chloro-2-(2-furyl)-(1,2,4]triazolo[1,5-c]quinazolin-5-amine) (15) was the gift of Dr. J. Francis of CIBA-Geigy, Summit, NJ. [3H]XAC and [3H]CGS21680 were obtained from Dr. E. Do of Dupont NEN (Boston, MA). CP-66,713 (4-amino-8-chloro-1-phenyl-[1,2,4]triazolo(4,3-a]quinoxaline) (16) was the gift of Dr. R. Sarges of Pfizer, Groton, CT.
Preparation of striatal membranes
Human striatal tissue was isolated by dissection from 1 cm thick coronal sections obtained from the Brain Bank at the University of California at lrvine School of Medicine. The age ranged from 45 to 85 years (median age 73), both male and female, and the samples had been frozen at −70°C between 2.5 and 9 hours post-mortem. The tissue was pooled at the time of preparation of membranes. A subject diagnosed with Alzheimer's disease was an 89 year-old male, and this sample was processed separately. Although none of the human subjects tested positive for infectious diseases, precautions against contact with infectious agents were observed. Caudate and putamen were processed together for most experiments, in the absence of protease inhibitors. Striatal membranes were homogenized in 20 volumes of ice cold 50 mM Tris (tris(hydroxymethyl)aminomethane) HCI at pH 7.4 using a polytron (Kinematica, Gmbh, Luzerne, Switzerland) at a setting of 2–3 for 10 sec. The membrane suspension was then centrifuged at 37,000 × g for 20 min at 4°C. The pellet was resuspended in the above buffer solution, and the membranes were again homogenized and centrifuged. Finally the pellet was suspended in buffer (100 mg wet weight per ml) and stored frozen at −70°C until used. Protein was determined using the BCA protein assay reagents (Pierce Chemical Co., Rockford, III).
Radioligand binding
[3H]CGS 21680 binding was carried out as described (17) using 20 μM 2-chloroadenosine to determine non-specific binding.
The binding of [3H]XAC (18–20) to human striatal membranes was measured in a total volume of 1 ml. An unlabeled competing ligand or NECA (at a final concentration of 100 μM, for determination of non-specific binding) was dissolved in 25 μl of dimethylsulfoxide. To this solution was added, 25 μl of 1 μl CPX to eliminate binding to A1-adenosine receptors, 750 μl of 50 mM Tris at pH 7.4 at room temperature, and 100 μl of radioligand to achieve a final concentration of 1 nM. Finally, 100 μl of a striatal tissue suspension was added to the assay medium, which contained 3 IU/ml adenosine deaminase, from calf intestine (Boehringer Mannheim GmbH, Germany). The mixture was incubated with shaking for 90 min at 24°C. All assays were done in duplicate or triplicate. The binding reaction was terminated by addition of 4 ml of ice cold 50 mM Tris, pH 7.4, followed by rapid filtration using a Brandel Cell Harvester (Brandel, Gaithersburg, MD). The contents of each tube were filtered through a Whatman GF/B filter which had been soaked in a flat pan in buffer containing 0.3% polyethyleneimine for 90 min. The addition of 0.01% CHAPS (3-[(3-cholamidopropyl)dimethylammnio]-1-propane sulfonate) to the TRIS buffer solution used for washing the membranes after adsorption to the glass fiber filters was found to enhance specific [3H]XAC binding. The filters were washed twice with 4 ml of ice cold 50 mM Tris, pH 7.4. Each filter disc was added to 7 ml of scintillation fluid, vortexed, and counted after at least 6 hours.
The minimum quantity of tissue required for reproducible binding was investigated, based on the ratio of specific to total binding. Specific [3H]XAC binding to A2-receptors relative to total binding increased linearly with increases in protein concentration over a range of 15–100 μg of protein per ml, suggesting that in this range non-specific binding represents in large part binding of [3H]XAC to the glass fiber filters. The degree of specific binding reached a plateau at quantities of protein in the range of 100–400 μg per ml. Thus, [3H]XAC binding experiments were carried out using 150–200 μg protein per ml.
IC50 values were computer-generated from competition binding data, using a non-linear regression formula on the GraphPAD program (Institute for Scientific Information), and were converted to Ki values using a Kd value for [3H]XAC of 2.98 nM, as determined in saturation experiments (see below) and the Cheng-Prusoff equation (21).
Photoaffinity labeling
For photoaffinity labeling, 125I-azido-PAPA-APEC was synthesized and purified by HPLC as described (22). Frozen human brain membranes were thawed, washed and suspended in 50 mM HEPES (N-2-hydroxyethylpiperazin-N'-2-ethanesulfonic acid) buffer (pH 6.8), containing 10 mM MgCl2 in either the absence or presence of protease inhibitors (5 mM EDTA, 0.1 mM phenylmethanesulfonyl fluoride, 0.1 mg/ml soybean trypsin inhibitor, 5 μg/ml leupeptin, 1 μg/ml pepstatin A). After pretreatment with 0.5 unit/ml adenosine deaminase, the membranes (approximately 1 mg protein/ ml / tube) were incubated in the dark at 37°C for 1 hour in the presence of 125I-azido-PAPA-APEC. Ice cold incubation buffer was then added followed by centrifugation at 43,000 μ g. The pellet was resuspended in 1 ml incubation buffer and irradiated for 4 min at a distance of 1 cm from the light source (UVCG-25 mineral light, 254 nm, UVP Inc., San Gabriel, CA). Membranes were then washed again and solubilized in 10% SDS/glycerol buffer prior to electrophoresis on a 10% acylamide gel. Radioactive bands were visualized by autoradiography.
RESULTS
Radioligand binding
Membranes prepared from pooled human striatal samples were subjected to saturation binding experiments at 25°C with [3H]CGS 21680, a high affinity and A2-selective adenosine agonist (3,17). A representative saturation isotherm for specific [3H]CGS21680 binding is shown in Figure 1. The amount of specific binding for 5 nM [3H]CGS21680 was approximately 65% of the total counts. Non-specific binding was defined with 20 μM 2-chloroadenosine. The same amount of nonspecific binding was obtained in the presence of 40 μM 2-chloroadenosine, 200 μM NECA, or 40 μM CGS21680. The Scatchard plot of the data is linear indicating a homogeneous class of non-cooperative binding sites. A Kd value of 17.8±1.1 nM and a Bmax value of 313±10 fmol/mg protein were determined.
Figure 1.

Saturation of [3H]CGS21680 binding to human striatal A2-adenosine receptors. Speciiic (o), non-specific (•), and total (Δ) binding were determined for 90 min at 24°C. Values are means of a typical experiment done in triplicate. The calculated Kd-value of 17.8±1.0 nM represents the mean of three separate experiments on membranes from pooled striatal tissue.
[3H]CGS21680 binding was also carried out in the presence of 100 μM GTP. The presence of the guanine nucleotide diminished the binding of the agonist radioligand at A2 adenosine binding sites, with a Kd value of 28.6±0.95 nM and a Bmax value of 185±22 fmol/mg protein observed.
Known adenosine receptor agonists competitively displaced specific [3H]CGS21680 binding to human striatal membranes, as shown in Table 1. A rank order of potency consistent with binding to human A2-adenosine receptors (12,23) was observed: DPMA = m-DITC-APEC > NECA > APEC > m-DITC-ADAC = CV1808 = CGS21680 = Biotinyl-APEC > R-PIA (N6-phenylisopropyladenosine) > ADAC > CCPA (2-chloro-N6-cyclopentyladenosine) > N-Acetyl-ADAC = CPA. CGS15943A, a potent non-xanthine adenosine receptor antagonist, was found to be the most potent compound to inhibit [3H]CGS21680 binding, with a Ki value of 0.96 nM. We found CGS21680 to inhibit the binding of [3H]CGS21680 with a Ki-value of 35 nM and to inhibit the binding of [3H]PIA to adenosine A1 receptors with a Ki-value of 4580 nM (both in human striatum), thus CGS21680 is approximately 132-fold selective. All compounds were found to interact with one binding component (i.e., Hill coefficients not significantly different from unity). A Ki for R-PIA of 84 nM in human striatal membranes was close to the value of 124 nM reported for rat striatum using [3H]NECA in the presence of 50 nM CPA (23).
TABLE 1.
Inhibition of Agonist Radioligand Binding at Human A2-Adenosine Receptors.
| Compound: | Ki (nM), Human A2-Receotors | Ki (nM), Rat A2-Receptorsb | Ratiod |
|---|---|---|---|
| agonists | |||
| DPMA | 5.46±1.97 | 4.4c | 1.2 |
| m-DITC-APEC | 6.78 ± 0.94 | 22 | 0.31 |
| NECA | 12.9 ± 3.7 | 12 | 1.1 |
| APEC | 22.3 ± 7.2 | 12 | 1.9 |
| m-DITC-ADAC | 28.2 ± 6.8 | 203 | 0.14 |
| CV1808 | 29.5±6.5 | 105 | 0.28 |
| CGS21680 | 34.7 ± 5.3 | 14 | 2.5 |
| Biotinyl-APEC | 38.8 ± 6.4 | 55 | 0.71 |
| R-PIA | 84.3±28.1 | 410 | 0.21 |
| ADAC | 151 ± 18 | 218 | 0.69 |
| CCPA | 212 ± 2 | 3900c | 0.05 |
| Ac-ADAC | 339 ± 63 | 700c | 0.48 |
| CPA | 362 ± 39 | 890 | 0.41 |
| antagonists | |||
| CGS15943 | 0.960±0.291 | 3 | 0.3 |
| XAC | 8.55±2.65 | 50 | 0.17 |
| theophylline | 4420± 150 | 21,000 | 0.21 |
Affinity, expressed as Ki values in nM, of adenosine receptor agonists and antagonists at human striatal A2-adenosine receptors (vs. [3H]CGS21680) and in rat (vs. [3H]NECA or [3H]CGS21680) striatum, unless noted. Binding of [3H]CGS21680 (5 nM) was carried out for 90 min at 24°C, in the presence of 0.05 M TRIS buffer at pH 7.4, 10 mM MgCl2. Ki values are the mean ± standard deviation of at least 3 experiments done in triplicate.
Ki values versus binding of [3H]NECA in the presence of 50 nM N6-cyclopentyladenosine are from refs 23, 31 or determined here (Ac-ADAC).
ratio of Ki-value at human A2-receptors versus Ki value at rat A2-receptors.
[3H]XAC (18–20) was examined as an antagonist radioligand at A2-adenosine receptors in human striatal membranes. Since XAC has a high affinity for A1-receptors, indeed in the rat (18) and mouse (24) it is A1-selective, it was necessary to add a sufficient quantity of an A1-selective antagonist to selectively block the A1 component. As in Bruns et al. (13) and in our previous study of [3H]XAC binding to A2-receptors in rabbit striatal membranes (20), CPX (8-cyclopentyl-1,3-dipropylxanthine) was selected for this purpose and added to the incubation medium. To determine the appropriate concentration of CPX to add to the assay medium, CPX inhibition of [3H]XAC binding to both A1 - and A2-receptors in human striatum was studied. The resulting inhibition curve (Figure 2) fit a 2-site model when analyzed by a computer-assisted curve-fitting program. The Ki-value for CPX at the high affinity (assumed to be A1) site was found to be 0.46±0.14 nM and at the low affinity (assumed to be A2) site 113±2.2 nM. The high affinity component represented 37% of the combined, specific [3H]XAC binding. In the presence of 25 nM CPX, 91% of [3H]XAC (1 nM) binding to human A1 receptors was eliminated, and 90% of binding to A2 receptors was preserved. Thus 25 nM CPX was used for subsequent A2 receptor binding experiments with [3H]XAC. The incubation was carried out at 25°C with non-specific binding defined in the presence of 100 μM NECA.
Figure 2.

Inhibition of binding of [3H]XAC to human striatal adenosine receptors by CPX. The biphasic curve represents binding of CPX to high affinity (A1) and low affinity (A2) sites. The binding was carried out at 24°C for 90 min. [3H]XAC was present at a concentration of 1.0 nM in 0.05 M Tris HCI buffer at pH 7.4.
1 nM [3H]XAC bound to human striatal membranes, incubated for 90 min at 24°C, with specific A2-binding amounting to approximately 50% of total binding. Scatchard analysis of the saturation curve showed that [3H]XAC exhibited saturable, specific binding to A2 binding sites with a Kd of 2.98±0.54 nM and a Bmax of 0.71±0.23 pmol/mg protein (Figure 3).
Figure 3.

Saturation curve of [3H]XAC binding to human striatal A2-adenosine receptors. The binding was determined for 90 min at 24°C. Values are means of a typical experiment done in triplicate. The calculated Kd-value of 2.98 nM represents the mean of three separate experiments using membranes from pooled striatal tissue.
Previously characterized adenosine receptor antagonists competitively displaced specific [3H]XAC binding to human striatal A2-adenosine receptors as shown in Figure 4. Inhibition of [3H]XAC binding by adenosine antagonists was consistent with affinities previously measured at rabbit A2-adenosine receptors (20) with a rank order of potency against 1 nM [3H]XAC: CGS15943A > XAC = PD115,199 > PAPA-XAC > CPX > HTQZ = XCC (8-[4-[(carboxymethyl)oxy]phenyl]-1,3-dipropylxanthine) = CP-66,713 > theophylline = caffeine, indicative of A2 receptors (Table 2). The antagonist inhibition curves also fit monophasic analysis with Hill coefficients in the range 1.0. The non-xanthine heterocycle HTQZ was not very potent at human A2 receptors, compared to its affinity at rat A2-adenosine receptors, at which the Ki value is 124 nM (14). In contrast, the non-xanthine heterocycle CGS15943 was more potent at human A2 receptors than at rat A2-adenosine receptors, at which the Ki value is 3 nM (17).
Figure 4.

Inhibition of binding of [3H]XAC to human striatal A2-adenosine receptors by various adenosine receptor ligands. The binding was carried out at 24°C for 90 min. [3H]XAC was present at a concentration of 1.0 nM in 0.05 M Tris HCI buffer at pH 7.4 in the presence of 25 nM CPX.
TABLE 2.
Inhibition of Antagonist Radioligand Binding at A2-Receptors from Several Species.a
| Compound: | Ki (nM), Human A2-Receptors | Ki (nM), Rabbit A2-Receptors | Ki (nM), Rat A2-Receptorsb |
|---|---|---|---|
| CGS15943 | 0.609±0.100 | n. d. | 3 |
| XAC | 5.93±2.78 | 7.5 | 50 |
| PD115,199 | 8.66±3.46 | 3.28 | 16c |
| PAPA-XAC | 57.5±7.9 | n. d. | n. d. |
| CPX | 110±35 | 179 | 490 |
| HTQZ | 742±55 | n. d. | 124c |
| XCC | 772±81 | 196 | 1130 |
| CP-66,713 | 832±62 | 365 | 12 |
| theophylline | 4720±1225 | 38,250 | 21,000 |
| caffeine | 5780±950 | n. d. | 37,000 |
Affinity, expressed as Ki values in nM, of adenosine receptor antagonists at human and rabbit striatal A2-adenosine receptors vs. [3H]XAC and in rat striatum vs. [3H]NECA in the presence of 50 nM N6-cyclopentyladenosine, unless noted. Binding of [3H]XAC (1.0 nM) was carried out for 90 min at 24°C, in the presence of 0.05 M Tris hydrochloride buffer at pH 7.4, 10 mM MgCl2 and 25 nM CPX. Ki values are the mean ± standard deviation of at least 3 experiments done in triplicate. Rabbit data from ref 20.
Adenosine agonists inhibited binding of [3H]XAC with Hill coefficients less than unity, indicative of complex binding to several affinity states. Thus, IC50 values only are reported in Table 3. The curves for inhibition by the agonist CGS21680 was analysed for conformity to a two site fit, but the results were variable, and no conclusion could be drawn.
TABLE 3.
Inhibition of [3H]XAC binding at human striatal A2-adenosine receptors y adenosine receptor agonists.a
| Compound: | IC50 (nM), Human A2-Receptors | Hill coefficient | Ki (nM), Rat A2-Receptorsa |
|---|---|---|---|
| NECA | 186±40 | 0.53 | 10.3 |
| R-PIA | 1410±94 | 0.40 | 124 |
| CV-1808 | 146±30 | 0.71 | 97 |
| CGS21680 | 992±551 | 0.48 | 15 |
Ki values versus binding of [3H]NECA in the presence of 50 nM N6-cyclopentyladenosine are from references 17, 23.
The effects of adding guanine nucleotides or cations to the incubation medium with single concentrations of agonist and antagonist radioligands were compared (Table 4). GTP (0.1 mM) had a significant effect on [3H]CGS21680 binding, resulting in a 37% decrease in binding, whereas Mg++ increased [3H]CGS21680 binding by 71%. Neither GTP nor Mg++ altered [3H]XAC binding significantly. [3H]XAC binding was sensitive to the presence of Na+ ions, with a decrease of 28%. The presence of 1 mM GTP did not have a clearly discernable effect on the curve for inhibition of [3H]XAC binding by CGS21680. The IC50 value was 2194±1363 nM in the presence and 992±551 (Table 3) in the absence of GTP.
TABLE 4.
| % of control | ||
|---|---|---|
| Agent | [3H]CGS21680(5 nM) | [3H]XAC (1 nM) |
| GTP (100 μM) | 63±7.7 | 102±4 |
| Mg++ (10 mM) | 171±14 | 103±6.5 |
| Na+ (150 mM) | n.d. | 72±4.5 |
Membranes were incubated with [3H]XAC (1.0 nM) and the appropriate concentration of the adenosine derivative for 60 min at 24°C, in 0.05 M TRIS buffer at pH 7.4 in the presence of 10 mM MgCl2 and 25 nM CPX. Ki values are the mean ± standard deviation of at least4 experiments done in triplicate, n.d. = not determined.
A comparison of A2-adenosine receptor density in caudate and putamen showed similar levels in these striatal regions (Table 5). Curiously, a distinct, low level of specific binding to A2-adenosine receptors in the cerebral cortex was found, as reported by Wan et al. (26). 5 nM [3H]CGS21680 binding to cerebral cortical A2-adenosine receptors revealed roughly one tenth of the level of specific binding found under the same conditions in the striatum, and this was unaffected by the addition of 50 nM CPX. In the cortex, [3H]XAC, in the presence of 25 nM CPX, also displayed one tenth of the level of specific binding relative to the striatum. In the absence of CPX, the level of specific [3H]XAC binding (1 nM) was 186±17 fmoVmg protein, as expected for a region rich in A1 adenosine receptors.
TABLE 5.
| Human brain region | [3H]CGS21680 (fmol/mg protein) | [3H]XAC binding (fmol/mg protein) |
|---|---|---|
| putamen | 67±5.5 (n=6) | 100±14 (n=8) |
| caudate | 39±0.5 (n=4) | 84±2.5 (n=4) |
| cortexa | 4.4±0.19 (n=4) | 9.5±4.8 (n=3) |
Specific binding was determined using 5 nM [3H]CGS21680 or 1 nM [3H]XAC, nonspecific binding was determined in the presence of 20 μM 2-chloroadenosine or 100 μM NECA. Each experiment was conducted in triplicate. Frontal and temporal cortex were combined.
Photoaffinitv labeling
Human striatal membranes prepared in the above manner, except that caudate and putamen were processed separately, were subjected to photoaffinity labeling experiments, using a method developed by us (7,22). PAPA-APEC was synthesized as reported (12) and iodinated by the chloramine-T/125I method. 125I-PAPA-APEC was purified using reverse phase HPLC (12). The aryl amine of 125I-PAPA-APEC was converted to an aryl azide through diazotization, and a second HPLC purification step was carried out. The resulting photoaffinity label was used to study the A2-receptor binding subunit and derived protein fragments separated on SDS polyacrylamide gels.
Photoaffinity labeling revealed several selectively labeled bands, the largest of which had a molecular weight of 45K (Figure 5), in agreement with that found for the bovine A2-receptor (7). In this receptor preparation, proteolytic cleavage was observed. resulting in fragments of MW 43K and 37K. In the absence of proteolytic inhibitors, the 37K fragment, which still bound 125I-PAPA-APEC, was predominant.
Figure 5.
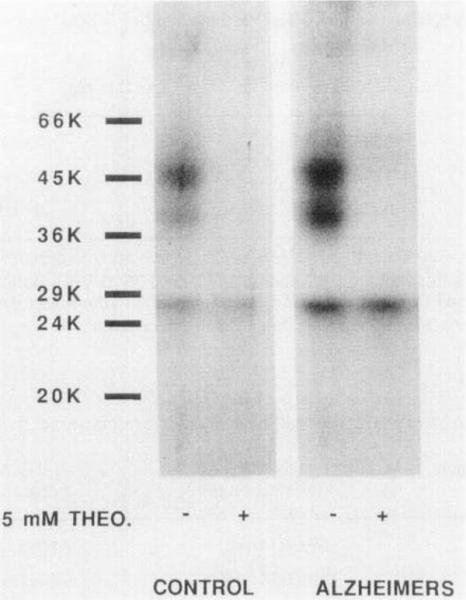
Photoaffinity labeling of A2-adenosine receptors in membranes (protease inhibitors added) from normal human striatum and from the striatum of an Alzheimer's patient. The control lanes each contain 105 μg protein, and the Alzheimers lanes each contain 173 μg protein. Membranes were incubated with the agonist photoaffinity probe 125I-azido-PAPA-APEC (1 nM) in the absence and presence of 5 mM theophylline. The membranes were irradiated with UV light and solubilized for sodium dodecylsulfate - polyacrylamide gel electrophoresis and autoradiography.
A preliminary comparison of A2 receptor photoaffinity labeling in striatal membranes (processed with protease inhibitors) obtained from normal human subjects and from a person diagnosed with Alzheimer's disease (Figure 5) showed no apparent difference in mobility of the receptor and fragments.
DISCUSSION
The human central A2 adenosine receptor has been studied using radioligand binding (25,26) with [3H]NECA or (3H]CGS21680. In spite of recent advances in characterization of A2 adenosine receptors through agonist binding, photoaffinity labeling, and cloning, a generally selective antagonist for this receptor is still lacking. A number of heterocyclic compounds (14–16) have been reported to have high affinity and/or modest selectivity for A2-adenosine receptors, yet none of these compounds are suitable as general A2-adenosine radioligands.
In this study we have explored the usefulness of [3H]XAC as an antagonist radioligand for the human A2-adenosine receptor. In other species, such as the rat, [3H]XAC is A1-selective and has successfully been used to label A1-receptors (18). However, considerable species differences exist, and 8-substituted xanthines have been shown to have particularly high affinity at human A2-receptors (27). Thus, it was reasonable to expect that XAC, structurally also an 8-phenyl substituted xanthine, might display high affinity for human A2-receptors, as was found to be the case for the rabbit (Kd 3.8 nM). Indeed, we found a Kd of 3 nM for [3H]XAC binding to human striatal A2-receptors. Since XAC also has considerable A1-affinity (nanomolar), and A1 -receptors are present in human striatum, it was necessary to add a sufficient quantity of an A1-selective ligand in order to selectivity block the A1 component. We have shown that in rabbit striatum [3H]XAC in the presence of 50 nM CPX is useful in an A2-binding assay (20). Similarly, we now show that this method is applicable to human A2-adenosine receptors, in the presence of 25 nM CPX. The adenosine agonist CCPA was reported to be more highly A1-selective than CPA in the rat (32). However, CCPA was not satisfactory for this purpose of blocking 13H]XAC to human striatal A1 receptors (data not shown), due to its unexpectedly high affinity at human striatal A2 receptors (Table I). Competition for [3H]XAC binding sites in the human striatum by the antagonist CPX fit a 2-site model. Nearly all of the [3H]XAC binding to A1 receptors was eliminated at 25 nM CPX. Thus, CPX was selected as more valuable for this purpose and added to the incubation buffer in subsequent binding experiments.
The rabbit A2-receptor was recently shown to undergo a specific proteolytic degradation, particularly in solubilized fractions, resulting in a change in molecular weight from 47,000 to 38,000. Both native and partially hydrolyzed rabbit A2-receptors bind the radioligand 125I-PAPA-APEC with high affinity. Similarly, the human A2-adenosine receptor forms proteolytic fragments that retain the radioligand binding properties. It is not known what enzyme is responsible for the cleavage or whether this proteolysis is relevant to the action of adenosine receptors in vivo. The proteolysis is diminished but not completely in the presence of proteolytic inhibitors (see Materials and Methods). Since significant proteolysis of the autopsy-derived human A2-receptor occurs, we treated the membranes in the absence of proteolytic inhibitors, resulting in a predominance of a 37K fragment that bound [3H]XAC and [3H]CGS21680 with high affinity. It is uncertain if this proteolytic fragment is identical to the native receptor in all pharmacological properties.
Inhibition of [3H]CGS21680 binding by agonists and antagonists (Table 1) and [3H]XAC binding by antagonists (Table 2) was as predicted in order of potency for A2-adenosine receptors. Bruns et al. (13) determined Ki values for inhibition of binding of [3H]PD115,199 in rat striatum by the A1-selective antagonist CPX and the non-selective PD115,199 to be 190 and 4.1 nM, respectively. This is in close agreement with our findings of Ki values against [3H]XAC binding in human striatum of 110 and 8.7 nM for CPX and PD115,199, respectively. In comparing [3H]XAC binding in human striatum to [3H]NECA or [3H]CGS21680 binding in rat striatum, CPX appeared to be 3-fold more potent at human A2-adenosine receptors. Theophylline and caffeine were considerably more potent at human A2-adenosine receptors than at rat A2-adenosine receptors as indicated in both [3H]CGS21680 and [3H]NECA binding. The apparently higher affinity of theophylline in human striatum raises the question whether A2-adenosine receptors may be particularly relevant to the therapeutic use of theophylline in man. A previous study of [3H]XAC binding to A2 adenosine receptors of human platelets (19) reported somewhat higher Ki values (nM) for antagonist competitors: XAC, 17; XCC, 1800; theophylline, 29,000; and caffeine, 51,000.
Among non-xanthine adenosine antagonists assayed versus [3H]XAC binding in human striatum, CGS15943 was at least as potent at human A2-adenosine receptors as at rat A2-adenosine receptors (Table 2). Thus, CGS15943 is the most potent reported A2 adenosine antagonist, although it is not very selective. Disappointingly, two recently reported putative A2-selective antagonists, CP-66,713 and HTQZ, were less potent than predicted from Ki values at rat A2-adenosine receptors.
The adenosine agonist DPMA was reported by the group at Parke Davis Warner-Lambert (31) as a moderately selective and high affinity agonist at A2-adenosine receptors. We found that DPMA is equally highly potent at human A2-adenosine receptors. CV1808, a slightly selective A2-adenosine receptor agonist, was moderately potent in human A2-adenosine receptors. CCPA (32), highly A1-selective agonist in the rat, was more potent at human A2-adenosine receptors (even more potent than CPA) than expected from data at rat A2-adenosine receptors. m-DITC-APEC and m-DITC-ADAC were designed as isothiocyanate-bearing affinity labels selective for A2-and A1-adenosine receptors, respectively (12). m-DITC-ADAC was more potent at human A2-adenosine receptors than in the rat. Biotinyl-APEC (12) is highly selective for A2-adenosine receptors (>90-fold, in rat brain). The high affinity of biotinyl-APEC in human brain suggests its use in conjunction with avidin complexation in affinity chromatography and histochemical studies of human A2-adenosine receptors.
The high affinity of XAC and its derivatives at human A2-adenosine receptors raises the possibility of their use as molecular probes for this receptor, for example in affinity labeling or affinity Chromatography. For example, XAC immobilized to an agarose chromatography matrix has been used for the affinity purification to homogeneity of A1-adenosine receptors from rat (29) and bovine brain (30). Such a column may be of use in purifying human A2-adenosine receptors.
Specific binding to A2-receptors in human cerebral cortex was detected using [3H]CGS21680. This result was also obtained by Wan et al, although the relative level in cortex compared to the striatum was somewhat higher in their study. In contrast measurable levels of A2-receptors in the rat cortex were not observed in previous studies (5) of adenosine receptor localization using [3H]CGS21680. The observed specific binding of [3H]XAC in the cortex is not as conclusive as is the [3H]CGS21680 binding, because of the low receptor density. The [3H]XAC binding may reflect the presence of A2 receptors in the cortex, but binding to residual A1 receptors cannot be ruled out. Similarly, Bruns et al (13) found [3H]PD115,199 in the rat cortex displayed 7% of the level of specific binding relative to the striatum. It is not known whether the apparent presence of the A2a receptor in a region in which it is commonly thought to be absent reflects differences in species or the detection limits of the assay methods used. There is evidence for physiological effects of A2-selective agonists when applied directly to brain regions other than those in which the receptor occurs in high density. In rat cortical neurons, iontophoretically applied CGS21680 depressed spontaneous and acetylcholine-evoked firing (4).
In drug screening, especially in the development of therapeutic entities to act at adenosine receptors, it is advisable to be cognizant of the species differences in affinities observed for adenosine antagonists. The differences are so great (up to 70-fold observed in this study) that a complete reversal of selectivity between the rat and human for a given drug is possible. For example, in Stone et al. (27), PACPX (8-(2-amino-4-chlorophenyl)-1,3-dipropylxanthine) was found to be 73-fold A1 selective in rat brain binding and nearly 2-fold A1 selective in human brain.
It has been shown that GTP regulates the binding of agonist radioligands to A1-receptors (reviewed in ref. 33). In the absence of GTP, two affinity states are detected, and in the presence of GTP most of the high affinity sites are converted to low affinity. At A2-receptors, GTP added to the assay medium affected the diminished the binding of the agonist radioligand [3H]CGS21680, but due to variability it was not possible to draw conclusions about the relative percentage of high and low affinity states. The enhancement of binding of [3H]CGS21680 by magnesium ions (Table 4) is consistent with the regulation of agonist binding by GTP-binding proteins, since for A1 receptors magnesium ions shift the distribution predominantly towards the high affinity form (33).
125I-PAPA-APEC photoaffinity labeling of A2-receptors was previously applied to bovine (7) and rabbit (9) striatum, the DDT1 MF-2 smooth muscle cell line (22), and PC12 pheochromocytoma cells (9). We have now shown that the human A2-receptor is nearly identical in molecular weight to the receptor in rat, calf, and other species (9).
ACKNOWLEDGEMENTS
We thank Dr. David Eveleth of Cortex Pharmaceuticals, Inc., Irvine Ca., for obtaining human brain tissue for this study, and the Brain Bank of the University of California at Irvine.
REFERENCES
- 1.Lohse MJ, Elger B, Lindenborn-Fotinos J, Klotz K-N, Schwabe U. Separation of solubilized A2 adenosine receptors of human platelets from non-receptor [3H]NECA binding sites by gel filtration. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;337:64–68. doi: 10.1007/BF00169478. [DOI] [PubMed] [Google Scholar]
- 2.Olsson RA, Pearson JD. Cardiovascular purinoceptors. Physiol. Rev. 1990;70:761–845. doi: 10.1152/physrev.1990.70.3.761. [DOI] [PubMed] [Google Scholar]
- 3.Hutchison AJ, Webb RL, Oei HH, Ghai GR, Williams M. CGS 21680C, an A2 selective adenosine receptor agonist with preferential hypotensive activity. J. Pharmacol. Exp. Ther. 1989;251:47–55. [PubMed] [Google Scholar]
- 4.Phillis JW. In: Purines in Cellular Signalling: Targets for New Drugs. Jacobson K, Daly J, Manganiello V, editors. Springer-Verlag; New York, N.Y.: 1990. pp. 41–47. [Google Scholar]
- 5.Jarvis MF, Jackson RH, Williams M. Autoradiographic characterization of high-affinity adenosine A2 receptors in the rat brain. Brain Res. 1989;484:111–118. doi: 10.1016/0006-8993(89)90353-3. [DOI] [PubMed] [Google Scholar]
- 6.Nikodijevic O, Daly JW, Jacobson KA. Characterization of the locomotor depression produced by an A2-selective adenosine agonist. FEBS Letters. 1990;261:67–70. doi: 10.1016/0014-5793(90)80638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barrington WW, Jacobson KA, Williams M, Hutchison AJ, Stiles GL. Identification of the A2 adenosine receptor binding subunit by photoaffinity crosslinking. Proc. Nat. Acad. Sci. USA. 1989;86:6572–6576. doi: 10.1073/pnas.86.17.6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barrington WW, Jacobson KA, Stiles GL. The glycoprotein nature of the A2 adenosine receptor binding subunit. Mol. Pharmacol. 1990;38:177–183. [PMC free article] [PubMed] [Google Scholar]
- 9.Nanoff C, Jacobson KA, Stiles GL. The A2 adenosine receptor: guanine nucleotide modulation of agonist binding is enhanced by proteolysis. Mol. Pharmacol. 1991;39:130–135. [PMC free article] [PubMed] [Google Scholar]
- 10.Maenhaut C, van Sande J, Libert F, Abramowicz M, Parmentier M, Vanderhaegen J-J, Dumont JE, Vassart G, Schiffmann S. RDC8 codes for an adenosine A2 receptor with physiological constitutive activity. Biochem. Biophys. Res. Comm. 1990;173:1169–1178. doi: 10.1016/s0006-291x(05)80909-x. [DOI] [PubMed] [Google Scholar]
- 11.Jacobson KA, Kirk KL, Padgett WL, Daly JW. Functionalized congeners of adenosine: Preparation of analogues with high affinity for adenosine receptors. J. Med. Chem. 1985;28:1341–1346. doi: 10.1021/jm00147a039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacobson KA, Barrington WW, Pannell LK, Jarvis MF, Ji X-D, Williams M, Hutchison AJ, Stiles GL. Agonist-derived molecular probes for A2-adenosine receptors. J. Mol. Recognition. 1989;2:170–178. doi: 10.1002/jmr.300020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bruns RF, Fergus JH, Badger EW, Bristol JA, Santay LA, Hays SJ. PD 115,199: an antagonist ligand for adenosine A2 receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1987;335:64–69. doi: 10.1007/BF00165038. [DOI] [PubMed] [Google Scholar]
- 14.Bruns RF. In: Purines in Cellular Signalling: Targets for New Drugs. Jacobson K, Daly J, Manganiello V, editors. Springer-Verlag; New York, N.Y.: 1990. pp. 126–135. [Google Scholar]
- 15.Williams M, Francis JE, Ghai GR, Braunwalder A, Psychyos S, Stone GA, Cash WD. Biochemical characterization of the triazobquinazoline, CGS 15943, a novel non-xanthine adenosine antagonist. J.Pharmacol. Exp. Ther. 1987;241:415–420. [PubMed] [Google Scholar]
- 16.Sarges R, Howard HR, Browne RG, Lebel LA, Seymour PA, Koe BK. 4-Amino[1,2,4]triazolo[4,3-a]quinoxalines - a novel class of potent adenosine receptor antagonists and potential rapid onset anti-depressants. J. Med. Chem. 1990;33:2240–2254. doi: 10.1021/jm00170a031. [DOI] [PubMed] [Google Scholar]
- 17.Jarvis MF, Schutz R, Hutchison AJ, Do E, Sills MA, Williams M. [3H]CGS 21680, an A2 selective adenosine receptor agonist directly labels A2 receptors in rat brain tissue. J.Pharmacol. Exp. Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 18.Jacobson KA, Ukena D, Kirk KL, Daly JW. [3H]Xanthine amine congener of 1,3-dipropyl-8-phenylxanthine: an antagonist radioligand for adenosine receptors. Proc. Natl. Acad. Sci. U.S.A. 1986;83:4089–4093. doi: 10.1073/pnas.83.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ukena D, Jacobson KA, Kirk KL, Daly JW. A [3H]amine congener of 1,3-dipropyl-8-phenyl xanthine - a new radioligand for A2 adenosine receptors of human platelets. FEBS Lett. 1986;199:269–274. doi: 10.1016/0014-5793(86)80493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ji X-D, Stiles GL, Jacobson KA. [3H]XAC (xanthine amine congener) is a radioligand for A2-adenosine receptors in rabbit striatum. Neurochem. Internat. 1991;18:207–213. doi: 10.1016/0197-0186(91)90187-i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (lC50) of an enzyme reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 22.Ramkumar V, Barrington WW, Jacobson KA, Stiles GL. Demonstration of both A1 and A2 adenosine receptors in DDT1 MF-2 smooth muscle cells. Mol. Pharrnacol. 1990;37:149–156. [PMC free article] [PubMed] [Google Scholar]
- 23.Bruns RF, Lu GH, Pugsley TA. Characterization of the A2 adenosine receptor labeled by [3H]NECA in rat striatal membranes. Mol. Pharmacol. 1986;29:331–346. [PubMed] [Google Scholar]
- 24.Deckert J, Morgan PF, Bisserbe JC, Jacobson KA, Kirk KL, Daly JW, Marangos PJ. Autoradiographic localization of mouse brain adenosine receptors with an antagonist ([3H]xanthine amine congener) ligand probe. Neurosci. Lett. 1988;86:121–126. doi: 10.1016/0304-3940(88)90557-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Reynaud D, Gharib A, Lagarde M, Sarda M. Characterization of A-2 receptors in postmortem human pineal gland. J. Neurochem. 1990;55:1316–1321. doi: 10.1111/j.1471-4159.1990.tb03141.x. [DOI] [PubMed] [Google Scholar]
- 26.Wan W, Sutherland GR, Geiger JD. Binding of the adenosine A2 receptor ligand [3H]CGS21680 to human and rat brain: Evidence for multiple affinity sites. J. Neurochem. 1990;55:1763–1771. doi: 10.1111/j.1471-4159.1990.tb04967.x. [DOI] [PubMed] [Google Scholar]
- 27.Stone GA, Jarvis MF, Sills MA, Weeks B, Snowhill EW, Williams M. Species differences in high-affinity adenosine A2 binding sites in striatal membranes from mammalian brain. Drug Devel. Res. 1988;15:31–46. [Google Scholar]
- 28.Ferkany JW, Valentine HL, Stone GA, Williams M. Adenosine A1 receptors in mammalian brain: species differences in their interactions with agonists and antagonists. Drug Dev. Res. 1986;9:85–93. [Google Scholar]
- 29.Nakata H. Purification of the A1-adenosine receptor from rat brain membranes. J. Biol. Chem. 1989;264:16545–16551. [PubMed] [Google Scholar]
- 30.Olah ME, Jacobson KA, Stiles GL. Purification and characterization of bovine cerebral cortex A1 adenosine receptor. Arch. Biochem. Biophys. 1991;283:440–446. doi: 10.1016/0003-9861(90)90665-l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bridges AJ, Bruns RF, Ortwine DG, Priebe SR, Szotek DL, Trivedi BK. N6-[2(3,5-dimethoxyphenyl)-2-(2-methylphenyl)-ethyl]-adenosine and its uronamide derivatives. Novel adenosine agonists with both high affinity and high selectivity for the adenosine A2 receptor. J. Med. Chem. 1988;31:1282–1285. doi: 10.1021/jm00402a004. [DOI] [PubMed] [Google Scholar]
- 32.Lohse MJ, Klotz K-N, Schwabe U, Cristalli G, Viitori S, Grifantini M. 2-Chloro-N6-Cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn-Schmiedeberg's Arch. Pharmacol. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- 33.Cooper DMF. In: Adenosine Receptors. Cooper D, Londos C, editors. Alan R. Liss, Inc.; New York, N.Y.: 1987. pp. 63–74. [Google Scholar]