

Abstract
Objective
Chronic demyelination can result in axonopathy and is associated with human neurological conditions such as multiple sclerosis (MS) in adults and cerebral palsy in infants. In these disorders myelin regeneration is inhibited by impaired differentiation of oligodendrocyte progenitors into myelin-producing oligodendrocytes. However, regulatory factors relevant in human myelin disorders and in myelin regeneration remain poorly understood. Here we have investigated the role of the transcription factor Nuclear Factor-I A (NFIA) in oligodendrocyte progenitor differentiation during developmental and regenerative myelination
Methods
NFIA expression patterns in human neonatal hypoxic-ischemic encephalopathy (HIE) and multiple sclerosis (MS), as well as developmental expression in mice were evaluated. Functional studies during remyelination were performed using a lysolecithin model, coupled with lentiviral misexpression of NFIA. The role of NFIA during oligodendrocyte lineage development was characterized using chick and mouse models and in vitro culture of oligodendrocyte progenitors. Biochemical mechanism of NFIA function was evaluated using chromatin immunopreciptation and reporter assays.
Results
NFIA is expressed in oligodendrocyte progenitors, but not differentiated oligodendrocytes during mouse embryonic development. Examination of NFIA expression in white matter lesions of human newborns with neonatal HIE, as well active MS lesions in adults, revealed that it is similarly expressed in oligodendrocyte progenitors and not oligodendrocytes. Functional studies indicate NFIA is sufficient to suppress oligodendrocyte progenitor differentiation during adult remyelination and embryonic development through direct repression of myelin gene expression.
Interpretation
These studies suggest that NFIA participates in the control of oligodendrocyte progenitor differentiation and may contribute to the inhibition of remyelination in human myelin disorders.
INTRODUCTION
Oligodendrocytes (OL) are responsible for the assembly of myelin sheaths that surround axons and provide insulation that facilitates saltatory nerve conduction. In humans, loss of these myelin sheaths due to white matter injury (WMI) or demyelinating disorders results in decreased neuronal activity and survival 1-3. Clinically, these insults manifest themselves in neurological conditions such as cerebral palsy (CP) in infants and multiple sclerosis (MS) in adults 1, 4, 5. Myelin sheaths are generated by oligodendrocyte progenitors (OLPs) that are recruited to developing axons or demyelinated lesions, where they differentiate in the immature brain (with a peak about P15 in mouse brain) or in a process called remyelination in the adult CNS, respectively 6-8.
In MS, autoimmune attack results in loss of OL and myelin in focal areas called plaques2. One component of disease progression in MS is failed remeylination of these plaques. This idea is supported by the finding of OLPs within MS plaques 1, 3, suggesting that the lesion environment lacks trophic signals or includes inhibitory signals preventing remyelination. Inhibition of myelin regeneration leads to chronic plaques, ongoing neurological dysfunction, axonal loss and disease progression. Failure of myelination has also been proposed in WMI of the newborn brain. Immature OLPs have been observed in preterm infant WMI areas 9, 10, suggesting an inability of OLPs to differentiate into mature OL and myelinate axons. Signaling pathways implicated in the inhibition of the OLP→ OL transition and/or OL myelination include Wnt, Notch, Lingo-1, hyaluronan, and these have been proposed to contribute to human myelin disorders 11-14.
A comprehensive understanding of the regulatory factors that control OLP differentiation during development might provide new insights into the pathogenesis and progression of human myelin disorders 6, 7. During embryonic development and adult remyelination, OLPs go through a series of differentiative steps that culminate in the generation of mature, myelin-producing OLs. The molecular mechanisms that control OLP differentiation in the developing central nervous system (CNS) are well characterized and a collection of recent findings indicate that a subset of these developmental processes are re-utilized during adult remyelination 6, 7, 15. Previously, we identified Nuclear Factor-IA (NFIA) as a key transcription factor in the specification of glial cell identity and the differentiation of astrocytes in the developing central nervous system (CNS) 16. NFIA is induced in ventricular zone (VZ) populations during the onset of gliogenesis in the developing spinal cord. Subsequent analysis found that its expression is maintained in mature astrocytes and is both necessary and sufficient for the generation of astrocyte precursors and their differentiation. In addition, NFIA is also expressed during glial specification in the pMN domain and subsequently in migrating OLPs during embryogenesis 16. Despite this knowledge and the extensive characterization of its role during CNS development, its expression and function during OL lineage development have not been examined.
Our understanding of how the transcriptional processes that directly suppress myelin gene expression are dysregulated in- and contribute to- the stalled differentiative state of OLPs in WMI and MS pathology remains incomplete. In this study we focused on expression and function of NFIA during OL lineage development, human disease and remyelination. We find NFIA is expressed in OLPs and is excluded from mature, myelin-gene expressing OLs in both the embryonic and post-natal spinal cord. Examination of NFIA expression in human MS and WMI lesions revealed that it is similarly expressed in OLPs and not OLs. Consistent with its pattern of expression, functional studies revealed that NFIA suppresses OLP differentiation during remyelination and in the embryonic spinal cord via direct transcriptional repression of several myelin genes. These functional studies, taken together with our characterization in human MS and HIE lesions, suggest that a dysregulation of NFIA may contribute to the suppression of remyelination in these human disorders.
Methods
Mice and In ovo electroporation
NFIA null mice were previously described 20. Embryos were collected at various stages, fixed in 4% paraformaldehyde, and analyzed. An HA tagged NFIA cDNA was cloned into the avian retrovirus RCAS (B) vector. In ovo electroporation was performed as described 16. Embryos were harvested at E11 and processed as described above.
OLP culture
Oligosphere cultures were performed as previously described 19. See Supplemental information for culture details. For viral infection of OLPs, cells were dissociated and plated on PDL-coated coverslips at a density of 1.5×104 cells/cm2 in OPM media subsequently infected with either NFIA-FUIGW or control GFP virus for 14 hours.
ChIP and Reporter Assays
Mouse E12.5 spinal cords were dissected, dissociated, and processed for ChIP assays. The samples were pre-cleared with protein G beads and immunoprecipitated using NFIA antibody (abcam) or control IgG (Santa Cruz). The DNA was purified and PCR was preformed using region specific primers. See supplemental information for primer sequences. HEK293 or HEK293T cell lines cells were transfected with pGL3-reporter constructs and a CMV-β-galactosidase vector using Superfect transfection reagent (Qiagen). Cells were harvested and analyzed for luciferase activity; β-galactosidase was used to normalize for transfection efficiency.
Induction of demyelination with lysolecithin in mouse spinal cord and viral misexpression in remyelinating lesions
Demyelinated lesions were produced in ventrolateral spinal cord white matter of 8-10-week-old mice 11,17. Mice were sacrificed at three survival time points: 5 dpl, 10 dpl and 14 dpl (see Fig. 3a) and perfused. For viral misexpression studies, adult spinal cord white matter was injected with lysolecithin at day 0, followed by secondary focal injection of the lesioned region with either an NFIA-containing lentivirus or control lentivirus at 3dpl. For these viral experiments, mice were perfused at 10dpl to assess the effect on OLP differentiation.
Figure 3. Misexpression of NFIA suppresses OLP differentiation during remyelination.

(A) Schematic of remyelination kinetics after lysolecithin lesioning of the spinal cord. 5 days post lesioning (5dpl) represents the peak of OLP recruitment into the lesion, 10dpl the onset of OLP differentiation and 14dpl the appearance of significant new myelin. (BE) NFIA expression in OLP populations within a lesion at 5- and 10-dpl. (B) NFIA expression, (C) Nkx2.2 expression, (D-E) co-expression of NFIA and Nkx2.2. (F-L) NFIA is not expressed in mature OLs in lesions at 10- and 14-dpl. (F) NFIA expression, (G-L) NFIA is not co-expressed with PLP. Filled arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization. (J-T) Viral overexpression of NFIA represses myelin gene expression in lysolecithin lesions. (J-L) Control lesion without viral injection, (M-O) lesion infected with NFIA virus, (P-R) lesion infected with GFP-control virus. (S-T) Quantification of the number of NFIA-expressing cells (S) and the number of Olig2 cells that co-express PLP mRNA (T). In J-R, dashed lines indicate the borders of the lesion. For cell counts, four animals per group were used. Cells were counted on three or more non-adjacent sections per mouse and presented as the average ± SD. ANOVA statistical analysis was performed in S (*p=0.0123) and T (*p=0.0067)
Human multiple sclerosis tissue
Human post-mortem tissue blocks were provided by the UK Multiple Sclerosis Tissue Bank at Imperial College London. All tissues were collected following fully informed consent by the donors via a prospective donor scheme following ethical approval by the London Multicentre Research Ethics committee (MREC 02/2/39). Multiple sclerosis lesions were characterized as described previously 11, 17, using LFB to assess demyelination, SMI-31 immunohistochemistry to assess preservation of axons, and LN3 immunohistochemistry to assess inflammatory cell activity. Lesions with florid parenchymal and perivascular inflammatory cell infiltration, myelin fragmentation, and demyelination with indistinct margins were classified as active plaques.
Human developmental white matter injury tissue
All human tissue was collected in accordance with guidelines established by the University of California San Francisco Committee on Human Research (H11170-19113-07). The procurement of tissue and its pathological diagnosis as hypoxic-ischemic encephalopathy (HIE) tissue has been described in detail previously 17.
RESULTS
NFIA expression is restricted to immature stages in the oligodendrocyte lineage
To define the expression dynamics of NFIA during OL lineage development we assessed the extent of its co-expression with the OLP markers Sox10, PDGFRα, and Nkx2.2 during the E12.5-E18.5 interval in the developing spinal cord. We found that NFIA is co-expressed with these markers throughout embryonic development, with nearly all Sox10- and Nkx2.2-expressing OLPs that occupy white matter regions demonstrating co-expression with NFIA at E18.5 (Fig.1A-C, F). Next we examined the extent of NFIA expression in mature OLs. Co-staining with mature OL markers MBP and PLP at E18.5 revealed that NFIA is not expressed in mature OLs (Fig.1D-E, G-H). Examination of post-natal stages indicates a similar expression pattern, where NFIA is highly co-expressed with OLP markers in white matter (Nkx2.2 and PDGFRα), but not mature OLs (CC1, MBP, PLP) (Fig.1I-Q). Furthermore, these expression patterns are maintained in the adult spinal cord (Fig. S1). These data indicate that NFIA is expressed in OLPs, but not mature OLs in the developing, post-natal, and adult mouse spinal cord.
Figure 1. NFIA expression during oligodendrocyte development.
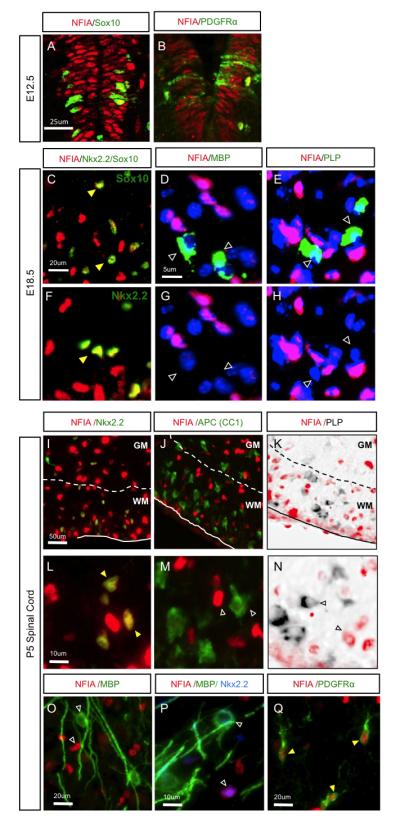
(A-B) NFIA is co-expressed with Sox10 and PDGFRa in the spinal cord at E12.5. (C-H) NFIA is co-expressed with OLP markers Sox10 and Nkx2.2 at E18.5 (C,F), but not markers of mature OLs MBP (D,G) or PLP mRNA (E,H). Images in C and F are from distinct areas and samples; Image sets in (D,G) and (E,H) are from the same image. (I Q) Analysis of NFIA expression in the post-natal spinal cord. NFIA is co-expressed with OLP marker Nkx2.2 (I,L,P), but not differentiation marker APC (J,M) or mature markers PLP (K,N) and MBP (O,P). In I-K, the dashed line denotes the border between the white matter (WM) and the grey matter (GM). Filled-yellow arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization.
NFIA expression in OL lineage is conserved in human demyelinating disorders
Next we investigated NFIA expression in the OL lineage in human WMI, by first examining its expression in subcortical white matter of human neonatal brain damaged by hypoxic-ischemic encephalopathy (HIE). HIE was diagnosed as previously described based on both clinical and neuropathological correlations 17. All HIE cases showed evidence of diffuse subcortical WMI, including astrogliosis and macrophage infiltration, along with ischemic neurons and variable degrees of neuronal loss. As shown (Fig. 2A-H), NFIA is expressed in areas of gliotic sub-cortical white matter where it demonstrates a conserved pattern of expression within the OL lineage. NFIA expression segregated from mature OL marker PLP mRNA, but was expressed in a proportion of the OLP expressing Nkx2.2 mRNA. Next we assessed areas of active MS lesions for NFIA expression. MS lesions were characterized as previously described 11 using LFB to assess demyelination, SMI-31 immunohistochemistry to assess preservation of axons, and LN3 immunohistochemistry to assess inflammatory activity. NFIA is expressed within the active edges of MS lesions in Nkx2.2 mRNA-expressing OLP (Fig.2I-L). These findings suggest that the developmental patterns of NFIA expression in the OLP lineage are conserved in various forms of human white matter injury.
Figure 2. Expression of NFIA in oligodendrocyte progenitors in human HIE and MS.
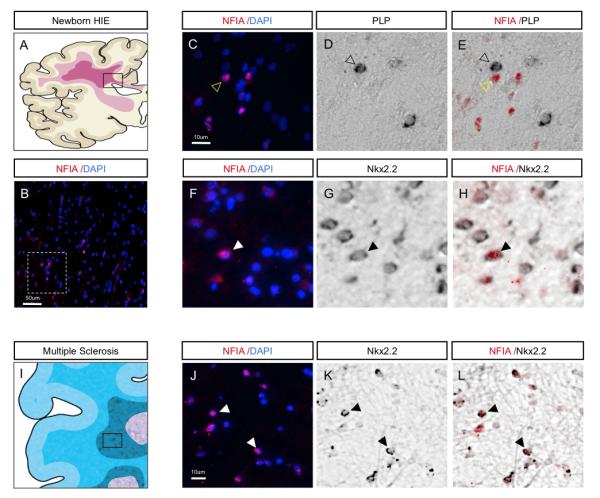
(A) Schematic illustrating white matter damage in pediatric HIE. Boxed area represents region of gliotic sub-cortical white matter and origins of the tissues used for analysis. (B) Expression of NFIA in human HIE; dashed box is region shown at higher magnification in C. (C-H) NFIA is not co-expressed with PLP-expressing cells (C-E), but is co-expressed with Nkx2.2-expressing cells (F-H), indicating expression in OLPs within areas of affected white matter. (I) Schematic demonstrating affected areas of white matter in human MS. Boxed area represents active lesion site and region used for analysis. (J-L) NFIA is co-expressed with Nkx2.2 within active MS lesions, indicating expression in OLPs. Filled arrowheads indicate colocalization. Unfilled arrowheads indicate lack of colocalization.
NFIA suppresses OLP differentiation during remyelination
That NFIA is expressed in OLP populations in human myelin disorders raised the possibility that it may contribute to impaired OLP differentiation and remyelination manifest in these conditions. Remyelination can be studied after adult murine lysolecithin injury, which removes OLPs and OLs from white matter while largely sparing axons. Such gliotoxic injury in spinal cord white matter has been extensively characterized 11, 18 and is repaired in a robust and highly reproducible manner in wild-type animals allowing for precise assessment of remyelination kinetics (Fig. 3A). Therefore, we first examined whether the expression pattern of NFIA in the OL lineage during murine remyelination is a recapitulation of its developmental and human expression. We found that NFIA is expressed in Nkx2.2-positive OLPs at the peak of OLP recruitment at 5 days post-lesion (dpl) and continues to be expressed in the OLP portion of the lineage at 10dpl (Fig. 3A-E). In contrast, NFIA is excluded from differentiating PLP mRNA-expression-positive cells at 10dpl, and mature OLs at 14dpl (Fig. 3F-I).
Because NFIA is expressed in OLPs and not mature OLs, we hypothesized that it functions to suppress OLP differentiation during remyelination. To address this possibility, we injected adult spinal cord white matter with lysolecithin at day 0, followed by secondary focal injection of the lesioned region with either an NFIA-containing lentivirus or control virus at 3dpl to catch the OLP as they are recruited into the lesion. We then assessed OL differentiation at 10dpl, when significant numbers of OLP have begun to differentiate in this paradigm (Fig.3A). Injection of NFIA-lentivirus led to a 50% increase (p<0.03) in the number of cells expressing NFIA within the area of the lesion compared to non-infected or control viral-infected lesions (Fig. 3J,M,P,S). In addition, OL differentiation was significantly impaired by the over-expression of NFIA in lesions, resulting in a 50% (p<0.02) reduction in the proportion of Olig2 cells expressing the mature OL marker PLP mRNA compared to control lesions (Fig. 3L,O,R,T). Moreover, ectopic NFIA expression in the lesion did not alter the total number of Olig2-expressing cells, indicating that the reduction in PLP-positive cells is not due to a loss of OLPs (Fig. 3N,Q). Additionally, the astrocyte response (GFAP) and inflammatory response (CD68) are unaltered by the NFIA lentiviral injection (Fig. S2). Together, our data indicate that NFIA is expressed in OLPs during experimental remyelination in mice and that its misexpression can suppress differentiation.
NFIA suppresses OLP differentiation during development
As a first step towards understanding the mechanism by which NFIA suppresses OLP differentiation, we determined whether NFIA suppresses OLP differentiation during normal development by misexpression in the embryonic chick spinal cord. Chick spinal cord was electroporated at E2, with RCAS(B) retroviral DNA containing NFIA and embryos were harvested at E11. Our analysis revealed that mis-expression of NFIA led to reduced numbers of MBP-, PLP-, and MAG-positive OLs in vivo (Fig. 4A-D, M), with no observable effect on the number of PDGFRα-expressing OLPs (Fig. 4E; Fig. S3). Additionally, numbers of GLAST-expressing astrocyte precursors appeared unaffected (Fig. 4F,M), suggesting that the reduction of mature OLs reflects suppressed OLP maturation rather than a conversion to the astrocyte lineage. To confirm that NFIA functions in OLP populations to suppress their differentiation we made use of in vitro OLP culture and mitogen withdrawal provoked differentiation 19. Misexpression of NFIA in OLP culture resulted in a significant decrease in the number of MBP-, PLP-, and MAG-expressing cells, without effecting expression of OLP markers (Fig 4G-L, N; Fig. S3). These data indicate that NFIA is sufficient to repress myelin gene expression during spinal cord development.
Figure 4. NFIA suppresses OLP differentiation during embryogenesis.

(A-F) Overexpression of NFIA in the embryonic chick spinal cord at E11. (A) Ectopic expression of RCAS/HA-NFIA, detected with α-HA. (B-D) Analysis of myelin gene expression in the presence of ectopic NFIA, arrows denote reduced expression. (E-F) Expression of astrocyte prescursor marker GLAST (F), and OLP marker PDGFRα E is unaffected by ectopic NFIA; quantification of MBP and GLAST (M). (G-L) Overexpression of NFIA in OLP culture suppresses myelin gene expression. (G,I,K) GFP-control virus, (H,J,L) NFIA virus; quantification of myelin gene expression (N). In M and N, *p<.005. (O-T) Analysis of OL differentiation in NFIA +/− and NFIA−/− embryos at E16.5; (CC) quantification of the number of MBP-expressing cells. (U-BB) In vitro analysis of OL differentiation using NFIA +/− (U-X) and NFIA−/−(Y-BB) OLP cultures. (DD) Quantification of number of cells expressing myelin genes. For A-F, “Ep” denotes the electroporated side and “Con” denotes the control side of the embryo. Quantification in M is from 3 embryos, 10 sections per embryo. Analysis of NFIA+/− and −/− embryos in O-T is derived from three litters for each timepoint and is comprised of at least three embryos per genotype. In vitro analysis was performed in triplicate on three sets of embryos derived from two litters. Quantification in N and DD is derived from experiments performed three times, in triplicate. In CC, **p=.017 and in DD, ***p<.003. All error bars indicate SD.
We next investigated OL lineage differentiation in the embryonic spinal cords of NFIA-null mice 20. As indicated in Figure 4O-T, E16.5 NFIA knockout embryos demonstrate an increase in the number of cells expressing the mature OL markers MBP and PLP; while, at E18.5 expression of MBP and PLP is comparable (Fig. 4CC; Fig. S3). This premature induction of MBP and PLP expression was observed without change in the number of OLPs expressing PDGFRα, indicating NFIA is necessary for the proper timing of OL maturation (Fig. 4Q-T; Fig. S3). Analysis of post-natal myelination in NFIA-null mice is precluded by lethality at birth 20. To confirm NFIA function in OLP populations we generated OLP cultures from NFIA heterozygous and null embryos. Their subsequent differentiation revealed an increase in the number of MBP-, MAG-, PLP-expressing cells in NFIA-null cultures three days after mitogen removal, while expression of OLP markers was unaffected (Fig. 4U-BB; Fig. S3). Significant differences in MAG-, and PLP-expression were no longer detected after seven days of differentiation, while increased MBP-expression in the NFIA-null cultures was maintained (Fig. 4DD). Collectively these observations show that NFIA function is required for the normal timing of OLP maturation into MBP-, MAG-, PLP-expressing OLs.
NFIA represses myelin gene expression via direct mechanisms
The forgoing regenerative and developmental studies indicate that NFIA can suppress OLP differentiation into mature OLs, therefore we sought to delineate the molecular mechanism by which it functions. Previously, conserved NFI binding sites had been identified in the promoters of the myelin genes MBP, PLP, and MAG 21-23, leading us to hypothesize that NFIA suppresses OLP differentiation through direct repression of these genes (Fig.5A-C). To determine whether NFIA associates with these identified binding sites, we performed chromatin immunoprecipitation assays (ChIP) on E12.5 mouse spinal cords (see methods). As shown in Figure 5D-F, we found that endogenous NFIA associates with regions of the MBP, PLP, and MAG promoters containing its binding site during spinal cord development at a stage when NFIA is expressed in OLPs. Next we cloned the promoter regions of these genes that encompass the NFI binding site and performed luciferase reporter assays. We found that NFIA represses activation of the MBP-, PLP-, and MAG-luciferase reporter constructs (Fig. 5G-I). Together these data indicate that MBP, PLP, and MAG are direct transcriptional targets of NFIA repression, suggesting that it suppresses OLP differentiation through direct repression of myelin gene expression.
Figure 5. NFIA associates with myelin gene promoters and represses their activity.
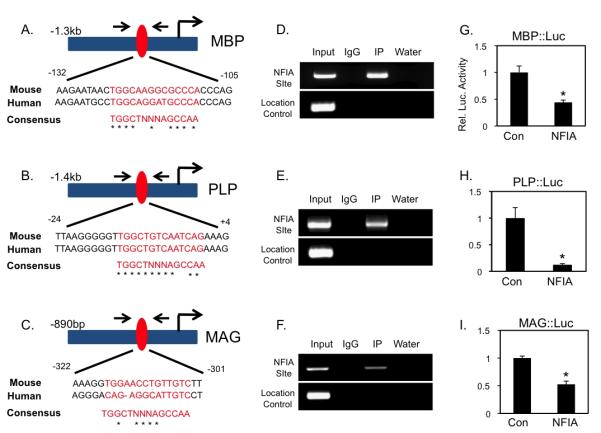
(A-C) Mouse/human alignment of consensus NFI-binding sites and schematic of MBP, PLP, and MAG promoters. (D-F) ChIP assay from E12.5 mouse spinal cord; arrows denote ChIP primers encompassing conserved NFI-site (red oval). (G-I) Reporter assays demonstrating NFIA repression of MBP, PLP, and MAG promoter activity. In G-I, *p<.01. All error bars indicate SD.
DISCUSSION
Our study describes, for the first time, the role of NFIA during OL lineage development and remyelination with implications for human WMI. We found that NFIA is expressed in OLPs, but not myelin gene-expressing OLs in the developing and post-natal spinal cord and that this pattern of expression is conserved during remyelination. Subsequent analysis of human MS and HIE, found that NFIA is expressed in OLP populations within demyelinated lesions. Consistent with its exclusion from mature OLs, functional studies revealed that NFIA suppresses OLP differentiation during development and adult remyelination via direct repression of myelin gene expression. These observations, coupled with our functional studies, suggest that a dysregulation of NFIA may contribute to the suppression of remyelination in these white matter disorders.
Analysis of NFIA expression and function suggest that its role in OLPs during remyelination is a recapitulation of development and point to a model where its downregulation stimulates OLP differentiation. One consideration when interpreting these studies is that the cellular constituency of a spinal cord lesion is comprised of numerous cell types, including, astrocytes, neurons, macrophages, and OLPs. The lentiviral injections utilized in our studies have the potential to infect all these cell types, therefore we cannot formally exclude the possibility that the effects on OLP differentiation in lesions are secondary to NFIA gene manipulation in these other cell populations, namely astrocytes. However there was no change in the astrocyte or macrophage response in these virally injected lesions (Fig. S2) suggesting that the effects of NFIA misexpression on OLP differentiation are not due to a change in the constituency of these populations. In addition, manipulation of NFIA expression in OLP cultures paralleled our in vivo developmental studies and is congruent with our misexpression studies during remyelination, suggesting that its effects on OLP differentiation in these in vivo systems are most likely the result of its function in OLP populations and not secondary to its role in astrocyte development.
Our studies demonstrate that NFIA contributes to the timing of OLP differentiation via direct repression of myelin gene expression. Indeed, delayed remyelination kinetics have been linked to chronic demyelination, which is consistent with recent studies showing that OLs initiate new myelin segments only during a temporally restricted developmental window 24, 25. However it remains unclear whether dysregulated NFIA expression is sufficient to account for stalled OLP maturation in human myelin disorders. Given the abnormal and diverse environment of a demyelinated axon tract and the multitude of regulatory factors present, it is likely that NFIA operates in conjunction with many of the signaling pathways implicated in the suppression of remyelination. The pathways that control the development of OLPs are often re-utilized in the adult during remyelination; among the key developmental pathways also implicated in remyelination are Wnt/Tcf4, LINGO, and Notch, each of which functions to suppress remyelination 11-13. That the effect of these pathways on remyelination is similar to that of NFIA, suggests that it could serve as a transcriptional effector of the global programs utilized by these signaling networks to suppress OLP differentiation and subsequent myelination. While formal regulatory relationships between NFIA and Wnt signaling or LINGO have yet to be established, Notch-ICD has been linked to the regulation of NFIA expression in cortical cultures 26.
A host of transcription factors have been linked to regulation of myelin gene expression, though relatively few have been implicated in direct repression 15. Hes5 is among the transcription factors implicated in myelin gene repression and associates with Sox10 to suppress its induction of MBP 27. Recently we found that NFIA can associate with Sox-family members, thus it is possible that NFIA mediates myelin gene repression through interactions with Hes5 and/or Sox10 (Kang, et al. submitted). Other key transcription factors implicated in myelin gene repression are Id2/4, which suppress OLP differentiation through direct repression of myelin genes and by interacting with Olig1/2 and preventing hetero-dimerization with E-box proteins 28-30. Interestingly, NFIA has been shown to associate with Olig2, thus its possible that it also associates with Id-genes and participates in Id2/4-mediated suppression of Olig1/2 gene function 16. Dissecting the functional inter-relationships between NFIA and other repressors of myelin gene expression will be critical in constructing a more comprehensive transcriptional framework of myelin gene repression during development and disease.
In both multiple sclerosis in the adult and white matter injury in the human newborn brain following HIE, inhibition of myelination after injury frequently contributes to ongoing neurological dysfunction. There are currently no therapies available targeted at promoting the repair of human white matter injury and this will be of paramount importance in the future to prevent progressive decline in these patients. Evidence suggests remyelination (adult MS patients) or developmental myelination (neonates) fails as a result of delayed and/or blocked OL differentiation. Thus a more complete understanding of the regulatory factors controlling this process and how they might become dysregulated in disease is critical to understanding (re)myelination failure in these human disorders. We have demonstrated a critical role for NFIA in controlling the timing of OLP differentiation via direct suppression of myelin genes, and a recapitulation of its expression in OLP in human white matter injury. A greater understanding of how NFIA is regulated in OL lineage and how it may become dysregulated in human white matter injury will lead to the identification of possible therapeutic targets to promote repair in these debilitating human disorders.
Supplementary Material
Acknowledgements
The NFIA mouse line was a gift from Richard Gronostajski. This work was supported by grants from the National Multiple Sclerosis Society (to DHR), the Gillson Longenbaugh Foundation (to BD), and the National Institutes of Health (R01 NS071153 to BD, 5-T32HL092332-08 to SG, and R01 NS040511 and R01 NS059893 to DHR). DHR is a Howard Hughes Medical Institute Investigator.
References
- 1.Chang A, Tourtellotte WW, Rudickm R, et al. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2000;346:165–173. doi: 10.1056/NEJMoa010994. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple Sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Kuhlmann T, Miron V, Cuo Q, et al. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain. 2008;131:1749–1758. doi: 10.1093/brain/awn096. [DOI] [PubMed] [Google Scholar]
- 4.Woodward LJ, Anderson PJ, Austin NC, et al. Neonatal MRI to Predict Neurodevelopmental Outcomes in Preterm Infants. NEJM. 2006;355:685–694. doi: 10.1056/NEJMoa053792. [DOI] [PubMed] [Google Scholar]
- 5.Khwaja O, Volpe JJ. Pathogenesis of cerebral white matter injury of permanturity. Arch. Dis. Child. Fetal Neonatal Ed. 2008;93:F153–F161. doi: 10.1136/adc.2006.108837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fancy SPJ, Chan JR, Barabzini SE, et al. Myelin Regeneration: A Recapitulation of Development? Annual Review of Neuroscience. 2011;34:21–43. doi: 10.1146/annurev-neuro-061010-113629. [DOI] [PubMed] [Google Scholar]
- 7.Miller R, Mi S. Dissecting myelination. Nat Neurosci. 2007;10:1351–1354. doi: 10.1038/nn1995. [DOI] [PubMed] [Google Scholar]
- 8.Emery B. Regulation of oligodendrocyte differentiation and myelination. Science. 2010;330:779–882. doi: 10.1126/science.1190927. [DOI] [PubMed] [Google Scholar]
- 9.Billiards SS, Haynes RL, Folkerth RD, et al. Myelin Abnormalities without Oligodendrocyte Loss in Periventricular Leukomalacia. Brain Pathology. 2008;18:153–163. doi: 10.1111/j.1750-3639.2007.00107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buser BS, Maire J, Riddle A, et al. Myelination failure in human perinatal white matter injury: Arrested pre-oligodendrocyye maturation contributes to myelination failure in premature infants. Annals of Neurology. 2011 doi: 10.1002/ana.22627. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fancy S, Baranzini SE, Zhao C, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mi S, Miller RH, Lee X, Scott ML, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8:745–751. doi: 10.1038/nn1460. [DOI] [PubMed] [Google Scholar]
- 13.John G, Shankar SL, Shafit-Zagardo B, et al. Multiple Sclerosis: Re-expression of a developmental pathway that restricts oligodendrocyte maturation. Nat Med. 2002;8:1115–1121. doi: 10.1038/nm781. [DOI] [PubMed] [Google Scholar]
- 14.Sloane JA, Batt C, Ma Y, et al. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. PNAS. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolay DJ, Doucette JR, Nazarali AJ. Transcriptional control of oligodendrogenesis. Glia. 2007;55:1287–1299. doi: 10.1002/glia.20540. [DOI] [PubMed] [Google Scholar]
- 16.Deneen B, Ho R, Lukaszewicz A, et al. The Transcription Factor NFIA Controls the Onset of Gliogenesis in the Developing Spinal Cord. Neuron. 2006;52:953–968. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 17.Fancy SPJ, Harrington EP, Yuen TJ, et al. Axin2 as regulatory and therapeutic target in newborn brain injury and remyelination. Nature Neuroscience. 2011;14:1009–1016. doi: 10.1038/nn.2855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnett HA, Fancy SPJ, Alberta JA, et al. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- 19.Pedraza CE, Monk R, Lei J, et al. Production, characterization, and efficient transfection of highly pure oligodendrocyte precursor cultures from mouse embryonic neural progenitors. Glia. 2008;56:1339–1352. doi: 10.1002/glia.20702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.das Neves L, Duchala C, Tolentino-Silva F, et al. Disruption of the murine nuclear factor I-A gene (Nfia) results in perinatal lethality, hydrocephalus, and agenesis of the corpus callosum. PNAS. 1999;96:11946–11951. doi: 10.1073/pnas.96.21.11946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Laszkiewicz I, Grubinska B, Wiggins RC, et al. Structural characterization of myelin-associated glycoprotein gene core promoter. Journal of Neuroscience Research. 1997;50:928–936. doi: 10.1002/(SICI)1097-4547(19971215)50:6<928::AID-JNR3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 22.Wight PA, Duchala C, Readhead C, et al. A myelin proteolipid protein-LacZ fusion protein is developmentally regulated and targeted to the myelin membrane in transgenic mice. J Cell Bio. 1993;123:443–454. doi: 10.1083/jcb.123.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H, Lu Y, Smith HK, et al. Olig1 and Sox10 Interact Synergistically to Drive Myelin Basic Protein Transcription in Oligodendrocytes. J. Neurosci. 2007;27:14375–14382. doi: 10.1523/JNEUROSCI.4456-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Watkins TA, Emery B, Mulinyawe S, et al. Distinct stages of myelination regulated by gamma-secretase and astrocytes in a rapidly myelinating CNS coculture system. Neuron. 2008;60:555–569. doi: 10.1016/j.neuron.2008.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Franklin RJM, ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neuroscience. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- 26.Namihira M, Kohyama J, Semi K, et al. Committed Neuronal Precursors Confer Astrocytic Potential on Residual Neural Precursor Cells. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- 27.Liu A LJ, Marin-Husstege M, Kageyama R, et al. A molecular insight of Hes5-dependent inhibition of myelin gene expression: old partners and new players. EMBO. 2006;25:4833–4842. doi: 10.1038/sj.emboj.7601352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kondo T, Raff M. The Id4 HLH protein and the timing of oligodendrocyte differentiation. EMBO. 2000;19:1998–2007. doi: 10.1093/emboj/19.9.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Marin-Husstege M, He Y, Li J, et al. Multiple roles of Id4 in developmental myelination: predicted outcomes and unexpected findings. Glia. 2006;54:285–296. doi: 10.1002/glia.20385. [DOI] [PubMed] [Google Scholar]
- 30.Wang S, Sdrulla A, Johnson JE, et al. A role for the helix-loop-helix protein Id2 in the control of oligodendrocyte development. Neuron. 2000;39:603–614. doi: 10.1016/s0896-6273(01)00237-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.