

Abstract
Adult-onset neurodegeneration and other protein conformational diseases are associated with the appearance, persistence, and accumulation of misfolded and aggregation prone proteins. To protect the proteome from long-term damage, the cell expresses a highly integrated protein homeostasis (proteostasis) machinery to ensure that proteins are properly expressed, folded, and cleared, and to recognize damaged proteins. Molecular chaperones have a central role in proteostasis as they have been shown to be essential to prevent the accumulation of alternate folded proteotoxic states as occurs in protein conformation diseases exemplified by neurodegeneration. Studies using invertebrate models expressing proteins associated with Huntington's disease, Alzheimer's disease, ALS, and Parkinson's disease have provided insights into the genetic networks and stress signaling pathways that regulate the proteostasis machinery to prevent cellular dysfunction, tissue pathology, and organismal failure. These events appear to be further amplified by aging and provide evidence that age-related failures in proteostasis may be a common element in many diseases.
I. Introduction
An increasing number of diseases are associated with the expression of proteins that misfold and interfere with diverse cellular processes. This is exemplified by neurodegenerative disorders including Parkinson's disease, amyotrophic lateral sclerosis (ALS), Alzheimer's disease, and polyglutamine (polyQ) diseases. These diseases are associated with the chronic expression of specific disease-associated proteins resulting in the accumulation of misfolded species in brain tissues of individuals diagnosed with neurodegeneration (Soto and Estrada, 2008). Despite the functional and structural diversity of proteins involved in these diseases, they all share common characteristics with the appearance of cytoplasmic, nuclear, or extracellular aggregates, inclusions, and amyloid-like material that has led to the protein misfolding hypothesis as a mechanism that leads to disease (Balch et al., 2008; Bucciantini et al., 2002).
To protect itself from the stress of misfolded proteins, all cells express cytoprotective machinery that includes molecular chaperones, a family of highly conserved proteins that recognize nascent polypeptides and folding intermediates to guide proteins to their native state. Molecular chaperones are also involved in the assembly and disassembly of multimeric complexes, translocation of proteins across cellular membranes, and regulating vesicular transport (Bukau and Horwich, 1998; Hartl, 1996; Hartl and Hayer-Hartl, 2002). Consequently, the genes that control these processes function as an integrated proteostasis network to maintain balance in protein biogenesis and to detect and prevent an imbalance leading to pathology and disease. Changes in the regulation of the proteostasis network or interference of chaperone function and clearance machineries are likely to have deleterious consequences in diseases of protein conformation and aging (Balch et al., 2008; Ben-Zvi et al., 2009).
Stress conditions that influence protein folding dynamics within a cell can lead to changes in expression of the components of the protein folding quality control system (Morimoto et al., 1997). These include: (i) environmental stress, such as fluctuations in temperature, hydration, nutrient balance, (ii) chemical stress, such as oxygen free radicals, transition heavy metals, and (iii) pathophysiological states, which in metazoans are associated with ischemia, viral or bacterial infection, and tissue injury. To protect itself, the cell activates the heat shock response and expresses genes encoding chaperones and other components of the protein quality control apparatus to reestablish cellular homeostasis. Despite the robust nature of the heat shock response and the capacity of chaperones to recognize misfolded proteins, chronic expression of disease-associated aggregation prone proteins escapes this vigilance, resulting in the accumulation of misfolded species and damaged proteins. The accumulation of alternate folded states and toxic species overburdens and functionally depletes the proteostasis machinery, which in turn amplifies protein damage (Gidalevitz et al., 2006; Gidalevitz et al., 2009). This suggests that the regulation of the protein quality control system is essential for proteostasis to monitor the state of the proteome throughout the lifetime of an organism.
C. elegans offers many advantages as a model system to establish the role of molecular chaperones in cellular and organismal responses to proteotoxic stress. This review will discuss the various C. elegans models for expression of neurodegenerative disease-associated proteins and the use of genetic approaches for identification of key regulators of chaperone networks that influence protein aggregation, stress responses and aging. We will describe the potential of C. elegans to examine how chaperone networks sense damaged proteins within specific tissues and the integration of this information at the level of the organism to control lifespan.
II. Molecular Chaperones and Protein Folding Quality Control
For proteins to function properly, they must fold and be stably maintained in their native conformation. Information contained within the primary amino acid sequence can dictate the three-dimensional shape of the protein (Anfinsen, 1973), which together with the environment of the cell ensures that proteins are assembled, processed, and transported. The pathway by which a protein achieves its unique folded state is complex and can involve an ensemble of intermediates and conformations (Wolynes et al., 1995). Proteins misfold when inappropriate yet energetically stable interactions occur, for example by self-association of hydrophobic residues leading to oligomerization and aggregation (Hartl and Hayer-Hartl, 2002).
Molecular chaperones are highly conserved and ubiquitously expressed in all subcellular compartments, cells, and tissues and are essential for the stability of the proteome under normal and stressful conditions (Frydman, 2001; Lindquist and Craig, 1988). The expression of many molecular chaperones is regulated by different forms of environmental and physiological stresses that can interfere with folding stability leading to a flux of misfolded proteins. Stress responsive molecular chaperones are referred to as heat shock proteins (Hsps) and classified by gene families according to their molecular mass as Hsp100, Hsp90, Hsp70, Hsp60, Hsp40 and small Hsps (sHsps). Each family is comprised of multiple members that share sequence identity and have common functional domains, and are expressed in different subcellular compartments and at different levels in tissues (Gething and Sambrook, 1992; Lindquist and Craig, 1988).
During protein biogenesis, nascent polypeptides emerge from the ribosome with exposed hydrophobic residues that are eventually buried within the interior of the folded protein. Among their properties, chaperones recognize hydrophobic stretches within nascent polypeptides to prevent inappropriate interactions that could result in aggregation (Deuerling et al., 1999; Teter et al., 1999; Thulasiraman et al., 1999). During translation, chaperones contribute to the maturation of nascent chains by assisting in folding events, assembly into a multimeric complex, or translocation into organelles (Deuerling et al., 1999; Hartl and Hayer-Hartl, 2002; Siegers et al., 1999; Teter et al., 1999). The association of molecular chaperones with client substrates provides kinetic and spatial partitioning of the nascent polypeptide to ensure orderly folding, to prevent misfolding, and to redirect folding of intermediates that accumulate in stressed cells (Figure 1). A member of the cytosolic Hsp70 family associated with the ribosome is involved in cotranslational protein folding whereas Hsp90 is largely involved in later events to assist folding of client proteins involved in signaling, transcription, and cell cycle control. Certain partially folded client proteins, for example those rich in WD repeats, preferentially interact with the TRiC chaperonin (Camasses et al., 2003; Siegers et al., 2003). The Hsp100 and small Hsp chaperone families recognize misfolded proteins that accumulate as amorphous and amyloid-like species (Patino et al., 1996; Raman et al., 2005). In cooperation with Hsp70, Hsp100 dissociates pre-formed aggregates, allowing reentry of these misfolded species into productive folding pathways (Ben-Zvi and Goloubinoff, 2001; Goloubinoff et al., 1999).
Figure 1. Multiple chaperone and co-chaperone families assist in protein folding.

Chaperones guide the protein fold starting with the initial steps of protein synthesis. As a nascent chain exits the ribosome channel, a series of chaperones bind the extended polypeptide, interacting with the protein throughout its maturation into the native folded state. Chaperone assisted folding involves the core chaperone families, Hsp70, Hsp90 and TRiC. Specific co-chaperones modulate the folding activities of core chaperones depending on substrate and the step in the folding pathway. Additionally, chaperones protect the native fold during denaturing stresses that unfold or damage cellular proteins. Hsp100s and small Hsps prevent protein aggregation and work with chaperones to refold denatured cellular proteins. Specific co-chaperones recognize substrates and cooperate with core chaperones, determining the fate of the damaged protein. The chaperone quality control system directs the protein either for refolding or degradation by the proteasome.
Chaperones function as machines with cycles of substrate binding and release regulated by ATP binding and hydrolysis. The efficiency of these folding machines is regulated by co-chaperones, a large class of proteins that interact with chaperones to modulate the nucleotide cycle of chaperones and to provide substrate selectivity. For example, members of the Hsp40/DnaJ co-chaperone family stimulate the ATPase activity of Hsp70 and stabilize substrate interactions (Freeman et al., 1995; Wittung-Stafshede et al., 2003). Other co-chaperones function as nucleotide exchange factors (NEFs) and release bound ADP from Hsp70. Among these co-chaperones are members of the Hsp40/DnaJ and cyclophilin family that interact directly with unfolded polypeptides to assist in folding (Bimston et al., 1998; Demand et al., 1998; Hohfeld and Jentsch, 1997; Sondermann et al., 2001; Takayama and Reed, 2001). For Hsp90 co-chaperones such as Cdc37, interaction with Hsp90 is essential for the activation of kinases while cyclophilins and p23 co-chaperones cooperate with Hsp90 in hormone receptor maturation (Cutforth and Rubin, 1994; Duina et al., 1996; Gerber et al., 1995; Kimura et al., 1997b; Pratt and Toft, 1997). Thus, the combination of co-chaperone regulation of the ATP cycle together with enhanced specificity and folding efficiency can influence the folded state of many proteins, each at a distinct stage in the folding cycle.
Another prominent role for co-chaperones is to coordinate interactions of chaperone machines between protein folding and degradation (Esser et al., 2004). The co-chaperone CHIP (carboxyl terminus of Hsp70 interaction protein) is a chaperone-associated E3 ubiquitin ligase that promotes ubiquitination and degradation of Hsp70 substrates (Connell et al., 2001; Esser et al., 2004; Meacham et al., 2001). CHIP inhibits the Hsp40-stimulated ATPase activity of Hsp70, thus reducing the chaperone activity of Hsp70 (Ballinger et al., 1999). The consequence of these events has been suggested to shift Hsp70 activity from folding to clearance, thus allowing abnormal proteins to be degraded by the proteasome. Therefore, the level and complement of co-chaperones expressed in a cell can modulate both the specificity and efficiency of folding and degradation of substrates by the core chaperone machines.
The regulation of genes encoding molecular chaperones is critical to maintain protein homeostasis and to restore it upon stress. The principal regulator of chaperone levels in eukaryotes is heat shock transcription factor (HSF1) that is activated by exposure of cells to numerous forms of physiological stress (Morimoto, 1993; Morimoto, 1998; Sorger, 1991). Stresses such as heat shock that increase the demand for the folding machinery are thought to titrate chaperones away from their interactions with HSF1, thus allowing HSF1 to become activated and transcribe high levels of multiple Hsp genes including the chaperones Hsp70, Hsp40, and Hsp90 (Kroeger and Morimoto, 1994; Mosser et al., 1988). The level of transcriptional activity is further regulated by a variety of post-translational modifications, including phosphorylation (Kline and Morimoto, 1997; Knauf et al., 1996; Pirkkala et al., 2001), sumoylation (Anckar et al., 2006) and acetylation of HSF1 (Westerheide et al., 2009). The balance of acetylation of HSF1 by p300/CBP and deaceylation by the NAD-dependent sirtuin, SIRT1, determines the duration of DNA binding activity and thus the persistence of the heat shock response (Westerheide et al., 2009). Once sufficient chaperone levels have been restored, Hsp70 and Hsp90 re-associate with HSF1 thereby repressing transcription (Shi et al., 1998), thus providing a negative feedback system to maintain sufficient levels of Hsps under different conditions (Morimoto, 1998).
Multiple factors are thought to be critical in the cellular response to protein misfolding and the subsequent ability of chaperones to protect the cell against protein damage. Although multiple chaperone families are ubiquitously expressed, it is likely that the folding requirements of the proteome in specialized cells require the expression of tissue-specific chaperones and co-chaperones. Therefore, the ability of specific differentiated cells to respond to protein misfolding events may be determined by the complement and levels of chaperones and co-chaperones expressed in each cell type. Because there are multiple and diverse stress conditions that result in protein misfolding, this suggests an equivalent capability to recognize various forms of stress-induced protein damage. However, the robustness of a cell's response to misfolded proteins may depend upon the development stage or age of an organism (Ben-Zvi et al., 2009). Furthermore, different types of stress may impose differential protein folding challenges that may influence a cell's ability to sense and remedy damage (Mosser et al., 1988). For example, aggregation prone disease proteins seem to escape the vigilance of the protein folding quality control system leading to cellular dysfunction. Overall, engineering a folding network that maintains protein homeostasis throughout the lifetime of an organism is complex. Studies using C. elegans have provided insight into the sensitivity of cells to the stress of misfolded proteins during aging and disease.
III. C. elegans models of diseases of protein folding
The nematode C. elegans provides an excellent in vivo system to evaluate chaperone networks in diseases of protein folding. The distinct tissue and cell types have been well characterized and behavioral assays that monitor their functionality are well described. The transparency of the organism allows the generation of transgenic lines expressing fluorescently tagged aggregation prone proteins that can be visualized in any tissue during development and throughout adulthood. With a relatively short lifespan of two-three weeks, it is feasible to perform experiments to assess the roles of cytoprotective pathways and chaperones on longevity. Furthermore, the availability of comprehensive RNAi libraries and genetic tools for tissue-specific overexpression of transgenes allow interrogation of the entire genome to identify genes affecting proteostasis. Based on these experimental strengths, C. elegans disease models provide a system to examine the cellular and organismal effects of expression of misfolded proteins such as those implicated in neurodegenerative disorders (Table 1).
Table 1. C. elegans disease models.
| Disease protein | Tissue expression pattern | Aggregation | Toxicity | Chaperone involvement | Reference |
|---|---|---|---|---|---|
|
| |||||
| Aβ42 | Body wall muscles | + | Paralysis | Yes | Link 1995 |
|
| |||||
| huntingtin fragment | Mechanosensory neurons | - | Touch insensitivity | Not Determined | Parker et al. 2001 |
| ASH and ASI sensory neurons | Age dependent | Nose touch defect | Not Determined | Faber et al. 1999 | |
|
| |||||
| polyglutamine tract | Body wall muscles | Age dependent | Motility defect | Yes | Morley et al. 2002 |
| Pan neuronal | + | Behavioral defect | Not Determined | Brignull et al. 2006 | |
| Intestine | Age dependent | Not Determined | Not Determined | Mohri-Shiomi et al. 2008 | |
|
| |||||
| α-synuclein | Body wall muscles | Age dependent | Not Determined | Yes | van Ham et al. 2008 |
| Motor neurons | - | Locomotion defect | Not Determined | Kuwahara et al. 2006 | |
| Dopaminergic neurons | - | Locomotion defect | Not Determined | Kuwahara et al. 2006 | |
|
| |||||
| superoxide dismutase | Body wall muscles | + | Motility defect | Not Determined | Gidalevitz et al. 2009 |
| Pan neuronal | Age dependent | Locomotion defect | Yes | Wang et al. 2009 | |
|
| |||||
| tau | Pan neuronal | Age dependent | Uncoordinated | Yes | Kraemer et al. 2003 |
Neurodegenerative diseases linked to hereditary genetic mutations can be modeled in C. elegans to learn about the mechanisms of aggregation and cellular toxicity. It is likely that mutations that alter the amino acid sequence or increase expression of disease-associated proteins affect folding stability and challenge cellular chaperone networks and clearance processes (Meredith, 2005). Misfolding, for example, is well established as a prominent contributor to the polyQ class of neurodegenerative disorders. There are at least nine neurodegenerative disorders, including Huntington's disease, Kennedy's disease, and Ataxias in which the disease locus encodes a protein containing an expanded glutamine tract. This polyQ expansion that occurs within the coding region alters the physical properties of the corresponding protein leading to aggregation that is dependent on age and length of the polyQ tract (Trottier et al., 1995). It is widely accepted that polyQ expansions create a deleterious “gain of function” mutation within the corresponding protein, that contributes significantly to disease pathology (Orr, 2001; Ross, 2002). Based on these observations, it is possible to study the toxic effects of polyQ expansions on cellular functions by expressing the polyQ tract alone or in the context of the disease protein.
Recapitulating aspects of polyQ diseases lends further support to the usefulness of C. elegans models for protein conformational diseases. This has been shown in multiple tissue specific models for length and age dependent aggregation and toxicity of C. elegans expressing various polyQ lengths in neurons, body wall muscle cells, and intestinal cells (Brignull et al., 2006; Faber et al., 1999; Mohri-Shiomi and Garsin, 2008; Morley et al., 2002; Parker et al., 2001; Satyal et al., 2000). In the body wall muscle model, different lengths of polyQ fused to the yellow fluorescent protein (YFP) under the control of the body wall muscle specific promoter, unc-54, are expressed early in development with the formation of aggregates correlating with the length of the polyQ (Figure 2a) (Morley et al., 2002). Likewise, expression of polyQ under the control of a pan neuronal promoter also leads to a length dependent aggregation in neurons (Figure 2b) (Brignull et al., 2006). Monitoring aggregate formation over the lifetime of the animal by dynamic imaging methods reveals that polyQ aggregates are immobile and increase with age (Figure 2c). The age dependence of polyQ aggregation correlates directly with toxicity that can be measured using motility to monitor muscle cell function (Figure 2d). Loss of motility is also age dependent and is further accelerated in animals expressing longer polyQ tracts (Morley et al., 2002). Length and age dependent aggregation correlating with toxicity provides a tractable system to further examine the cellular and organismal effects of chronic expression of a misfolded protein.
Figure 2. C. elegans models of protein misfolding.
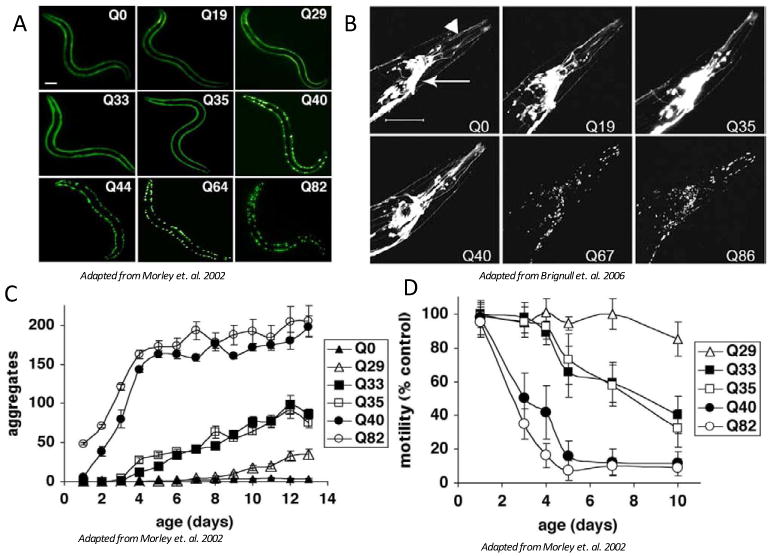
Length-dependent aggregation of polyQ-YFP fusion proteins expressed in body wall muscle cells (A) or neurons (B). Epifluoresence micrographs of 3- to 4-day-old animals expressing different lengths of polyQ-YFP. Scale bar = 0.1 mm (A), or scale bar = 50 um (B). Influence of aging on polyQ aggregation (C) and toxicity (D). (C) Number of aggregates in Q82 (○), Q40 (•), Q35 (□), Q33 (■), Q29 (△), and Q0 (▲) that accumulate during aging. Data are mean +/- SEM. Each data point represents at least five animals. (D) Motility index as a function of age for the same cohorts of animals described in C. Data are mean +/- SD as a percentage of age-matched Q0 animals.
Age dependent loss of functionality and aggregation has also been observed in C. elegans models for the aggregation prone peptide Aβ42 that is generated by processing of the amyloid precursor protein (APP) and associated with Alzheimer's disease pathology (Link, 1995). Transgenic animals that express Aβ42 in the body wall muscles using the unc-54 promoter accumulate aggregates and exhibit age dependent muscle toxicity (Link, 1995). In models for Familial Amyotrophic Lateral Sclerosis (ALS) or Parkinson's disease, expression of mutant superoxide dismutase (SOD1) and α-synuclein, respectively, also results in aggregation of disease proteins in the body wall muscle (Gidalevitz et al., 2009; van Ham et al., 2008). Furthermore, pan neuronal expression of mutant SOD1 or neuronal specific expression of mutant α-synuclein causes locomotion defects (Kuwahara et al., 2006; Wang et al., 2009). Pan neuronal expression of mutant tau that is associated with frontotemporal dementia and Parkinsonism leads to both age dependent aggregation and neuronal dysfunction (Kraemer et al., 2003) (Table 1). An advantage of these C. elegans transgenic models is their common genetic background (N2 Bristol), which makes it possible to compare different misfolded proteins in different tissues and to identify genetic modifiers.
In addition to comparing the expression of different aggregation prone proteins among transgenic lines, the ability to generate tissue specific expression allows a comparison of the same disease related protein in different tissue types, ie., body wall muscle, neurons, and intestinal cells expressing polyQ tracts (Brignull et al., 2006; Mohri-Shiomi and Garsin, 2008; Morley et al., 2002). Generally, longer polyQ tracts were necessary for aggregation and toxicity in the neuronal model. Expression of a lower number of glutamines, Q40 in body wall muscle, exhibited a clear aggregation and functional phenotype in young adults while detection of aggregates along with the variable range of behavioral deficits in the pan neuronal expression of Q40 suggest differential robustness of proteostasis networks between tissue types. Furthermore, fluorescence recovery after photobleaching (FRAP) revealed different sensitivity of specific neuronal subtypes to expanded polyQ tracts unlike body wall muscle cells that did not exhibit any heterogeneity. For example, Q40 did not aggregate in interneurons whereas ventral cord neurons expressed aggregated polyQ (Brignull et al., 2006). In intestinal cells, only 25% of the Q40 expressing animals develop aggregates throughout the life of the animals (Mohri-Shiomi and Garsin, 2008). Differential aggregation patterns within neuronal subtypes were also noted in the C. elegans model expressing a mutant form of SOD1 using a different pan neuronal promoter (Wang et al., 2009). Mutant SOD1 aggregates within the ventral nerve cord were detected earlier during development than in lateral body wall neurons. Specific mechanosensory neurons (PVDR) and specific interneurons (SDQR) consistently contained aggregates. Furthermore, expression of α-synuclein in the motor or dopaminergic neurons did not result in the appearance of aggregates (Kuwahara et al., 2006). These differences reveal that the cellular context strongly influences the aggregation phenotypes, however the fundamental aspects of protein misfolding diseases of age dependence of aggregate accumulation and cellular dysfunction are recapitulated.
Multiple facets within the protein quality control system may contribute to a cell's ability to respond to chronic expression of disease proteins. Misfolded disease proteins place a burden on the existing cellular folding network which, over time, leads to a collapse of proteostasis as other conformationally challenged proteins misfold (Gidalevitz et al., 2006). Depending on the level and subset of chaperones and co-chaperones that are available, the recognition and processing of these misfolded substrates may lead to deleterious consequences. For example, SOD1 or Q40 expressed in the ventral nerve cord consistently accumulated aggregates resulting in loss of neuronal function (Brignull et al., 2006; Wang et al., 2009). This suggests that the neurons of the ventral nerve cord express a chaperone network that either deals poorly with polyQ or mutant SOD1 or is limited in availability of chaperones perhaps due to competing requirements for normal neuronal function. The limited capability of the chaperone network to recognize these misfolded proteins may limit the initiation of a proper stress response and the subsequent elevation of specific chaperones. The accumulation of protein aggregates may also result from inefficiencies in transport and clearance, thus an enhancement of any of these steps could restore protein homeostasis.
IV. Chaperone networks that influence protein misfolding
The initial genome wide RNAi screen for modifiers of protein aggregation was based on a C. elegans model that expresses Q35 (Nollen et al., 2004) that takes advantage of the distinctive age dependence of aggregate formation associated with the physiological loss of motility (Figure 2c and 2d). Two candidate genes were used to validate this screen; downregulation of hsf-1, the principle regulator of chaperone expression, and hsp-1, that corresponds to Hsp70, with knockdown of both genes leading to an earlier onset aggregation phenotype consistent with the expected role for chaperones to suppress polyQ aggregation. The genome-wide screen for genes that resulted in early onset aggregation identified a specific subset of chaperones including two Hsp70s, one Hsp40, six subunits of the TRiC complex, and two TPR domain containing co-chaperones (Table 2). Another genome-wide RNAi screen employing a model for α-synuclein expression identified two chaperones, a different member of the Hsp70 family and an Hsp90 family member that when knocked-down, enhanced aggregation (Roodveldt et al., 2009; van Ham et al., 2008) (Table 2). In a biochemical approach to identify chaperones that associate with Aβ42 (Fonte et al., 2002), a combination of co-immunoprecipitation and mass spectrometry identified a subset of chaperones corresponding to two Hsp70 family members, three sHsps, and one TPR domain containing protein (Table 2). Overexpression of one of the sHsps, hsp-16.2, was shown to suppress Aβ42 toxicity (Fonte et al., 2008). Comparison of models expressing different aggregation-prone proteins using either RNAi or biochemical approaches provides new insights on the specificity of chaperone networks on aggregation and toxicity.
Table 2. Chaperones that respond to protein misfolding.
| Disease protein | Tissue expression pattern | Analysis | Chaperones identified | Reference |
|---|---|---|---|---|
| Aβ42 | Body wall muscles | CoIP of Aβ42 | HSF1 HSP70 (hsp-1, hsp-3) sHSPs (hsp-16.11, hsp-16.48, hsp-16.2) Co-chaperone-TPR (R05F9.10) |
Fonte et al. 2002 |
| polyglutamine tract | Body wall muscles | RNAi screen of Age dependent aggregation | HSF1 HSP70 (hsp-1, hsp-6) HSP40 (rme-8) TRiC (cct-1, cct-2, cct-4, cct-5, cct-6, cct-7) Co-chaperone-TPR (C50F2.3, MO3F8.3) |
Nollen et al. 2004 |
| α-synuclein | Body wall muscles | RNAi screen of Age dependent aggregation | HSP70 (hsp-70) HSP90 (R151.7) Co-chaperone-TPR (hip-1) |
van Ham et al. 2008 Roodveldt et al. 2009 |
| superoxide dismutase | Pan neuronal | RNAi screen of Age dependent aggregation | HSF1 HSP40 (dnj-19) sHSPs (F08H9.4) TRiC (cct-4, cct-5) Co-chaperone-Nucleotide exchange (C30C11.4, stc-1) |
Wang et al. 2009 |
| tau | Pan neuronal | RNAi screen ofAge dependent behavioral defect | HSF1 HSP70 (hsp-1) Co-chaperone-FKBP (fkb-6) Co-chaperone-TPR (T09B4.10) |
Kraemer et al. 2006 |
The threshold for polyQ aggregation and toxicity differs when expressed in neurons and muscle cells, which suggests that each tissue expresses a distinct complement of chaperones. Screens on pan-neuronal models expressing mutant SOD1 or tau identified subsets of chaperones that protect neurons from aggregation prone proteins (Table 2). In both models, RNAi against hsf-1 led to an enhancement in the age related phenotype that affects movement (tau) or aggregation (SOD1). Whereas the SOD1 screen identified one Hsp40, one sHsp, and two subunits of the TRiC complex together with two nucleotide exchange factors (Wang et al., 2009), the tau model identified the same Hsp70, hsp-1, as in the Q35 screen, but different co-chaperones, one Fkbp that may regulate Hsp90 and one TPR domain containing protein (Kraemer et al., 2006). The differences between the screens for neuronal models and between neuronal and muscle models suggests that related but non-identical chaperone components are involved. This suggests that efforts to identify the chaperones expressed in each cell type and tissue may provide insights on mechanisms of cytoprotection and susceptibility to disease associated proteins.
Overexpression of individual chaperones in Drosophila melanogaster models for Ataxin-3 prevents aggregation and progressive retinal degeneration (Bonini, 2002; Warrick et al., 1998). The inclusions contain Hsp70 and overexpression of human Hsp70 suppresses polyQ dependent degeneration (Warrick et al., 1999). Overexpression of a Hsp70 co-chaperone of the Hsp40 family, Hdj1, suppresses polyQ associated neurodegeneration while another member, Hdj2 had no effect (Chan et al., 2000). As a complement to this candidate gene approach, a genome wide screen employing overexpression lines using the Ataxin-3 Drosophila model identified the Hsp70 chaperone homologous to hsp-1 in C. elegans and two Hsp40s, one sHsp, and one TPR domain containing co-chaperone (Bilen and Bonini, 2007). These studies in a Drosophila model for a related aggregation prone protein reinforces the central role of chaperone networks in the susceptibility to misfolding stress.
In contrast to candidate approaches to identify modifiers for misfolding and aggregation, the majority of these ~400 genes identified in the genome-wide RNAi screens sort into multiple functional categories, including RNA processing, signal transduction, metabolism and transport (Nollen et al. 2004; van Ham et al. 2008; Wang et al. 2009; Kraemer et al. 2006). In these studies, only a small number of chaperones were identified. Chaperones act in a concerted fashion in vivo, therefore RNAi against a specific chaperone could influence the level or activity of other chaperones as expected from a tightly regulated network. Moreover, within the cell, chaperones perform unique, redundant or compensatory functions, to ensure proper folding and to prevent misfolding. Therefore, reducing the level of a single chaperone by RNAi treatment in vivo may not be sufficient to disrupt the cellular folding network. Nevertheless, the screens in C. elegans using multiple models for protein misfolding reveal a central role for HSF1 and Hsp70 in delaying aggregation and toxicity associated with expression of different disease proteins in different tissues. It is noteworthy that distinct members of the Hsp40, sHsp and co-chaperones were identified in each screen suggesting a basis for tissue-specificity associated with each of these aggregation prone substrates. Based on these findings, we speculate that specific chaperone networks may cooperate to maintain normal physiology in response to different protein folding challenges. Therefore, the availability of chaperones and co-chaperones expressed in specific cell types becomes critical to protect the cell and tissue from misfolded proteins. Understanding the regulation of chaperones levels and specificity is integral to mechanisms of age related aggregation and toxicity and the failure to sense and respond to misfolded disease proteins.
V. Regulation of chaperone networks during proteotoxic stress and aging
A hallmark for many neurodegenerative diseases is the age-associated onset of phenotypes due to aggregation and toxicity. These characteristics are observed in C. elegans models of protein misfolding; moreover it has been established that genes that regulate longevity suppress misfolding, in part by enhancing chaperone levels (Cohen et al., 2006; Morley et al., 2002). C. elegans has been an invaluable model organism for the discovery of pathways that modulate lifespan, including the insulin/insulin-like growth factor-1 signaling (ILS) pathway (Kenyon et al., 1993; Kimura et al., 1997a), that is conserved in higher organism such as flies and mice (Holzenberger et al., 2003; Piper et al., 2005). The C. elegans daf-2 gene encodes an insulin-like receptor (Apfeld and Kenyon, 1998; Dorman et al., 1995), which upon signaling activates a kinase cascade including phosphatidyl inositol 3-kinase (AGE-1), AKT/PKB, and PDK. This results in phosphorylation and inhibition of at least two transcription factors, the FOXO orthologue, DAF-16 (Lin et al., 1997), and the Nrf orthologue, SKN-1 (Tullet et al., 2008) that together positively regulate determinants of longevity (Figure 3). In addition to a doubling of lifespan compared to wild-type animals (Kenyon et al., 1993), mutations in daf-2 or age-1 exhibit increased resistance to various stresses, such as increased temperature (Lithgow et al., 1995), pathogens (Garsin et al., 2003) and oxidative stress (Honda and Honda, 1999). Reduction of ILS also delays the onset of polyQ aggregation (Morley et al., 2002); likewise this has been observed for Aβ42 induced paralysis in C. elegans (Cohen et al., 2006), and in Aβ mouse models of Alzheimer's disease (Cohen et al., 2009). These results reveal that pathways that determine lifespan and stress resistance are genetically connected to the regulation of proteostasis and the suppression of proteotoxicity.
Figure 3. Integration of stress responses and aging in C. elegans.

Stress responses are mediated through at least three transcription factors, HSF-1, DAF-16 and SKN-1. Ability of the animals to respond to various stresses decreases dramatically with age. Dietary restriction (DR), reduced insulin-like signaling (ILS) and various stresses activate cellular pathways leading to transcription of specific chaperone networks and other proteostasis machinery. Integration of these signals leads to coordination of the proper response that facilitates stress resistance and longevity in addition to suppression of proteotoxicity.
If the functionality of chaperone networks declines during aging, the compensatory upregulation of chaperones should increase longevity and enhance stress resistance. Indeed, long-lived C. elegans ILS mutants exhibit elevated levels of sHsps (Hsu et al., 2003; Walker et al., 2001), likely through activation of DAF-16. Moreover, studies on large isogenic C. elegans populations have demonstrated a clear predictive correlation between transcriptional activity of the hsp-16.2 promoter and the lifespan of the individual animal (Rea et al., 2005). Furthermore, overexpression of hsp-6 (Yokoyama et al., 2002) or extra copies of hsp-16 genes (Walker and Lithgow, 2003), can extend lifespan of C. elegans, and similarly lifespan extensions by increased chaperone levels have been reported in Drosophila (Tatar et al., 1997). Taken together, these observations indicate a direct involvement of chaperones in longevity. Whether this is the consequence of specific interactions of these chaperones with substrates or that upregulation of these chaperones feeds back into the chaperone network to enhance other components of the stress response remains unclear.
Molecular interactions between genetic pathways regulating lifespan and chaperone networks are linked by the activities of transcription factors such as HSF-1 that detect and respond to the flux of misfolded proteins. For example, C. elegans over expressing hsf-1 are both stress resistant and long lived (Hsu et al., 2003; Morley and Morimoto, 2004). In contrast, knockdown of hsf-1 results in a dramatically decreased lifespan as well as early formation of age pigments and several other markers of aging (Garigan et al., 2002). hsf-1 was identified in genome wide RNAi screens as a modifier of age dependent phenotypes associated with expression of disease proteins (Table 2). Knockdown of hsf-1 also decreases the lifespan of ILS mutants to the same extent as daf-16 knockdown, indicating that HSF-1 may be an important downstream transcription factor in the ILS pathway (Figure 3) (Hsu et al., 2003; Morley and Morimoto, 2004). Genetic analysis has revealed that DAF-16 and HSF-1 are essential transcriptional activators for a subset of chaperones, mostly sHsps, including hsp-16.1, hsp-16.49, hsp-12.6 and sip-1, which have HSF-1 and DAF-16 consensus DNA binding elements (Hsu et al., 2003). RNAi-mediated knockdown of each of these sHsps significantly reduces the lifespan of ILS-mutants, but none to the extent of the transcriptional activators daf-16 or hsf-1 (Hsu et al., 2003; Morley and Morimoto, 2004), indicating that sHsps may compensate for each other or have a concerted effect. Additional stress responsive transcription factors, such as SKN-1, may also regulate chaperone expression, either alone or in conjunction with DAF-16 or HSF-1 (Figure 3) (Oliveira et al., 2009; Park et al., 2009). The differential regulation of stress responsive transcription factors may be essential for cells and organisms to respond to various stresses by upregulation of specific chaperone networks.
The activity of stress responsive transcription factors are essential both for the ILS pathway and Dietary Restriction (DR) to extend lifespan and delay onset of age associated diseases in organisms from yeast to mammals (Masoro, 2005). Many of these studies have linked the effects of DR to the nicotinamide adenine dinucleotide (NAD+) dependent histone/protein deacetylase Sir2 (Imai et al., 2000; Kaeberlein and Powers, 2007). The role of C. elegans SIR-2.1 in DR is controversial (Greer and Brunet, 2009), but overexpression of sir-2.1 has been shown to extend lifespan (Tissenbaum and Guarente, 2001) and activation of SIR-2.1 with resveratrol results in rescue of polyQ toxicity (Parker et al., 2005). Consistently, the genome wide RNAi screens identified Sir2 family members as modifiers of age dependent phenotypes associated with expression of disease proteins (Kraemer et al., 2006; van Ham et al., 2008). Bacterial deprivation, i.e. the complete removal of bacteria, the food source for C. elegans, after adulthood results in long-lived animals and suppression of the toxicity of both Aβ and Q35 in an HSF-1 dependent manner (Steinkraus et al., 2008). These observations are consistent with the recent demonstration that the transcriptional activity of mammalian HSF-1 is positively regulated by the human Sir2 orthologue, SIRT1 (Westerheide et al., 2009), which also regulates the FOXO transcription factor (Brunet et al., 2004). Studies in mammalian cell lines have demonstrated that aging and cellular senescence reduces an organism's ability to respond to stress and maintain homeostasis even though HSF-1 protein levels remain constant with age (Fawcett et al., 1994). In contrast, SIRT1 protein levels appear to dramatically decrease with age (Sasaki et al., 2006), providing a possible regulatory link between a compromised heat shock response and aging (Westerheide et al., 2009).
The importance of a rapid and robust activation of stress pathways to ensure an appropriate response to proteotoxic stress and restoration of cellular health becomes evident in the face of various insults. Studies in C. elegans have shown that stress responsiveness of both the DAF-16 and the SKN-1 pathways is greatly reduced in older animals (Przybysz et al., 2009). The ability of C. elegans to maintain folding of metastable proteins becomes compromised early in adulthood (Ben-Zvi et al., 2009). While the folding machinery expressed during larval development is able to maintain correct folding and function of a variety of proteins harboring temperature-sensitive mutations, each of these proteins misfold and lose function shortly after adulthood in all somatic tissues examined. At the same age, wild-type animals show significantly decreased activation of the cytoprotective heat shock response and unfolded protein response (UPR) (Ben-Zvi et al., 2009). This suggests that the capacity of the proteostasis machinery is limited, and that persistent challenge and the accumulation of damaged proteins can tip the balance between the load of damaged proteins and the proteostasis machinery (Gidalevitz et al., 2006). However, overexpression of DAF-16 or HSF-1 restores the ability of aged cells to maintain a functional proteome suggesting that activation of transcriptional networks can restore cellular and organismal health (Figure 3) (Ben-Zvi et al., 2009).
VI. Concluding remarks
The symptoms of many neurodegenerative diseases associated with the expression of aggregation prone proteins begin later in life suggesting that aged cells are more susceptible to proteotoxic stresses (Figure 4). During the aging process, these disease-associated proteins continually interfere with normal chaperone function. Over time, this leads to the accumulation of damaged proteins that further challenges the capacity of the proteostasis machinery. Efforts by the cellular quality control machinery to respond to this cumulative damage can be enhanced by reduction of insulin signaling and stimulating dietary restriction and cytoprotective stress response pathways that activate transcriptional networks leading to expression of key chaperone networks. The positive outcome of rebalancing cellular protein homeostasis and restoring functionality of the proteome has the potential for novel approaches to treat neurodegenerative diseases and other diseases associated with missense mutations and protein instability. Therapeutic avenues that target proteostasis regulators may identify strategies for protection from toxic protein species and restore healthy aging (Figure 4) (Balch et al., 2008; Powers et al., 2009).
Figure 4. Proteostasis networks maintain functionality throughout lifespan.

Under normal conditions, the proteostasis machinery, which includes molecular chaperones, maintains a properly folded proteome in young animals. However, chronic expression of aggregation prone proteins or accumulation of damaged proteins during aging can lead to depletion of chaperones and components of the proteostasis machinery resulting in protein misfolding. Reduction of insulin signaling and dietary restriction activate transcriptional networks leading to expression of key chaperone components. These components facilitate chaperone mediated refolding and clearance of damaged proteins, rebalancing cellular protein homeostasis. As a consequence, functionality is restored, increasing the healthspan of the organism.
Acknowledgments
We thank members of the Morimoto lab for critical discussions and reading of the manuscript. J.S.P. was supported by an individual postdoctoral fellowship from the Carlsberg Foundation; research in the laboratory of R.I.M. was supported by grants from the National Institutes of Health (NIGMS and NIA), the HDSA Coalition for the Cure, and the ALS Association.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anckar J, et al. Inhibition of DNA binding by differential sumoylation of heat shock factors. Mol Cell Biol. 2006;26:955–64. doi: 10.1128/MCB.26.3.955-964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anfinsen CB. Principles that govern the folding of protein chains. Science. 1973;181:223–30. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- Apfeld J, Kenyon C. Cell nonautonomy of C. elegans daf-2 function in the regulation of diapause and life span. Cell. 1998;95:199–210. doi: 10.1016/s0092-8674(00)81751-1. [DOI] [PubMed] [Google Scholar]
- Balch WE, et al. Adapting proteostasis for disease intervention. Science. 2008;319:916–9. doi: 10.1126/science.1141448. [DOI] [PubMed] [Google Scholar]
- Ballinger CA, et al. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol. 1999;19:4535–45. doi: 10.1128/mcb.19.6.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi A, et al. Collapse of proteostasis represents an early molecular event in Caenorhabditis elegans aging. Proc Natl Acad Sci U S A. 2009;106:14914–9. doi: 10.1073/pnas.0902882106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Zvi AP, Goloubinoff P. Review: mechanisms of disaggregation and refolding of stable protein aggregates by molecular chaperones. J Struct Biol. 2001;135:84–93. doi: 10.1006/jsbi.2001.4352. [DOI] [PubMed] [Google Scholar]
- Bilen J, Bonini NM. Genome-wide screen for modifiers of ataxin-3 neurodegeneration in Drosophila. PLoS Genet. 2007;3:1950–64. doi: 10.1371/journal.pgen.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bimston D, et al. BAG-1, a negative regulator of Hsp70 chaperone activity, uncouples nucleotide hydrolysis from substrate release. Embo J. 1998;17:6871–8. doi: 10.1093/emboj/17.23.6871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonini NM. Chaperoning brain degeneration. Proc Natl Acad Sci U S A. 2002;99(4):16407–11. doi: 10.1073/pnas.152330499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brignull HR, et al. Polyglutamine proteins at the pathogenic threshold display neuron-specific aggregation in a pan-neuronal Caenorhabditis elegans model. J Neurosci. 2006;26:7597–606. doi: 10.1523/JNEUROSCI.0990-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–5. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- Bucciantini M, et al. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 2002;416:507–11. doi: 10.1038/416507a. [DOI] [PubMed] [Google Scholar]
- Bukau B, Horwich AL. The Hsp70 and Hsp60 chaperone machines. Cell. 1998;92:351–66. doi: 10.1016/s0092-8674(00)80928-9. [DOI] [PubMed] [Google Scholar]
- Camasses A, et al. The CCT chaperonin promotes activation of the anaphase-promoting complex through the generation of functional Cdc20. Mol Cell. 2003;12:87–100. doi: 10.1016/s1097-2765(03)00244-2. [DOI] [PubMed] [Google Scholar]
- Chan HY, et al. Mechanisms of chaperone suppression of polyglutamine disease: selectivity, synergy and modulation of protein solubility in Drosophila. Hum Mol Genet. 2000;9:2811–20. doi: 10.1093/hmg/9.19.2811. [DOI] [PubMed] [Google Scholar]
- Cohen E, et al. Opposing activities protect against age-onset proteotoxicity. Science. 2006;313:1604–10. doi: 10.1126/science.1124646. [DOI] [PubMed] [Google Scholar]
- Cohen E, et al. Reduced IGF-1 Signaling Delays Age-Associated Proteotoxicity in Mice. Cell. 2009;139:1157–1169. doi: 10.1016/j.cell.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, et al. The co-chaperone CHIP regulates protein triage decisions mediated by heat-shock proteins. Nat Cell Biol. 2001;3:93–6. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- Cutforth T, Rubin GM. Mutations in Hsp83 and cdc37 impair signaling by the sevenless receptor tyrosine kinase in Drosophila. Cell. 1994;77:1027–36. doi: 10.1016/0092-8674(94)90442-1. [DOI] [PubMed] [Google Scholar]
- Demand J, et al. The carboxy-terminal domain of Hsc70 provides binding sites for a distinct set of chaperone cofactors. Mol Cell Biol. 1998;18:2023–8. doi: 10.1128/mcb.18.4.2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuerling E, et al. Trigger factor and DnaK cooperate in folding of newly synthesized proteins. Nature. 1999;400:693–6. doi: 10.1038/23301. [DOI] [PubMed] [Google Scholar]
- Dorman JB, et al. The age-1 and daf-2 genes function in a common pathway to control the lifespan of Caenorhabditis elegans. Genetics. 1995;141:1399–406. doi: 10.1093/genetics/141.4.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duina AA, et al. A cyclophilin function in Hsp90-dependent signal transduction. Science. 1996;274:1713–5. doi: 10.1126/science.274.5293.1713. [DOI] [PubMed] [Google Scholar]
- Esser C, et al. Cooperation of molecular chaperones with the ubiquitin/proteasome system. Biochim Biophys Acta. 2004;1695:171–88. doi: 10.1016/j.bbamcr.2004.09.020. [DOI] [PubMed] [Google Scholar]
- Faber PW, et al. Polyglutamine-mediated dysfunction and apoptotic death of a Caenorhabditis elegans sensory neuron. Proc Natl Acad Sci U S A. 1999;96:179–84. doi: 10.1073/pnas.96.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett TW, et al. Effects of neurohormonal stress and aging on the activation of mammalian heat shock factor 1. J Biol Chem. 1994;269:32272–8. [PubMed] [Google Scholar]
- Fonte V, et al. Interaction of intracellular beta amyloid peptide with chaperone proteins. Proc Natl Acad Sci U S A. 2002;99:9439–44. doi: 10.1073/pnas.152313999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonte V, et al. Suppression of in vivo beta-amyloid peptide toxicity by overexpression of the HSP-16.2 small chaperone protein. J Biol Chem. 2008;283:784–91. doi: 10.1074/jbc.M703339200. [DOI] [PubMed] [Google Scholar]
- Freeman BC, et al. Identification of a regulatory motif in Hsp70 that affects ATPase activity, substrate binding and interaction with HDJ-1. Embo J. 1995;14:2281–92. doi: 10.1002/j.1460-2075.1995.tb07222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J. Folding of newly translated proteins in vivo: the role of molecular chaperones. Annu Rev Biochem. 2001;70:603–47. doi: 10.1146/annurev.biochem.70.1.603. [DOI] [PubMed] [Google Scholar]
- Garigan D, et al. Genetic analysis of tissue aging in Caenorhabditis elegans: a role for heat-shock factor and bacterial proliferation. Genetics. 2002;161:1101–12. doi: 10.1093/genetics/161.3.1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA, et al. Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science. 2003;300:1921. doi: 10.1126/science.1080147. [DOI] [PubMed] [Google Scholar]
- Gerber MR, et al. Cdc37 is required for association of the protein kinase Cdc28 with G1 and mitotic cyclins. Proc Natl Acad Sci U S A. 1995;92:4651–5. doi: 10.1073/pnas.92.10.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gething MJ, Sambrook J. Protein folding in the cell. Nature. 1992;355:33–45. doi: 10.1038/355033a0. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, et al. Progressive disruption of cellular protein folding in models of polyglutamine diseases. Science. 2006;311:1471–4. doi: 10.1126/science.1124514. [DOI] [PubMed] [Google Scholar]
- Gidalevitz T, et al. Destabilizing protein polymorphisms in the genetic background direct phenotypic expression of mutant SOD1 toxicity. PLoS Genet. 2009;5:e1000399. doi: 10.1371/journal.pgen.1000399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goloubinoff P, et al. Sequential mechanism of solubilization and refolding of stable protein aggregates by a bichaperone network. Proc Natl Acad Sci U S A. 1999;96:13732–7. doi: 10.1073/pnas.96.24.13732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer EL, Brunet A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell. 2009;8:113–27. doi: 10.1111/j.1474-9726.2009.00459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU. Molecular chaperones in cellular protein folding. Nature. 1996;381:571–9. doi: 10.1038/381571a0. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: from nascent chain to folded protein. Science. 2002;295:1852–8. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- Hohfeld J, Jentsch S. GrpE-like regulation of the hsc70 chaperone by the anti-apoptotic protein BAG-1. Embo J. 1997;16:6209–16. doi: 10.1093/emboj/16.20.6209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzenberger M, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421:182–7. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- Honda Y, Honda S. The daf-2 gene network for longevity regulates oxidative stress resistance and Mn-superoxide dismutase gene expression in Caenorhabditis elegans. Faseb J. 1999;13:1385–93. [PubMed] [Google Scholar]
- Hsu AL, et al. Regulation of aging and age-related disease by DAF-16 and heat-shock factor. Science. 2003;300:1142–5. doi: 10.1126/science.1083701. [DOI] [PubMed] [Google Scholar]
- Imai S, et al. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Kaeberlein M, Powers RW., 3rd Sir2 and calorie restriction in yeast: a skeptical perspective. Ageing Res Rev. 2007;6:128–40. doi: 10.1016/j.arr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Kenyon C, et al. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–4. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Kimura KD, et al. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997a;277:942–6. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- Kimura Y, et al. Cdc37 is a molecular chaperone with specific functions in signal transduction. Genes Dev. 1997b;11:1775–85. doi: 10.1101/gad.11.14.1775. [DOI] [PubMed] [Google Scholar]
- Kline MP, Morimoto RI. Repression of the heat shock factor 1 transcriptional activation domain is modulated by constitutive phosphorylation. Mol Cell Biol. 1997;17:2107–15. doi: 10.1128/mcb.17.4.2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauf U, et al. Repression of human heat shock factor 1 activity at control temperature by phosphorylation. Genes Dev. 1996;10:2782–93. doi: 10.1101/gad.10.21.2782. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, et al. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum Mol Genet. 2006;15:1483–96. doi: 10.1093/hmg/ddl067. [DOI] [PubMed] [Google Scholar]
- Kraemer BC, et al. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc Natl Acad Sci U S A. 2003;100:9980–5. doi: 10.1073/pnas.1533448100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeger PE, Morimoto RI. Selection of new HSF1 and HSF2 DNA-binding sites reveals difference in trimer cooperativity. Mol Cell Biol. 1994;14:7592–603. doi: 10.1128/mcb.14.11.7592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuwahara T, et al. Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem. 2006;281:334–40. doi: 10.1074/jbc.M504860200. [DOI] [PubMed] [Google Scholar]
- Lin K, et al. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278:1319–22. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–77. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- Link CD. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc Natl Acad Sci U S A. 1995;92:9368–72. doi: 10.1073/pnas.92.20.9368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lithgow GJ, et al. Thermotolerance and extended life-span conferred by single-gene mutations and induced by thermal stress. Proc Natl Acad Sci U S A. 1995;92:7540–4. doi: 10.1073/pnas.92.16.7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masoro EJ. Overview of caloric restriction and ageing. Mech Ageing Dev. 2005;126:913–22. doi: 10.1016/j.mad.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Meacham GC, et al. The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol. 2001;3:100–5. doi: 10.1038/35050509. [DOI] [PubMed] [Google Scholar]
- Meredith SC. Protein denaturation and aggregation: Cellular responses to denatured and aggregated proteins. Ann N Y Acad Sci. 2005;1066:181–221. doi: 10.1196/annals.1363.030. [DOI] [PubMed] [Google Scholar]
- Mohri-Shiomi A, Garsin DA. Insulin signaling and the heat shock response modulate protein homeostasis in the Caenorhabditis elegans intestine during infection. J Biol Chem. 2008;283:194–201. doi: 10.1074/jbc.M707956200. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–10. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- Morimoto RI. Regulation of the heat shock transcriptional response: cross talk between a family of heat shock factors, molecular chaperones, and negative regulators. Genes Dev. 1998;12:3788–96. doi: 10.1101/gad.12.24.3788. [DOI] [PubMed] [Google Scholar]
- Morimoto RI, et al. The heat-shock response: regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 1997;32:17–29. [PubMed] [Google Scholar]
- Morley JF, et al. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2002;99:10417–22. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley JF, Morimoto RI. Regulation of longevity in Caenorhabditis elegans by heat shock factor and molecular chaperones. Mol Biol Cell. 2004;15:657–64. doi: 10.1091/mbc.E03-07-0532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosser DD, et al. Coordinate changes in heat shock element-binding activity and HSP70 gene transcription rates in human cells. Mol Cell Biol. 1988;8:4736–44. doi: 10.1128/mcb.8.11.4736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollen EA, et al. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci U S A. 2004;101:6403–8. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RP, et al. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009;8:524–41. doi: 10.1111/j.1474-9726.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr HT. Beyond the Qs in the polyglutamine diseases. Genes Dev. 2001;15:925–932. doi: 10.1101/gad.888401. [DOI] [PubMed] [Google Scholar]
- Park SK, et al. Oxidative stress and longevity in Caenorhabditis elegans as mediated by SKN-1. Aging Cell. 2009;8:258–69. doi: 10.1111/j.1474-9726.2009.00473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JA, et al. Resveratrol rescues mutant polyglutamine cytotoxicity in nematode and mammalian neurons. Nat Genet. 2005;37:349–50. doi: 10.1038/ng1534. [DOI] [PubMed] [Google Scholar]
- Parker JA, et al. Expanded polyglutamines in Caenorhabditis elegans cause axonal abnormalities and severe dysfunction of PLM mechanosensory neurons without cell death. Proc Natl Acad Sci U S A. 2001;98:13318–23. doi: 10.1073/pnas.231476398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino MM, et al. Support for the prion hypothesis for inheritance of a phenotypic trait in yeast. Science. 1996;273:622–6. doi: 10.1126/science.273.5275.622. [DOI] [PubMed] [Google Scholar]
- Piper MDW, et al. Models of insulin signalling and longevity. Drug Discovery Today: Disease Models. 2005;2:249–256. [Google Scholar]
- Pirkkala L, et al. Roles of the heat shock transcription factors in regulation of the heat shock response and beyond. Faseb J. 2001;15:1118–31. doi: 10.1096/fj00-0294rev. [DOI] [PubMed] [Google Scholar]
- Powers ET, et al. Biological and chemical approaches to diseases of proteostasis deficiency. Annu Rev Biochem. 2009;78:959–91. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- Pratt WB, Toft DO. Steroid receptor interactions with heat shock protein and immunophilin chaperones. Endocr Rev. 1997;18:306–60. doi: 10.1210/edrv.18.3.0303. [DOI] [PubMed] [Google Scholar]
- Przybysz AJ, et al. Increased age reduces DAF-16 and SKN-1 signaling and the hormetic response of Caenorhabditis elegans to the xenobiotic juglone. Mech Ageing Dev. 2009;130:357–69. doi: 10.1016/j.mad.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman B, et al. AlphaB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid beta-peptide and beta2-microglobulin. Biochem J. 2005;392:573–81. doi: 10.1042/BJ20050339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea SL, et al. A stress-sensitive reporter predicts longevity in isogenic populations of Caenorhabditis elegans. Nat Genet. 2005;37:894–8. doi: 10.1038/ng1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roodveldt C, et al. Chaperone proteostasis in Parkinson's disease: stabilization of the Hsp70/alpha-synuclein complex by Hip. Embo J. 2009;28:3758–70. doi: 10.1038/emboj.2009.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington's disease and related disorders. Neuron. 2002;35:819–22. doi: 10.1016/s0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- Sasaki T, et al. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell. 2006;5:413–22. doi: 10.1111/j.1474-9726.2006.00235.x. [DOI] [PubMed] [Google Scholar]
- Satyal SH, et al. Polyglutamine aggregates alter protein folding homeostasis in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2000;97:5750–5. doi: 10.1073/pnas.100107297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, et al. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 1998;12:654–66. doi: 10.1101/gad.12.5.654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegers K, et al. TRiC/CCT cooperates with different upstream chaperones in the folding of distinct protein classes. Embo J. 2003;22:5230–40. doi: 10.1093/emboj/cdg483. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Siegers K, et al. Compartmentation of protein folding in vivo: sequestration of non-native polypeptide by the chaperonin-GimC system. Embo J. 1999;18:75–84. doi: 10.1093/emboj/18.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sondermann H, et al. Structure of a Bag/Hsc70 complex: convergent functional evolution of Hsp70 nucleotide exchange factors. Science. 2001;291:1553–7. doi: 10.1126/science.1057268. [DOI] [PubMed] [Google Scholar]
- Sorger PK. Heat shock factor and the heat shock response. Cell. 1991;65:363–6. doi: 10.1016/0092-8674(91)90452-5. [DOI] [PubMed] [Google Scholar]
- Soto C, Estrada LD. Protein misfolding and neurodegeneration. Arch Neurol. 2008;65:184–9. doi: 10.1001/archneurol.2007.56. [DOI] [PubMed] [Google Scholar]
- Steinkraus KA, et al. Dietary restriction suppresses proteotoxicity and enhances longevity by an hsf-1-dependent mechanism in Caenorhabditis elegans. Aging Cell. 2008;7:394–404. doi: 10.1111/j.1474-9726.2008.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayama S, Reed JC. Molecular chaperone targeting and regulation by BAG family proteins. Nat Cell Biol. 2001;3:E237–41. doi: 10.1038/ncb1001-e237. [DOI] [PubMed] [Google Scholar]
- Tatar M, et al. Chaperoning extended life. Nature. 1997;390:30. doi: 10.1038/36237. [DOI] [PubMed] [Google Scholar]
- Teter SA, et al. Polypeptide flux through bacterial Hsp70: DnaK cooperates with trigger factor in chaperoning nascent chains. Cell. 1999;97:755–65. doi: 10.1016/s0092-8674(00)80787-4. [DOI] [PubMed] [Google Scholar]
- Thulasiraman V, et al. In vivo newly translated polypeptides are sequestered in a protected folding environment. Embo J. 1999;18:85–95. doi: 10.1093/emboj/18.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–30. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- Trottier Y, et al. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–6. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- Tullet JM, et al. Direct inhibition of the longevity-promoting factor SKN-1 by insulin-like signaling in C. elegans. Cell. 2008;132:1025–38. doi: 10.1016/j.cell.2008.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Ham TJ, et al. C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet. 2008;4:e1000027. doi: 10.1371/journal.pgen.1000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker GA, Lithgow GJ. Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin-like signals. Aging Cell. 2003;2:131–9. doi: 10.1046/j.1474-9728.2003.00045.x. [DOI] [PubMed] [Google Scholar]
- Walker GA, et al. Heat shock protein accumulation is upregulated in a long-lived mutant of Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci. 2001;56:B281–7. doi: 10.1093/gerona/56.7.b281. [DOI] [PubMed] [Google Scholar]
- Wang J, et al. An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 2009;5:e1000350. doi: 10.1371/journal.pgen.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrick JM, et al. Suppression of polyglutamine-mediated neurodegeneration in Drosophila by the molecular chaperone HSP70. Nat Genet. 1999;23:425–8. doi: 10.1038/70532. [DOI] [PubMed] [Google Scholar]
- Warrick JM, et al. Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell. 1998;93:939–49. doi: 10.1016/s0092-8674(00)81200-3. [DOI] [PubMed] [Google Scholar]
- Westerheide SD, et al. Stress-inducible regulation of heat shock factor 1 by the deacetylase SIRT1. Science. 2009;323:1063–6. doi: 10.1126/science.1165946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittung-Stafshede P, et al. The J-domain of Hsp40 couples ATP hydrolysis to substrate capture in Hsp70. Biochemistry. 2003;42:4937–44. doi: 10.1021/bi027333o. [DOI] [PubMed] [Google Scholar]
- Wolynes PG, et al. Navigating the folding routes. Science. 1995;267:1619–20. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- Yokoyama K, et al. Extended longevity of Caenorhabditis elegans by knocking in extra copies of hsp70F, a homolog of mot-2 (mortalin)/mthsp70/Grp75. FEBS Lett. 2002;516:53–7. doi: 10.1016/s0014-5793(02)02470-5. [DOI] [PubMed] [Google Scholar]