

Abstract
Lateral gene transfer (LGT) between bacteria constitutes a strong force in prokaryote evolution, transforming the hierarchical tree of life into a network of relationships between species. In contrast, only a few cases of LGT from eukaryotes to prokaryotes have been reported so far. The distal animal intestine is predominantly a bacterial ecosystem, supplying the host with energy from dietary polysaccharides through carbohydrate-active enzymes absent from its genome. It has been suggested that LGT is particularly important for the human microbiota evolution. Here we show evidence for the first eukaryotic gene identified in multiple gut bacterial genomes. We found in the genome sequence of several gut bacteria, a typically eukaryotic glycoside-hydrolase necessary for starch breakdown in plants. The distribution of this gene is patchy in gut bacteria with presence otherwise detected only in a few environmental bacteria.
We speculate that the transfer of this gene to gut bacteria occurred by a sequence of two key LGT events; first, an original eukaryotic gene was transferred probably from Archaeplastida to environmental bacteria specialized in plant polysaccharides degradation and second, the gene was transferred from the environmental bacteria to gut microbes.
Keywords: eukaryote-to-prokaryote LGT, DPE2, Bacteroides sp, gut microbiota, GH77
Introduction
LGT allows for rapid transfer of genes under strong selection and represents one way that members of the microbiota could share metabolic capabilities. It has been shown that LGT is particularly important for the microbiota evolution in the human distal gastrointestinal tract.1 Polysaccharide utilization is an important activity in the lower intestine and the ability of resident bacteria to utilize different polysaccharides provides a distinct competitive advantage.2 Recently, it has been demonstrated that Bacteroides have acquired new useful genes from environmental microbes.3 Bacteroides are the most frequent bacteria in the human gut microbiota, and harvest a vast array of dietary and host-derived glycans via outer membrane protein complexes. Genes encoding these proteins are clustered together in similarly patterned Polysaccharide utilization loci (PUL). Notably, Bacteroides thetaiotaomicron, a prototypic Bacteroides, possesses 88 PULs, differing in polysaccharide specificity.2 Intriguingly, we have found in the genome of various species of Bacteroidales, an isolated glycoside-hydrolase coding gene that belongs to CAZy family GH77. The top-scoring BLASTp hit of a characterized protein was Arabidopsis thaliana DPE2 (Disproportionating Enzyme 2). Plant DPE2 are modular glycoside hydrolases consisting of a GH77 domain interrupted by an insertion of ~150 amino acids and two carbohydrate binding modules (CBM20) at the N-terminal extension (Fig. 1). The DPE2 gene codes for a 4-α-glucanotransferase (EC 2.4.1.25) essential for maltose metabolism during the conversion of transitory starch to sucrose in the cytosol of plant cells.4 Previous phylogenetic analyses support the eukaryotic origin of DPE2-like coding gene.5 Moreover, DPE2-like genes are present only in Bacteroidales but absent in all others groups of Bacteria, including Cyanobacteria. This further argues for an origin in the nuclear genomes of eukaryotes and not from past endosymbiosis and transfer from an ancestral plastid.
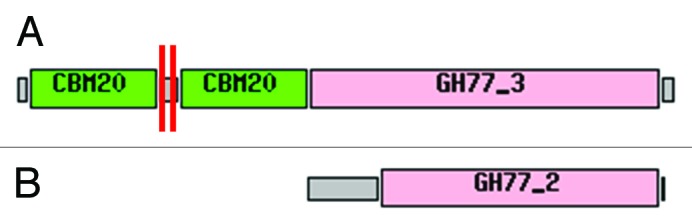
Figure 1. Reference modular structures of the (A) DPE2-like protein and (B) bacteria-like GH77 protein as represented in CAZy database. Horizontal red bars represent the position of the conserved introns.
Results
To elucidate the ancestry and evolutionary history of bacterial DPE2-like proteins, we constructed phylogenetic trees. DPE2-like genes were identified in a few eukaryotic taxa and in a small group of gut and environmental bacteria (Table 1). The phylogenetic analysis showed that bacterial DPE2-like enzymes form a highly supported group branching with their eukaryotic orthologs (Fig. 2). The cluster of bacterial enzymes was positioned inside the eukaryotic cluster. Interestingly, DPE2-like enzymes of environmental bacteria were positioned at the base of the bacterial cluster. The tree topology suggests that one LGT event occurred from eukaryota, probably Archaeplastida, to ancient environmental bacterium similar to Haliscomenobacter hydrossis or Paludibacter propionicigenes. H. hydrossis is sporadically observed in aeration tanks of sewage treatment plants and in paper industry wastewater treatment plants and P. propionicigenes is a fermentative anaerobe from plant residue and rice roots dwelling in irrigated rice-field soil. Both bacteria are specialized in the degradation of plant polysaccharides and could potentially be in contact in a common environment. Later, another LGT event has occurred, from these environmental bacteria to gut Bacteroidales, possibly using food as vector. Interestingly, we identified only one bacteria species that possess two DPE2-like genes. Succinatimonas hippei, a human gut Gammaproteobacteria, presents a DPE2-like gene (Fig. 2) but also a prototypic bacterial GH77 (Fig. 1). We propose that S. hippei has acquired this gene recently from Bacteroidales bacteria inside the human gut, and has also retained the original bacterial GH77 gene. An alternative scenario cannot be excluded; it is also possible that the gut community has acquired DPE2 gene directly from plants or other eukaryote dwelling in the animal intestine and later environmental bacteria have acquired it by LGT from gut bacteria released in the environment. However, the branching order in the topology we have reconstructed places environmental bacteria in a basal position and thus tend to support more a first transfer to environmental bacteria then to gut bacteria.
Table1. List of sequences included in the DPE2 phylogenetic analysis, accession numbers and full species names are included. Sequences are sorted by taxonomic group, then by species name. DPE2-like sequences that possess introns in the genomic sequences are indicated.
| Species | Accession | Introns | Taxon |
|---|---|---|---|
|
Arabidopsis lyrata |
XP_002879894.1 |
yes |
chloroplastida |
|
Arabidopsis thaliana |
AAL91204.1 |
yes |
chloroplastida |
|
Carica papaya |
evm.TU.supercontig_92.60 |
yes |
chloroplastida |
|
Chlamydomonas reinhardtii |
XP_001701179.1 |
yes |
chloroplastida |
|
Micromonas sp RCC299 |
ACO70268.1 |
yes |
chloroplastida |
|
Oryza sativa Japonica Group |
NP_001060547.1 |
yes |
chloroplastida |
|
Ostreococcus lucimarinus CCE9901 |
ABO98795.1 |
yes |
chloroplastida |
|
Physcomitrella patens subsp patens |
XP_001779217.1 |
yes |
chloroplastida |
|
Populus trichocarpa |
XP_002323208.1 |
yes |
chloroplastida |
|
Populus trichocarpa |
XP_002308854.1 |
yes |
chloroplastida |
|
Ricinus communis |
XP_002523669.1 |
yes |
chloroplastida |
|
Selaginella moellendorffii |
XP_002979331.1 |
yes |
chloroplastida |
|
Selaginella moellendorffii |
XP_002988641.1 |
yes |
chloroplastida |
|
Sorghum bicolor |
XP_002461165.1 |
yes |
chloroplastida |
|
Vitis vinifera |
XP_002278329.1 |
yes |
chloroplastida |
|
Volvox carteri f. nagariensis |
XP_002956849.1 |
yes |
chloroplastida |
|
Dictyostelium discoideum AX4 AX4 |
EAL65318.1 |
yes |
amoebozoa |
|
Dictyostelium purpureum |
XP_003286541.1 |
yes |
amoebozoa |
|
Polysphondylium pallidum PN500 |
EFA84397.1 |
yes |
amoebozoa |
|
Entamoeba dispar SAW760 |
EDR24789.1 |
no |
amoebozoa |
|
Entamoeba histolytica HM-1:IMSS |
EAL52093.1 |
no |
amoebozoa |
|
Giardia intestinalis ATCC 50581 |
EET00671.1 |
no |
metamonada |
|
Giardia lamblia ATCC 50803 |
XP_001709888.1 |
no |
metamonada |
|
Trichomonas vaginalis G3 ATCC PRA-98 |
EAX97809.1 |
no |
metamonada |
|
Cyanidioschyzon merolae |
CMT204C |
no |
rhodophyte |
|
Cyanidioschyzon merolae |
CMP352C |
no |
rhodophyte |
|
Galderia sulphuraria |
Gs34050.1 |
no |
rhodophyte |
|
Calliarthron tuberculosum |
contig_2_67557 |
no |
rhodophyte |
|
Porphyridium cruentum |
Contig14123 |
no |
rhodophyte |
|
Porphyridium cruentum |
Contig9269 |
no |
rhodophyte |
|
Alistipes shahii WAL 8301 |
CBK62792.1 |
- |
bacteroidetes (gut) |
|
Bacteroides caccae ATCC 43185 |
ZP_01961413.1 |
- |
bacteroidetes (gut) |
|
Bacteroides coprocola DSM 17136 |
ZP_03011722.1 |
- |
bacteroidetes (gut) |
|
Bacteroides coprophilus DSM 18228 |
ZP_03643552.1 |
- |
bacteroidetes (gut) |
|
Bacteroides eggerthii DSM 20697 |
ZP_03457421.1 |
- |
bacteroidetes (gut) |
|
Bacteroides finegoldii DSM 17565 |
ZP_05415079.1 |
- |
bacteroidetes (gut) |
|
Bacteroides fragilis NCTC 9343 |
YP_213214.1 |
- |
bacteroidetes (gut) |
|
Bacteroides ovatus ATCC 8483 |
ZP_02066204.1 |
- |
bacteroidetes (gut) |
|
Bacteroides stercoris ATCC 43183 |
ZP_02436972.1 |
- |
bacteroidetes (gut) |
|
Bacteroides thetaiotaomicron VPI-5482 |
AAO77253.1 |
- |
bacteroidetes (gut) |
|
Bacteroides uniformis ATCC 8492 |
EDO54757.1 |
- |
bacteroidetes (gut) |
|
Bacteroides vulgatus ATCC 8482 |
ABR39076.1 |
- |
bacteroidetes (gut) |
|
Bacteroides xylanisolvens XB1A |
CBK66288.1 |
- |
bacteroidetes (gut) |
|
Haliscomenobacter hydrossis DSM 1100 |
AEE47959.1 |
- |
bacteroidetes (environmental) |
|
Paludibacter propionicigenes WB4 |
ADQ79431.1 |
- |
bacteroidetes (environmental) |
|
Flavobacteriaceae bacterium 3519–10 |
ACU06866.1 |
- |
bacteroidetes (environmental) |
|
Chryseobacterium gleum ATCC 35910 |
ZP_07084258.1 |
- |
bacteroidetes (vagina) |
|
Parabacteroides distasonis ATCC 8503 |
ABR41798.1 |
- |
bacteroidetes (gut) |
|
Parabacteroides johnsonii DSM 18315 |
ZP_03478180.1 |
- |
bacteroidetes (gut) |
|
Parabacteroides merdae ATCC 43184 |
ZP_02031326.1 |
- |
bacteroidetes (gut) |
|
Porphyromonas gingivalis W83 |
BAG33312.1 |
- |
bacteroidetes (gut) |
|
Prevotella bergensis DSM 17361 |
ZP_06005569.1 |
- |
bacteroidetes (gut) |
|
Prevotella bivia JCVIHMP010 |
ZP_06267844.1 |
- |
bacteroidetes (gut) |
|
Prevotella bryantii |
ZP_07061030.1 |
- |
bacteroidetes (gut) |
|
Prevotella buccae D17 |
ZP_06419222.1 |
- |
bacteroidetes (gut) |
|
Prevotella copri DSM 18205 |
ZP_06252955.1 |
- |
bacteroidetes (gut) |
|
Prevotella disiens FB035–09AN |
ZP_07323182.1 |
- |
bacteroidetes (gut) |
|
Prevotella marshii DSM 16973 |
ZP_07366273.1 |
- |
bacteroidetes (gut) |
|
Prevotella melaninogenica ATCC 25845 |
ADK95815.1 |
- |
bacteroidetes (gut) |
|
Prevotella oris F0302 |
ZP_06254330.1 |
- |
bacteroidetes (gut) |
|
Prevotella timonensis CRIS 5C-B1 |
ZP_06289177.1 |
- |
bacteroidetes (gut) |
|
Prevotella veroralis F0319 |
ZP_05857248.1 |
- |
bacteroidetes (gut) |
| Succinatimonas hippei YIT 12066 | ZP_08077820.1 | - | bacteroidetes (gut) |

Figure 2. Phylogenetic analyses of the DPE2-like sequences. Representation of the phylogenetic tree was generated by FigTree (http://tree.bio.ed.ac.uk/software/figtree/), the different branch colors correspond to the different taxa further detailed in Table 1. The Bayesian topology was chosen as a reference and, overall, it corresponds to that of the maximum likelihood (ML) consensus trees. Supporting posterior probability values are indicated at each node and the corresponding bootstrap support values from the ML analysis are reported in parentheses. The orange arrow indicates the LGT from the eukaryotes to environmental bacteria, the yellow arrow the LGT from environmental bacteria to gut bacteria and the red arrow the LGT from Bacteroides to Proteobacteria.
In eukaryotes, we studied the conservation of intron position. Two introns localized between the two CBM20 modules are shared by the eukaryotic lineages supporting the common origin of DPE2-like genes (Fig. S1). The possible eukaryotic progenitors are Rhodophyta DPE2 genes. Interestingly, Rhodophyta coding genes, including DPE2, are mostly intronless, which could make easier the transfer of the eukaryotic gene to recipient bacteria.6,7 The phylogeny of the two CBM20 reflects the GH77 module phylogeny (Fig. 3), indicating their presence in the common ancestor of eukaryotic DPE2-like proteins.

Figure 3 (See previous page). Phylogenetic analyses of the two CBM20 identified on DPE2-like sequences. CBM20_1 corresponds to the carbohydrate binding domain located at the N-terminal region. Bayesian phylogenetic tree generated by FigTree (http://tree.bio.ed.ac.uk/software/figtree/), different branch colors correspond to the different taxa. The overall topology of the Bayesian tree corresponds to that of the ML consensus trees (data not shown).
Discussion
Many complex plant polysaccharides are resistant to digestion due to either insolubility or lack of host-encoded hydrolytic enzymes. These carbohydrates are not absorbed in the upper gastrointestinal tract but serve as a major source of carbon and energy for the distal gut microbial community. These “nondigestible” dietary carbohydrate substrates include the so-called resistant starch fraction, plant cell wall material and oligosaccharides. Polysaccharide degradation is one of the core functions encoded by the human gut microbiota and the ability to target these substrates resides in many different PULs.8 The starch utilization system (SUS) was the first PUL to be described. Although the SUS system is essential for the growth of B. thetaiotaomicron on starch, SUS genes are not required for growth on maltose, a typical byproduct of the starch breakdown9 (and other publications by Salyers and co-workers). Furthermore, until now it is not proven that any of the SUS enzymes can degrade maltose. Here we show that the gut bacteria DPE2-like gene was most likely acquired by a LGT of eukaryotic origin and we suspect that it is probably involved in maltose degradation. MalQ, another GH77, is indispensable to the maltose regulon of Escherichia coli transferring maltosyl and longer dextrinyl residues onto glucose, maltose and longer maltodextrins. This operon is absent in the genome of gut bacteria belonging to the order bacteroidales. It has been shown that E. coli mutants lacking MalQ amylomaltase can no longer grow on maltose, but this ability can be restored by A. thaliana DPE2.10 It is possible that DPE2 represents a substantial competitive advantage to the host and the microbiota, providing gut bacteria with the capacity to degrade resistant starch byproducts. We speculate that the LGT event leading to the acquisition of DPE2 by gut microbiota has been crucial in the host-bacterial relationship establishment with animals during evolution, and other similar gene transfers can certainly be expected.
Materials and Methods
Sequence similarity search and retrieval
The putative GH77 sequences were identified using the CAZy annotation pipeline,11 and the DPE2 proteins homologues identified in various different databases (DB). Protein sequence data were retrieved by BLASTp searches against the NCBI non-redundant database, EUpathDB (http://eupathdb.org/eupathdb/), Phytozome v7.0 (http://www.phytozome.net/) and the Galderia sulphuraria DB (http://genomics.msu.edu/cgi-bin/galdieria/blast.cgi) using the Bacteroidales DPE2-like sequences and Arabidopsis thaliana DPE2 (AT2G40840) as queries. Only sequences that aligned the entire length of the GH77 module at the protein level with an e-value not higher than e-50 were kept for multiple sequence alignment in order to keep as much as possible informative sites for phylogenetic reconstruction. Sequences of a same species that were 100% identical to one another or entirely included in a longer one were eliminated to remove redundancy. There is a small amount of public Rhodophyte sequences available. For this reason three additional Rhodophyte DPE2 fragmentary sequences were included in the analysis: two Porphyridium cruentum and one Calliarthron tuberculosum protein sequences. These sequences were obtained from a non-public database hosted at Rutgers University, which is part of a current sequencing project. DPE2-like sequences were aligned using MUSCLE with default parameters12 and multiple sequence alignments were manually examined using JALVIEW.13,14
Phylogenetic reconstruction
We performed phylogenetic analyzes using two different approaches, Bayesian estimation and bootstrapped maximum likelihood, as described by Danchin.15 We have rooted the tree using as out group four bacterial GH77 proteins that non-controversially belong to a different CAZy subfamily: YP_003248951.1 (Fibrobacter succinogenes ssp. succinogenes S85), YP_004699220.1 (Spirochaeta caldaria DSM 7334), CAN92536.1 (Sorangium cellulosum “So ce 56”) and ABS24534.1 (Anaeromyxobacter sp Fw109-5). Blast and a HMM libraries are used as complementary comparison tools for family and sub-family division in CAZy database. These bacterial-type GH77 sequences (GH77_2, Fig. 1) were chosen because they are the closest GH77 that are not DPE2-like sequences (GH77_3, Fig. S2). Bayesian phylogenetic reconstructions were done using MrBayes software16 with a mixture of models, an estimated gamma distribution of rates of evolution and an estimation of the proportion of invariable sites. By default 100,000 generations were run for each phylogeny reconstruction. In case the average standard deviation of split frequencies was not inferior to 0.05 after 100,000 generations, additional generations were launched until congruence was reached (<0.05). Consensus trees and statistics were obtained after systematically “burning” 25% of generated trees. Posterior probability support values are reported for each node in Figure 2. To obtain support from a second independent method, we also performed phylogenetic analyses using maximum likelihood (ML) estimation with the RAxML software.17 We systematically ran 100 bootstrap replicates followed by a ML search for the best‐scoring tree. For DPE2 and CBM20 phylogeny we selected the WAG model of amino acids evolution because it returned the best posterior probability score in corresponding Bayesian phylogenetic analysis. We used a model with four categories of estimated gamma rates of evolution as well as an estimate of the proportion of invariable sites. The overall topology of the ML consensus trees corresponds to that of the Bayesian trees and the values between parentheses in Figure 2 correspond to bootstrap values. Trees were generated using FigTree (http://tree.bio.ed.ac.uk/software/figtree/).
Intron position determination
Exon-intron structures were predicted based on alignment of corresponding protein sequences with genome assemblies, using the online tool WEBSCIPIO.18 Positions of introns were reported on the protein sequences by inserting the “XXXXX” characters at the junction between two consecutive exons. We generated multiple alignments to determine conservation of intron positions between species and clades.
Supplementary Material
Acknowledgments
We thank D. Bhattacharya for C. tuberculosum and P. cruentum sequences, and A. Weber for the G. sulphuraria sequence.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/mge/article/20375
References
- 1.Xu J, Mahowald MA, Ley RE, Lozupone CA, Hamady M, Martens EC, et al. Evolution of symbiotic bacteria in the distal human intestine. PLoS Biol. 2007;5:e156. doi: 10.1371/journal.pbio.0050156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonnenburg ED, Zheng HJ, Joglekar P, Higginbottom SK, Firbank SJ, Bolam DN, et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell. 2010;141:1241–52. doi: 10.1016/j.cell.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464:908–12. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- 4.Lu Y, Sharkey TD. The role of amylomaltase in maltose metabolism in the cytosol of photosynthetic cells. Planta. 2004;218:466–73. doi: 10.1007/s00425-003-1127-z. [DOI] [PubMed] [Google Scholar]
- 5.Deschamps P, Colleoni C, Nakamura Y, Suzuki E, Putaux JL, Buléon A, et al. Metabolic symbiosis and the birth of the plant kingdom. Mol Biol Evol. 2008;25:536–48. doi: 10.1093/molbev/msm280. [DOI] [PubMed] [Google Scholar]
- 6.Rogers MB, Patron NJ, Keeling PJ. Horizontal transfer of a eukaryotic plastid-targeted protein gene to cyanobacteria. BMC Biol. 2007;5:26. doi: 10.1186/1741-7007-5-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet. 2008;9:605–18. doi: 10.1038/nrg2386. [DOI] [PubMed] [Google Scholar]
- 8.Martens EC, Chiang HC, Gordon JI. Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe. 2008;4:447–57. doi: 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shipman JA, Cho KH, Siegel HA, Salyers AA. Physiological characterization of SusG, an outer membrane protein essential for starch utilization by Bacteroides thetaiotaomicron. J Bacteriol. 1999;181:7206–11. doi: 10.1128/jb.181.23.7206-7211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lu Y, Steichen JM, Yao J, Sharkey TD. The role of cytosolic alpha-glucan phosphorylase in maltose metabolism and the comparison of amylomaltase in Arabidopsis and Escherichia coli. Plant Physiol. 2006;142:878–89. doi: 10.1104/pp.106.086850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for Glycogenomics. Nucleic Acids Res. 2009;37(Database issue):D233–8. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–91. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clamp M, Cuff J, Searle SM, Barton GJ. The Jalview Java alignment editor. Bioinformatics. 2004;20:426–7. doi: 10.1093/bioinformatics/btg430. [DOI] [PubMed] [Google Scholar]
- 15.Danchin EGJ, Rosso MN, Vieira P, de Almeida-Engler J, Coutinho PM, Henrissat B, et al. Multiple lateral gene transfers and duplications have promoted plant parasitism ability in nematodes. Proc Natl Acad Sci U S A. 2010;107:17651–6. doi: 10.1073/pnas.1008486107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–5. doi: 10.1093/bioinformatics/17.8.754. [DOI] [PubMed] [Google Scholar]
- 17.Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 18.Odronitz F, Pillmann H, Keller O, Waack S, Kollmar M. WebScipio: an online tool for the determination of gene structures using protein sequences. BMC Genomics. 2008;9:422. doi: 10.1186/1471-2164-9-422. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.