

Abstract
Recent research suggests that microtubule-associated protein 1 light chain 3B (LC3B) confers protection against hypoxia-induced pulmonary hypertension (HPH) by inhibiting proliferation of pulmonary artery (PA) wall cells. We recently demonstrated that 17β-estradiol (E2), a sex hormone with known protective properties in HPH, increases lung LC3-II expression in chronically hypoxic male Sprague-Dawley rats. Stimulatory E2 effects on LC3-II were recapitulated in isolated hypoxic (1% O2 for 48 h), but not room air-exposed primary rat PA endothelial cells (PAECs), and were accompanied by hypoxia-specific inhibitory effects on other parameters involved in proproliferative signaling (MAPK3/ERK1-MAPK1/ERK2 activation, VEGF secretion), as well as inhibitory effects on PAEC proliferation. Taken together, these results suggest that E2 mediates hypoxia-specific antiproliferative effects in PAECs, and that stimulation of autophagy may be one of the underlying mechanisms of E2-mediated protection in HPH. Viewed in the context of previously published data, these results indicate that LC3 1) exerts protective effects in the pathogenesis of HPH, and 2) may represent a potential target for future therapeutic interventions in HPH.
Keywords: 17-beta estradiol, estrogen, hypoxia, pulmonary artery endothelial cells, pulmonary vascular remodeling, cell proliferation, MAPK3/ERK1, MAPK1/ERK2
Hypoxia-induced pulmonary hypertension is a common clinical problem in patients with chronic hypoxic lung disease, sleep disordered breathing, or high-altitude exposure. If left untreated, HPH frequently leads to right ventricular failure and death. However, no pharmacologic treatment options for HPH exist, and pulmonary vasodilators used for other types of pulmonary hypertension, if used in HPH, may worsen ventilation/perfusion mismatch and oxygenation. New treatment strategies for HPH are therefore needed. Ideally, such strategies should be directed at the exaggerated and uncontrolled proliferation of pulmonary vascular wall cells that underlies the progressive narrowing of the lumen of the pulmonary vasculature and that represents the morphological correlate for the clinically observed increases in PA pressures and right ventricular afterload.
Recent research suggests that LC3B confers protection against HPH by inhibiting proliferation of PA endothelial cells and smooth muscle cells. Therapies aimed at enhancing autophagy may therefore be of merit in HPH. E2 attenuates HPH through an unknown mechanism that includes inhibitory effects on PA wall cell proliferation. A better understanding of the mechanisms involved in E2-mediated antiproliferative effects may allow for targeted, nonhormonal therapeutic strategies in HPH. However, the effects of E2 on autophagy in HPH have not yet been described. We therefore sought to determine whether E2-mediated protection in HPH is associated with stimulatory effects on LC3 expression.
We exposed male Sprague-Dawley rats to hypobaric hypoxia (Patm = 382 mmHg for 2 weeks; equivalent to 10% FiO2) while being treated with E2 (75 µg/kg/day via subcutaneous osmotic minipumps) or vehicle. E2 treatment was associated with improved cardiopulmonary hemodynamics, decreased PA remodeling, decreased whole lung MAPK3-MAPK1 activation, and increased whole lung expression of LC3-II. Stimulatory E2 effects on LC3-II were recapitulated in isolated hypoxic (1% O2 for 48 h), but not room air-exposed PAECs. Interestingly, disparate, O2-concentration-dependent E2 effects were also observed with regards to other parameters involved in PAEC proliferation. In particular, in hypoxic PAECs, E2 decreased MAPK3-MAPK1 activation, vascular endothelial growth factor secretion, and cell proliferation, and increased expression of the cell cycle-inhibitor CDKN1B/p27Kip1, while no such effects were noted in PAECs exposed to room air. Mechanistic cell culture experiments demonstrated inhibitory effects of MAPK3-MAPK1 on PAEC LC3-II expression, suggesting that E2 may stimulate autophagy by decreasing MAPK3-MAPK1 activation. While E2 effects on several endpoints were attenuated by estrogen receptor (ER) blockade, E2 action on LC3-II did not appear to be ER-mediated. This suggests a potential role of nontraditional ERs or additional, ER-independent E2 metabolites in mediating E2 effects on autophagy.
Viewed in the context of previously published data, these results indicate that (1) LC3 exerts beneficial regulatory effects in the pathogenesis of HPH, (2) the protective effects of E2 in HPH may be mediated at least in part by hypoxia-specific, stimulatory effects on LC3-II expression, and (3) LC3 may represent a potential target for future therapeutic interventions in HPH (Fig. 1). The latter point is of particular interest, since the mechanistic target of rapamycin (MTOR) inhibitor rapamycin attenuates HPH in rodents. Interestingly, rapamycin administration decreases chronic hypoxia-induced human and rat PA smooth muscle cell proliferation. While the effects of rapamycin on autophagy in HPH have not yet been evaluated, it is conceivable that stimulation of autophagy may at least in part be responsible for rapamycin’s antimitogenic effects. Stimulation of autophagy is currently being explored as a therapeutic option in cancer, and it is conceivable that enhancing autophagy may also counteract the uncontrolled proliferation of vascular wall cells in HPH and other types of pulmonary hypertension. Clearly, further studies are needed to investigate the role of autophagy as a therapeutic target in pulmonary hypertension, but the encouraging results observed with E2 and rapamycin suggest that in the future such investigations may translate into improved patient outcomes.
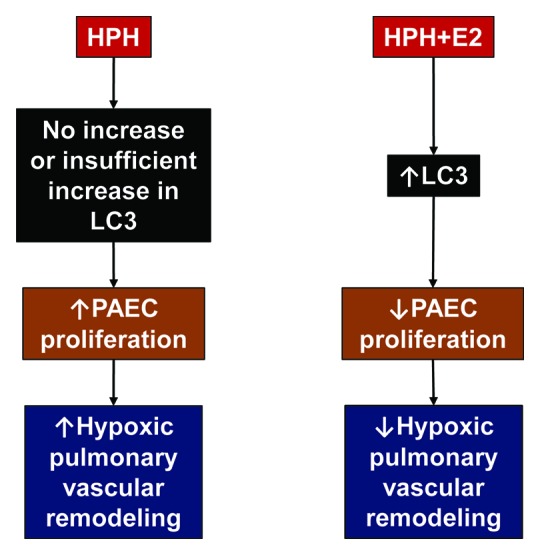
Figure 1. Potential role of autophagy in attenuation of hypoxia-induced pulmonary hypertension (HPH). Lack of increase or insufficient increase in LC3 in HPH may allow for pulmonary artery endothelial cell (PAEC) activation and proliferation with subsequent hypoxic pulmonary vascular remodeling. PAECs play a critical role in HPH development by secreting growth factors and pro-inflammatory cytokines that further stimulate PA smooth muscle cell proliferation and recruitment of inflammatory as well as progenitor cells. Hypoxia-specific stimulatory effects of 17β-estradiol (E2) on autophagy may attenuate PAEC activation and proliferation, thereby contributing to attenuated hypoxic pulmonary vascular remodeling. Note that, in addition to autophagy of PAECs, autophagy of PA smooth muscle cells may also represent a therapeutic target in HPH. Similar effects may be observed with other inducers of autophagy (e.g., rapamycin).
Footnotes
Previously published online: www.landesbioscience.com/journals/autophagy/article/20520