

Abstract
The purpose of this study was to investigate the in vivo distribution and antitumor activity of irinotecan (camptothecin (CPT)-11)-loaded folate-targeted liposome (F-Lip) in tumor-bearing mice following i.v. administration. Folate–poly(ethylene glycol)–distearoylphosphatidylcholine (FA–PEG–DSPE) was synthesized by amide reaction of DSPE–PEG–NH2 and FA. F-Lip modified by FA–PEG–DSPE was prepared by an ammonium sulfate gradient. The mean particle size and entrapment efficiency of F-Lip with negative charge were 197.8 ± 4.58 nm and 91.39 ± 2.34 %, respectively. The distributions of CPT-11 and SN-38 in the tumor after i.v. administration of F-Lip, CPT-11-loaded liposomes (C-Lip), and CPT-11 injection (C-Inj) were far greater with the F-Lip group in comparison to the C-Inj and C-Lip, which might contribute to folate-meditated targeting uptake by the folate receptor on the surface of the tumor cells. The uptake of CPT-11 in the liver and rectum for two liposome groups were all markedly increased as compared to the C-Inj. Moreover, F-Lip exhibited a dose-dependent tumor growth inhibition and superior anticancer activity to C-Lip and C-Inj after i.v. administration. It also showed no significant body weight loss and much lower toxicity on the center immune organs. Therefore, F-Lip may be presented as potential candidates for tumor targeting drug delivery.
KEY WORDS: cancer targeting, CPT-11, folate, liposomes, SN-38
INTRODUCTION
Irinotecan (camptothecin (CPT)-11), 7-ethyl-10-[4-(1-piperidino)-1-piperidino]-carbonyloxy-camptothecin, is a water-soluble camptothecin derivative. It is currently the first-line drug of choice for the treatment of the cancers including colorectal cancer, refractory cervical cancer, and other gynecological cancers (1,2). CPT-11 is a pro-drug, converted in vivo to its active metabolite, SN-38 (7-ethyl-10-hydroxycamptothecin). SN-38 exhibits up to 100–1,000-fold more cytotoxic activity than CPT-11 although the plasma concentrations of SN-38 are usually one to two orders of magnitude lower than those of CPT-11 (3). Therefore, it is necessary to monitor the concentrations of SN-38 in plasma and tissues to actually evaluate antitumor effect of CPT-11.
Some drawbacks including lactone ring instability at physiological pH and severe side toxicity have prevented the clinical application of CPT-11 in cancer therapy. Moreover, although CPT-11 is converted to SN-38 in the liver and tumor, the metabolic conversion rate is less than 10 % (4,5), which might be related with the carboxyesterase (CEs) activity. One of the reasons is that the saturation action of the CEs by overdose drugs released from the common preparation of CPT-11 might inhibit the further conversion of CPT-11 (6).
Some reports have studied the liposome formulation of camptothecin (CPT) (6) and its derivatives including 9-nitro-camptothecin (7) and CPT-11(8,9). It has been shown that the liposome can enhance the drug solubility, reduce toxicity associated with free antitumor drugs, and improve stability of the drugs. The method of CPT-11 encapsulation by liposome can effect on drug retention following in vivo administration and that better drug retention was associated with significant improvement in therapeutic efficacy (9). Sadzuka et al. (10) reported that the tumor accumulation of CPT-11 and SN-38 and the antitumor activity of CPT-11 were increased by poly(ethylene glycol) (PEG)-modified liposome. Furthermore, the intestinal disorder, a side effect of CPT-11, was decreased by liposomalization. However, insufficient uptake at tumor sites will decrease the therapeutic benefit, and nonspecific association with healthy tissues can lead to toxic side effects (8). Some targeting ligands such as transferring (11,12), folate (FA; or folic acid) (13), and arginine–glycine–aspartic acid peptide (14) were widely used in the tumor targeting drug delivery systems to improve the uptake of the drugs in the tumor.
Folate, a water-soluble vitamin, can induce receptor-mediated endocytosis. The FA receptors (FR) have served as an attractive target for tumor-specific drug delivery since they are overexpressed on the surface of many cancer cells, but only minimally distributed in normal tissues. Particularly interesting is the observation that FR-targeted liposomal delivery can reverse multidrug resistance to doxorubicin (15). FA–PEG–distearoylphosphatidylcholine (DSPE) synthesized by amide reaction of FA–PEG–amine and DSPE was used for a liposome formulation of paclitaxel targeting the FR. Compared to the non-targeted liposome, FR-targeted liposome containing paclitaxel showed 3.8-fold greater cytotoxicity activity with longer terminal half-life (16). Han et al. (8) described that polymer micelles prepared with FA–PEG–DSPE and mPEG–DSPE had a higher antitumor activity than both mPEG–DSPE polymer micelle and free drugs. The best molar weight of FA–PEG–DSPE and mPEG–DSPE was 1:100.
In this study, a CPT-11-loaded folate-targeted liposome (F-Lip) was prepared with FA–PEG–DSPE to increase the targeting efficiency of CPT-11 to the tumor as well as higher stability and lower toxicity side effect. The plasma elimination and biodistribution of CPT-11 and its metabolite SN-38 after i.v. administration was investigated. Furthermore, the therapeutic efficacy of F-Lip was evaluated in S-180 tumor-bearing mice.
MATERIALS AND METHODS
Materials
Irinotecan (CPT-11, purity 99.5 %; Lot: 071203) and SN-38 (purity 99.2 %; Lot: 071128) were purchased from Tianranyuan Co. (Chengdou, China). CPT-11 injection (40 mg) was obtained from HengRui Co (Lianyungang, China). Folate acid was from Wuhan Yuancheng Technology Development Co. Ltd. (Wuhan, China). The n-hydroxy-succinamide (NHS) was purchased from Qiyun Biochemistry Co Ltd (Guangzhou, China), and dicyclohexylcarbodiimide (DCC) was purchased from Yanchang Biochemistry Co Ltd (Shanghai, China). Amino–polyethylene glycol (Amino–PEG; Mr ~ 3,350) was purchased from Sigma. Di-tert-butyl dicarbonate (Boc2O) was purchased from Jinxiang Chemical Factory (Danyang, China). Soybean phosphatidylcholine (S100) and DSPE were obtained from Lipoid GmbH (Ludwigshafen, Germany). Cholesterol was purchased from Huixing Biochemistry Co Ltd (Shanghai, China). All other materials were of analytical grade and obtained from commercial company.
Synthesis of FA–PEG–DSPE
DSPE–PEG–NH2 was synthesized as described in a previous report (17). The products were determined by 1H NMR (Avace AV-500, Bruker, Germany). FA–PEG–DSPE conjugate was synthesized by the modified method (18). The synthesis scheme was shown in Fig. 1. Briefly, folate was activated with DCC and NHS at a 1:1:2 molar ratios in 40 mL DMSO for 18 h in dark place and filtered to remove the DCU. DSPE–PEG–NH2 was added to the resulting solution with activated folate. After staying overnight in dark place, the reactants were isolation in a gel column (Sephadex G-50) eluted by 0.1 M NaHCO3 and 0.05 M acetic acid solution, respectively, to remove DMSO and unreacted folate. The resulting solution was then lyophilized and stored at 4°C until use. The product was a yellow dry powder. The composition of the prepared FA–PEG–DSPE conjugate was analyzed by 1H NMR (Avace300, Bruker, Germany).
Fig. 1.

Synthetic scheme of FA–PEG–DSPE
Preparation of CPT-11-Loaded Folate-Targeted Liposome
F-Lip was prepared by an ammonium sulfate gradient according to the following procedures (19). Briefly, lipid mixture (soybean phosphatidylcholine/cholesterol/sodium deoxycholate = 10:1:0.125, mass ratio) was dissolved in dehydrated alcohol and dried under vacuum at 50°C. Lipid film was hydrated by adding 10 mL ammonium sulfate solution (0.1 M) and then vortex extensively at 50°C for 0.5 h. The lipid suspension was extruded at least two times through a filter (50 mm, Minipore) applied with polycarbonate membranes (Whatman 0.2 μm) to obtain desirable size distribution. The FA–PEG–DSPE was added and incubated at 60°C for 1.5 h. The resulting liposome suspension was dialyzed twice by dialysis bag (10,000 MWCO) in 0.9 % sodium chloride to remove uncapsulated ammonium sulfate. Then CPT-11 was dissolved in citric acid of 10.4 mg/mL and added to the above suspension. The F-Lip suspension was obtained by incubating at 45°C for 1 h after adjusting pH to 7.0 using 1 M NaOH solution. CPT-11-loaded liposomes (C-Lip) were prepared by the same procedure without FA–PEG–DSPE. Entrapment efficiency (percent) of the liposome was performed by ultra-filtration method (19). Briefly, 1.0 mL liposome suspensions was spiked into dialysis tube (Whatman, MW 100,000) and then ultracentrifuged at 40,000 rpm for 30 min to remove untrapped CPT-11. The concentrations of free CPT-11 in the filtrate (Cuntrapped) and CPT-11 in the liposomes (Ctotal) were measured by high-performance liquid chromatography (HPLC). The entrapment efficiency was calculated as follows:
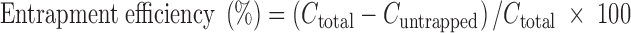 |
Characterization of the F-Lip
The particle size and zeta potential of the liposomes was measured by photocorrelation spectrometer (Zetasizer 3000HS, Malvern Instruments Corp., UK) following dilution with double-distilled water. The shape and surface morphology of the liposomes were observed by using a Hitachi H-700 transmission electron microscope (TEM). Negative staining with 1 % phosphotungstic acid solution was performed to enhance image quality.
Stability of CPT-11 in Plasma
A 0.1 mL of the sample solution (1 mg/mL of CPT-11) was added to 1 mL rat plasma and incubated for 2 h. Then the CPT-11 was extracted from plasma by adding the 100 μL of phosphate buffered saline (PBS; pH 1.3), 50 μL aliquot of camptothecin (IS) working solution (2.0 μg/mL in acetonitrile), 100 μL of methanol, and 400 μL of acetonitrile. After centrifugation, the supernatant was analyzed by previously described HPLC method (20).
Determination of CPT-11 and SN-38 in Plasma and Tissues by HPLC
A HPLC method was developed and validated for simultaneous determination of CPT-11 and SN-38 in plasma and tissues of the mice. A Shimadzu RF-5301 HPLC system consisted of a pump (LC-10ATVP, Shimadzu, Japan), a Fluori-Monitor RF-10Axl fluorescence detector (Shimadzu, Japan). A stainless steel analytical column packed with Hypersil ODS material (5 μm, 250 × 4.6 mm Diamaonsil™) was used for chromatographic separation and was maintained at a temperature of 25°C. The mobile phase was composed of acetonitrile–50 mM phosphate buffered saline (containing 0.2 % sodium heptanesulfonate with the pH adjusted to 4.0 with phosphoric acid) (30:70, v/v), and it was degassed by ultrasonication and filtered through a 0.22-μm cellulosic acetate filter (Millipore). The mobile phase was delivered at a flow rate of 1.0 ml/min, and the column effluent was monitored at an excitation wavelength of 380 nm and an emission wavelength of 540 nm.
The standard curves of peak area ratio as a function of CPT-11 or SN-38 concentration over an investigated range were linear with regression coefficients (r) of >0.998 for plasma and tissues. The extraction and method recoveries were greater than 80 %. The within-day and between-day relative standard deviations were less than 12 %.
In Vivo Distribution Studies in Tumor-Bearing Mice
S180 cells were collected from the intraperitoneal cavity of S180-bearing ICR mice. The mice (6 weeks old) were injected subcutaneously in the right armpit with S180 cells suspensions (2 × 107 cell/mL). After 4 days, the S180-bearing mice were randomly divided into three groups. CPT-11 injection (control group), C-Lip, and F-Lip were, respectively, administered by tail vein injection to the mice at a dose of 20 mg/kg. At 0.5, 1, 4, and 8 h after administration, the blood samples were collected from the ocular artery. The mice were then sacrificed, and the tumor, liver, spleen, lung, heart, rectum, and kidney were carefully excised. Each tissue was quickly rinsed with saline and blotted with filter paper. After weighing, the tissue samples were homogenized with the saline. Measurements were made using five mice at each time point.
Blood samples were anticoagulated with heparin and centrifuged at 5,000 rpm for 10 min to obtain plasma. A 100-μL volume of plasma was immediately transferred into a 1.5-mL centrifuge tube with 10 μL sodium dodecyl sulfate solution of 0.4 g/mL and then homogenized rapidly by vortex mixing. A 100 μL of PBS (pH 1.3) was added and vortex-mixed. Next 50-μL aliquot of IS working solution (2.0 μg/mL in acetonitrile), 100 μL of methanol, and 400 μL of acetonitrile were added into the resulting plasma samples. The mixtures were vortex-mixed for 3 min and centrifuged at 16,000 rpm for 3 min. For 0.5-mL tissues, homogenates were extracted by similar procedures. An aliquot of 50 μL was injected into LC system for analysis. All experiments on animals were carried out in strict accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals.
In Vivo Antitumor Activity
The in vivo anticancer activity was evaluated against S180 solid tumors in mice. The S180-bearing mice were randomly divided into ten groups. The negative control group was treated with saline. CPT-11 injection (10, 20, or 40 mg/kg dose), C-Lip (5, 10, or 20 mg/kg dose), and F-Lip (5, 10, or 20 mg/kg dose) were, respectively, administrated by tail vein injection to the mice. The administration was performed at 0, 3, 5, and 7 days, respectively. The body weights were measured every 2 days. At 24 h after the last administration, the mice were then sacrificed after weighting, and the tumor, thymus gland, and spleen were carefully excised and weighted.
Data and Statistical Analysis
Pharmacokinetic parameters in plasma and tissues were calculated using a statistical moment algorithm according to measured values for infinite time (21). The statistical differences among group means were assessed using the two-way unweighted means analysis of variance test, and a value of p < 0.05 was considered statistically significance.
RESULTS AND DISCUSSION
Synthesis and Characterization of FA–PEG–DSPE
The composition of the product was analyzed by 1H-NMR (Fig. 2). The proton peaks of PEG (–CH2CH2O–) from the products appeared at 3.3–3.6 ppm, and the new amide linkage between FA and DSPE–PEG–NH2 was observed at 8.04 ppm. Besides, the products had the characteristic peaks of arene groups of FA at 6.6–8.7 ppm (22) and a series of peaks of DSPE at 0.85 ppm (–CH3–), at 1.19 ppm (–CH2–), at 2.28 ppm (–CH2C=O), at 1.51 ppm (–CH2CH2C=O), and at 4.19 ppm (CH2–O–C=O).
Fig. 2.

1H NMR spectra of HO–PEG–NH2 (a), amino–PEG–DSPE (b), and FA–PEG–DSPE (c) in DMSO
Characterization of F-Lip
The CPT-11 liposomes were prepared by pH gradient method. As shown in Table I, the mean particle size and entrapment efficiency of F-Lip were 197.8 ± 4.6 nm and 91.4 ± 2.3 %, respectively, which were slightly decreased as compared to C-Lip with no significant difference. Moreover, F-Lip and C-Lip carried negative charge. The representative TEM images are shown in Fig. 3. The results showed that the F-Lip had almost spherical in shape. Only less than 2.5 % leak amount of CPT-11 in F-Lip at a week after preparation was observed, which was also about three times lower than that in C-Lip. Moreover, there was no significant change of the particle size, polydispersity index, and zeta potential over time (data not shown). Nonetheless, the F-Lip suspension solution should be lyophilized to keep the long-term stability.
Table I.
The Encapsulation Efficiency %, Size, and Zeta Potential of F-Lip and C-Lip (n = 3)
| Samples | EE (%) | Size (nm) | Zeta potential (mV) |
|---|---|---|---|
| F-Lip | 91.4 ± 2.3 | 197.8 ± 4.6 | −11.7 ± 3.0 |
| C-Lip | 95.3 ± 3.1 | 206.6 ± 2.2 | −9.0 ± 2.0 |
EE encapsulation efficiency, F-Lip CPT-11-loaded folate-targeted liposome, C-Lip CPT-11-loaded liposome
Fig. 3.

Representative TEM images of irinotecan-loaded folate-targeted liposomes (F-Lip)
Stability of CPT-11 in Plasma
The stability of CPT-11 in the rat plasma was investigated. The results in Fig. 4 showed that the degradation percent of CPT-11 in two liposomes after incubation with plasma for 2 h were significantly lower than that in the C-Inj, which suggested that the liposomes could increase the stability of CPT-11 in rat plasma.
Fig. 4.

Degradation percent of CPT-11 in different formulations after incubation with the rat plasma for 2 h (**p < 0.01)
In Vivo Distribution of F-Lip
The mean concentration–time profiles of CPT-11 and SN-38 in plasma and typical tissues after i.v. administration are presented in Fig. 5, and the non-compartment pharmacokinetic parameters calculated according to the measured values for infinite time are given in Tables II and III. Plasma concentration of CPT-11 after i.v. administration of two liposome groups, especially at 0.5 h, was increased relative to the C-Inj (the control). F-Lip and C-Lip yielded higher level where the concentrations of CPT-11 at 0.5 h were 2.733 and 3.221 times greater than the control, respectively. Such high plasma concentrations of CPT-11 for two liposome groups might be owing to the improved stability of CPT-11 by protection the drug from chemical degradation and the sustained release of CPT-11 from the liposome. Some studies showed that the liposome, especially prepared at lower pH condition by pH gradient method, resulted in stabilization of the lactone form of camptothecins (such as CPT and CPT-11) (23,24).
Fig. 5.

Biodistribution of CPT-11 and SN-38 in plasma, liver, tumor, and intestinum rectum after i.v. administration of CPT-11 injection (C-Inj), CPT-11-loaded liposome (C-Lip), and CPT-11-loaded folate-targeted liposome (F-Lip)
Table II.
Pharmacokinetic Parameters of CPT-11 in the Plasma and Tissues After i.v. Administration of C-Inj, C-Lip, and F-Lip, Respectively
| Parameters | Tissues | C-Inj | C-Lip | F-Lip |
|---|---|---|---|---|
| AUC (ng/h/g) | Plasma | 1,035.1 | 13,357.3 | 12,952.0 |
| Liver | 9,670.1 | 18,834.9 | 21,056.2 | |
| Rectum | 6,486.4 | 6,784.6 | 10,370.7 | |
| Tumor | 1,249.4 | 1,537.7 | 2,506.2 | |
| C max (ng/g) | Plasma | / | / | / |
| Liver | 3,988.5 ± 808.5 | 5,612.0 ± 145.7* | 6,079 ± 528.8* | |
| Rectum | 1,455.1 ± 367.2 | 1,191.9 ± 46.6 | 2,108.0 ± 306.6**,*** | |
| Tumor | 610.6 ± 28.0 | 710.1 ± 27.6* | 774.9 ± 46.0** |
Results of C max are given as mean ± SD, and the others are given as mean (n = 5). AUC values are calculated for 0–8 h. AUC value in plasma is given as nanogram hour per milliliter
AUC area under the curve, C-Inj CPT-11 injection, C-Lip CPT-11-loaded liposome, F-Lip CPT-11-loaded folate-targeted liposome
*p < 0.05 vs control group (C-Inj); **p < 0.01 vs control group (C-Inj); ***p < 0.01 vs C-Lip group
Table III.
Pharmacokinetic Parameters of SN-38 in the Plasma and Tissues After i.v. Administration of C-Inj, C-Lip, and F-Lip, Respectively
| Parameters | Tissues | C-Inj | C-Lip | F-Lip |
|---|---|---|---|---|
| AUC (ng/h/g) | Plasma | 192.2 | 225.8 | 276.7 |
| Liver | 327.3 | 659.2 | 844.2 | |
| Rectum | 159.0 | 172.1 | 334.2 | |
| Tumor | 374.3 | 462.4 | 449.4 | |
| C max (ng/g) | Plasma | 61.8 ± 8.6 | 66.1 ± 7.0 | 95.6 ± 16.2*,*** |
| Liver | 61.1 ± 12.4 | 338.7 ± 198.1* | 223.5 ± 36.5** | |
| Rectum | 26.9 ± 6.4 | 39.3 ± 12.8 | 53.3 ± 10.4* | |
| Tumor | 125.2 ± 11.1 | 187.3 ± 5.5** | 316.3 ± 10.5**,**** |
Results of C max are given as mean ± SD, and the others are given as mean (n = 5). AUC values are calculated for 0–8 h. AUC value in plasma is given as nanogram hour per milliliter
AUC area under the curve, C-Inj CPT-11 injection, C-Lip CPT-11-loaded liposome, F-Lip CPT-11-loaded folate-targeted liposome
*p < 0.05 vs control group (C-Inj); **p < 0.01 vs control group (C-Inj); ***p < 0.05 vs C-Lip group; ****p < 0.01 vs C-Lip group
Based on the area under the curve (AUC)0 − t values, the uptake of CPT-11 in the tumor was far greater with the F-Lip group in comparison to the control and C-Lip group. The Cmax of F-Lip was also significantly higher than that of the control (p < 0.01). Especially, the concentration of CPT-11 in the tumor at 1 h was 3.27 times higher than that of the C-Inj, yielding a comparable efficacy with previous studies by Sadzuka et al., where CPT-11 concentration in the tumor 8 h after administration in liposomes with DSPC (S-Lip) and PEG-coated S-Lip (S-PEG) groups increased by 3.2- and 4.5-fold, respectively, compared to that in the Sol (10). Moreover, the concentration of CPT-11 in the tumor at 1 h was 2.73 times higher than that of the C-Lip, although the Cmax of F-Lip and C-Lip was similar with the same peak time at 0.5 h, which suggested that the drug clearance would be slower as to the F-Lip group. These results showed that F-Lip resulted in an increased distribution of CPT-11 to the tumor. FA, as a targeting ligand, offers many advantages including high specificity for tumors, non-immunogenicity, relatively good stability, and available in large quantities (25). In this study, F-Lip achieved the highest uptake of CPT-11 in the tumor as compared to C-Inj and C-Lip, which was attributed to effective FA receptor-mediated endocytosis. It is consistent with previous studies of folate-targeted liposome for other antitumor drugs such as doxorubicin (26–28), zoledronic acid (29), and paclitaxel (30). Other possible mechanism might be related to several factors: (1) The sizes of the F-Lip were less than 200 nm. Some studies reported that the particles of 100–200 nm might aggregate and localize in solid tumor with enhanced permeability and retention (EPR) effect (11,31–33). (2) Some studies indicated that folate-targeted liposome had a potential role in overcoming drug resistance because of the bypass of the P-glycoprotein efflux pump (15,26). The previous studies also showed that FR-mediated uptake of the liposome loading doxorubicin into a multidrug-resistant subline of M109-HiFR cells was unaffected by P-glycoprotein efflux pump. Gabizon et al. also described that FR-targeted liposomal delivery can reverse multidrug resistance to doxorubicin (15). In addition, the AUC0 − t and Cmax values of C-Lip were higher than those of the control, which was attributed to the improved stability of CPT-11 and the passive tumor targeting based on the EPR effect due to the C-Lip sizes of ~200 nm (23,33).
Moreover, compared to the control group, the kidney, heart, and lung uptake of the F-Lip group were all decreased, and the spleen uptake was increased. The uptake of CPT-11 in the tumor, liver, and spleen after i.v. administration of the C-Lip were increased compared to the control, although it was less than those of the F-Lip group (data not shown).
The concentration of SN-38 in the plasma and tissues was also investigated. The results were listed in Table III. The Cmax of SN-38 in the tumor was far greater with F-Lip than those of the control and C-Lip although the AUC for F-Lip was only slightly greater than that of the control. These data suggest that the targeting of CPT-11 to the tumor by liposomalization may elevate the SN-38 level in the tumor, which could increase the antitumor activity of CPT-11. Moreover, the AUC and Cmax of SN-38 in the liver for two liposome groups were all markedly increased as compared to the C-Inj, and the amplification was also greater than those in the tumor. The sustained release of CPT-11 from the liposome could prevent the saturation of carboxyesterase enzyme in the liver (34), which may provide higher conversion efficiency of CPT-11 to SN-38 in liver. Some studies also reported that CPT-11 after i.v. administration of free CPT-11 was converted to SN-38 mainly in the liver. The amount of SN-38 converted from CPT-11 in the tumor homogenate at 2 h was only 37 % of that in the liver (7).
These studies also demonstrated that the distribution level of CPT-11 and SN-38 in the rectum with the F-Lip group was obviously increased as compared to the C-Inj. Previous study on the pharmacokinetics showed that the CPT-11 could be reabsorbed by the hepato-enteral circulation (35). Therefore, for F-Lip group, the uptake of CPT-11 in the rectum was increased because higher concentration of CPT-11 in the liver facilitated the hepato-enteral circulation of CPT-11. It suggested that the F-Lip had a potential for the therapy of the intestinal cancer.
Interesting, we also found that the expected long circulation effect was not observed although the FA–PEG–DSPE, a PEGylated lipid, was added in the formulation of F-Lip. On the contrary, compared to C-Inj and C-Lip, F-Lip showed faster clearance and shorter retention based on plasma concentration–time profiles and MRT values (MRT values of C-Inj, C-Lip, and F-Lip calculated using a statistical moment algorithm were 1.53, 1.11, and 1.07 h, respectively). It might be due to the following reasons: The amount of FA–PEG–DSPE in F-Lip formulation is too low to affect the surface characteristics of the liposome (such as surface hydrophilicity and stereospecific blockade), so F-Lip cannot avoid the uptake of RES cells. Moreover, the FA residues in F-Lip can bind to the plasma folate binding protein in the blood, which results in faster removal of F-Lip by RES due to nonspecific mechanisms (36). The in vivo distribution results also showed that the uptake of CPT-11 in the liver and spleen after i.v. administration of F-Lip were slightly higher than that obtained after i.v. administration of C-Lip. In addition, compared to the control, the increased uptake in the liver after i.v. administration of C-Lip might result from the passive targeting effect of liposomes due to the phagocytosis by RES cells.
Some studies showed that long circulation in blood could increase accumulation of antitumor drugs in the tumors by EPR effects (6). Therefore, the tumor targeting efficiency of F-Lip could be further improved by adding the PEGylated lipid such as mPEG–DSPE in the formulation to prolong the circulation time in the blood. However, PEG coating interferes with the uptake of F-Lip (36). Therefore, the amount of PEGylated lipid and PEG length must be noticed to reduce the interference with folate receptor-mediated cell uptake (14).
In Vivo Antitumor Activity
The in vivo antitumor activity of the C-Lip and F-Lip was investigated. The saline was as the negative control, and C-Inj was as the positive control. The results in Table IV showed that a dose-dependent tumor growth inhibition was observed with increasing dose of CPT-11 in three treatment groups compared to the negative control. To obtain the significant tumor growth inhibition effect, the required drug doses in C-Lip and F-Lip were 10 and 5 mg/kg, which were much lower than that in C-Inj group (40 mg/kg). Furthermore, the tumor growth inhibition rate of two liposome groups at the dose of 20 mg/kg was significantly higher than those of C-Inj at the dose of 40 mg/kg (p < 0.01 for F-Lip and p < 0.05 for C-Lip). The tumor growth inhibition rate of two liposome groups at the low dose of 10 mg/kg yielded comparable efficacy with that of C-Inj at medium dose of 20 mg/kg. Moreover, the F-Lip was found to be most efficient, obtaining higher tumor growth inhibition rate than C-Lip at the same dose.
Table IV.
Tumor Weight and Tumor Inhibitory Rate After i.v. Administration of F-Lip, C-Lip, and C-Inj in S180 Tumor-Bearing Mice (mean ± SD, n = 11)
| Groups | Dose (mg/kg) | Weight of tumor (g) | Tumor inhibitory rate (%) |
|---|---|---|---|
| Blank | / | 0.97 ± 0.39 | 0 |
| F-Lip | 20 | 0.37 ± 0.22** | 61.84 |
| 10 | 0.63 ± 0.38* | 35.05 | |
| 5 | 0.70 ± 0.28* | 27.83 | |
| C-Lip | 20 | 0.43 ± 0.38** | 55.64 |
| 10 | 0.65 ± 0.49* | 32.72 | |
| 5 | 0.72 ± 0.36 | 25.73 | |
| C-Inj | 40 | 0.49 ± 0.25* | 48.48 |
| 20 | 0.63 ± 0.25 | 35.05 | |
| 10 | 0.69 ± 0.46 | 28.87 |
F-Lip CPT-11-loaded folate-targeted liposome, C-Lip CPT-11-loaded liposome, C-Inj CPT-11 injection
*p > 0.05 vs. control group (the blank); **p > 0.01 vs. control group (the blank)
The toxicity of the formulation was evaluated by the body weight loss, thymus index, and spleen index. Compared to the negative control, there was no remarkable significant body weight loss in mice after i.v. administration four times of the F-Lip and C-Lip (p > 0.05), while obvious body weight loss for C-Inj group was observed. It suggested that the liposome could decrease the toxicity of CPT-11. Moreover, based on thymus index and spleen index (Table V), the F-Lip also showed much lower toxicity on the center immune organs than C-Inj and C-Lip.
Table V.
Thymus Index and Spleen Index After i.v. Administration of F-Lip, C-Lip, and C-Inj in S180 Tumor-Bearing Mice (Mean ± SD, n = 11)
| Groups | Dose (mg/kg) | Thymus index (g/kg) | Spleen index (g/kg) |
|---|---|---|---|
| Blank | / | 1.27 ± 0.56 | 14.41 ± 4.58 |
| 20 | 0.68 ± 0.25* | 12.30 ± 2.82 | |
| F-Lip | 10 | 0.70 ± 0.50 | 14.84 ± 3.61 |
| 5 | 1.01 ± 0.15 | 16.39 ± 4.23 | |
| 20 | 0.51 ± 0.25** | 12.83 ± 3.56 | |
| C-Lip | 10 | 0.86 ± 0.21** | 14.78 ± 2.10 |
| 5 | 0.65 ± 0.50** | 16.09 ± 3.99 | |
| 40 | 0.70 ± 0.35** | 10.34 ± 2.92* | |
| C-Inj | 20 | 0.75 ± 0.44** | 14.82 ± 3.69 |
| 10 | 0.65 ± 0.49** | 11.53 ± 2.30* |
F-Lip CPT-11-loaded folate-targeted liposome, C-Lip CPT-11-loaded liposome, C-Inj CPT-11 injection
*p > 0.05 vs. control group (the blank); **p > 0.01 vs. control group (the blank)
CONCLUSION
FA–PEG–DSPE was synthesized by amide reaction of DSPE–PEG–NH2 and FA. F-Lip modified by FA–PEG–DSPE was successfully prepared by an ammonium sulfate gradient with higher entrapment efficiency of 91.39 ± 2.34 % and the mean particle size of less than 200 nm. The distributions of CPT-11 and SN-38 in the tumor were far greater with the F-Lip group in comparison to the C-Inj and C-Lip, which might contribute to folate-meditated targeting uptake by the folate receptor on the surface of the tumor cells. The uptake of CPT-11 in the liver and rectum for two liposome groups was all markedly increased as compared to the C-Inj. Moreover, F-Lip exhibited a dose-dependent tumor growth inhibition, superior anticancer activity, and lower toxicity as compared to C-Lip and C-Inj after i.v. administration. Therefore, F-Lip may be presented as potential candidates for tumor targeting drug delivery.
ACKNOWLEDGMENTS
This study was supported by the Major Project of National Science and Technology of China for New Drugs Development (no. 2009ZX09310-004) and the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (no. JKGQ201107).
REFERENCES
- 1.Houghton PJ, Cheshire PJ, Hallman JC, Bissery MC, Mathieu-Boue A, Houghton JA. Therapeutic efficacy of the topoisomerase I inhibitor 7-ethyl-10-(4-[1-piperidino]-1-piperidino)-carbonyloxy-camptothecin against human tumor xenografts: lack of cross-resistance in vivo in tumors with acquired resistance to the topoisomerase I inhibitor 9-dimethylaminomethyl- 10-hydroxycamptothec. Cancer Res. 1993;53:2823–2829. [PubMed] [Google Scholar]
- 2.Gerrits CJ, De Jonge MJ, Schellens JH, Stoter G, Verweij J. Topoisomerase I inhibitors: the relevance of prolonged exposure for present clinical development. Br J Cancer. 1997;76:952–962. doi: 10.1038/bjc.1997.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rowinsky EK, Grochow LB, Ettinger DS, Sartorius SE, Lubejko BG, Chen T-L, Rock MK, Donehower RC. Phase I and pharmacological study of the novel topoisomerase I inhibitor 7-ethyl-lo-[−4-(l-piperidino)-l-piperidino]-carbonyloxy camptothecin (CPT-11) administered as a ninety-minute infusion every 3 weeks. Cancer Res. 1994;54:427–436. [PubMed] [Google Scholar]
- 4.Rothenberg ML, Kuhn JG, Burris HA, III, Nelson J, Eckardt JR, Tristan-Morales M, Hilsenbeck SG, Weiss GR, Smith LS, Rodriguez GI, Rock MK, Von Hoff DD. Phase I and pharmacokinetic trial of weekly CPT-11. J Clin Oncol. 1993;11:2194–2204. doi: 10.1200/JCO.1993.11.11.2194. [DOI] [PubMed] [Google Scholar]
- 5.Slatter JG, Schaaf LJ, Sams JP, Feenstra KL, Johnson MJ, Bombardt PA, Cathcart KS, Verburg MT, Pearson LK, Compton LD, Miller LL, Baker DS, Pesheck CV, Lord RS., III Pharmacokinetics, metabolism, and excretion of irinotecan (CPT-11) following I.V. infusion of [(14)C]CPT-11 in cancer patients. Drug Metab Dispos. 2000;28:423–433. [PubMed] [Google Scholar]
- 6.Hattori Y, Shi L, Ding W, Koga K, Kawano K, Hakoshima M, Maitani Y. Novel irinotecan-loaded liposome using phytic acid with high therapeutic efficacy for colon tumors. J Control Release. 2009;136:30–37. doi: 10.1016/j.jconrel.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Watanabe M, Kawano K, Toma K, Hattori Y, Maitani Y. In vivo antitumor activity of camptothecin incorporated in liposomes formulated with an artificial lipid and human serum albumin. J Control Release. 2008;127:231–238. doi: 10.1016/j.jconrel.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 8.Han X, Liu J, Liu M, Xie C, Zhan C, Gu B, Liu Y, Feng L, Lu W. 9-NC-loaded folate-conjugated polymer micelles as tumor targeted drug delivery system: preparation and evaluation in vitro. Int J Pharm. 2009;372:125–131. doi: 10.1016/j.ijpharm.2008.12.035. [DOI] [PubMed] [Google Scholar]
- 9.Ramsay E, Alnajim J, Anantha M, Zastre J, Yan H, Webb M, Waterhouse D, Bally M. A novel liposomal irinotecan formulation with significant anti-tumor activity: use of the divalent cation ionophore A23187 and copper-containing liposomes to improve drug retention. Eur J Pharm Biopharm. 2008;68:607–617. doi: 10.1016/j.ejpb.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 10.Sadzuka K, Hirotsu S, Hirota S. Effect of liposomalization on the antitumor activity, side-effects and tissue distribution of CPT-11. Cancer Lett. 1998;127:99–106. doi: 10.1016/S0304-3835(98)00031-7. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki R, Takizawa T, Kuwata Y, Mutoh M, Ishiguro N, Utoguchi N, Shinohara A, Eriguchi M, Yanagie H, Maruyama K. Effective anti-tumor activity of oxaliplatin encapsulated in transferring-PEG-liposome. Int J Pharm. 2008;346:143–150. doi: 10.1016/j.ijpharm.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 12.Hong M, Zhu S, Jiang Y, Tang G, Pei Y. Efficient tumor targeting of hydroxycamtothecin loaded PEGylated niosomes modified with transferring. J Control Release. 2009;133:96–102. doi: 10.1016/j.jconrel.2008.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Gabizon A, Horowitz AT, Goren D, Tzemach D, Mandelbaum-Shavit F, Qazen MM, Zalipsky S. Targeting folate receptor with folate linked to extremities of poly(ethyleneglycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10:289–298. doi: 10.1021/bc9801124. [DOI] [PubMed] [Google Scholar]
- 14.Zako T, Nagata H, Terada N, Utsumi A, Sakono M, Yohda M, Ueda H, Soga K, Maeda M. Cyclic RGD peptide-labeled upconversion nanophosphors for tumor cell-targeted imaging. Biochem Biophys Res Commun. 2009;381:54–58. doi: 10.1016/j.bbrc.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9:6551–6559. [PubMed] [Google Scholar]
- 16.Wu J, Liu Q, Lee RJ. A folate receptor-targeted liposomal formulation for paclitaxel. Int J Pharm. 2006;316:148–153. doi: 10.1016/j.ijpharm.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 17.Zalipsky S, Brandeis E, Newman MS, Woodle MC. Long circulating, cationic liposomes containing amino-PEG-phosphatidylethanolamine. FEBS Lett. 1994;353:71–74. doi: 10.1016/0014-5793(94)01013-7. [DOI] [PubMed] [Google Scholar]
- 18.Chan P, Kurisawa M, Chung JE, Yang YY. Synthesis and characterization of chitosan-g-poly(ethylene glycol)-folate as a non-viral carrier for tumor-targeted gene delivery. Biomaterials. 2007;28:540–549. doi: 10.1016/j.biomaterials.2006.08.046. [DOI] [PubMed] [Google Scholar]
- 19.Zhang ZQ, Zhu JB, Yao J, Li Z, He HY. Preparation of irinotecan liposomes by pH gradient method. J Chin Pharm Univ. 2008;39:312–316. [Google Scholar]
- 20.Zhang ZQ, Yao J, Wu X, Zou J, Zhu J. An accurate assay for simultaneous determination of irinotecan and its active metabolite SN-38 in rat plasma by LC with fluorescence detection. Chromatographia. 2009;70:399–405. doi: 10.1365/s10337-009-1161-5. [DOI] [Google Scholar]
- 21.Yao J, Zhou JP, Ping QN, Lu Y, Chen L. Distribution of nobiletin chitosan-based microemulsion in brain following i.v. injection in mice. Int J Pharm. 2008;352:256–262. doi: 10.1016/j.ijpharm.2007.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Paranjpe PV, Chen Yu, Kholodovych V, Welsh W, Stein S, Sinko PJ. Tumor-targeted bioconjugate based delivery of camptothecin: design, synthesis and in vitro evaluation. J Control Release. 2004;100:275–292. doi: 10.1016/j.jconrel.2004.08.030. [DOI] [PubMed] [Google Scholar]
- 23.Chou TH, Chen SC, Chu IM. Effect of composition on the stability of liposomal irinotecan prepared by a pH gradient method. J Biosci Bioeng. 2003;95:405–408. [PubMed] [Google Scholar]
- 24.Vaage J, Donovan D, Uster P, Working P. Tumour uptake of doxorubicin in polyethylene glycol-coated liposomes and therapeutic effect against a xenografted human pancreatic carcinoma. Br J Cancer. 1997;75:482–486. doi: 10.1038/bjc.1997.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sudimack J, Lee RJ. Targeted drug delivery via the folate receptor. Adv Drug Deliv Rev. 2000;41:147–162. doi: 10.1016/S0169-409X(99)00062-9. [DOI] [PubMed] [Google Scholar]
- 26.Goren D, Horowitz AT, Tzemach D, Tarshish M, Zalipsky S, Gabizon A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin Cancer Res. 2000;6:1949–1957. [PubMed] [Google Scholar]
- 27.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 28.Gabizon A, Tzemach D, Gorin J, Mak L, Amitay Y, Shmeeda H, Zalipsky S. Improved therapeutic activity of folate-targeted liposomal doxorubicin in folate receptor-expressing tumor models. Cancer Chemother Pharmacol. 2010;66:43–52. doi: 10.1007/s00280-009-1132-4. [DOI] [PubMed] [Google Scholar]
- 29.Shmeeda H, Amitay Y, Gorin J, Tzemach D, Mak L, Ogorka J, Kumar S, Zhang JA, Gabizon A. Delivery of zoledronic acid encapsulated in folate-targeted liposome results in potent in vitro cytotoxic activity on tumor cells. J Control Release. 2010;146:76–83. doi: 10.1016/j.jconrel.2010.04.028. [DOI] [PubMed] [Google Scholar]
- 30.Stevens PJ, Lee RJ. A folate receptor-targeted emulsion formulation for paclitaxel. Anticancer Res. 2003;23:4927–4932. [PubMed] [Google Scholar]
- 31.Ishida O, Maruyama K, Sasaki K, Iwatsuru M. Size-dependent extravasation and interstitial localization of polyethyleneglycol liposomes in solid tumor-bearing mice. Int J Pharm. 1999;190:49–56. doi: 10.1016/S0378-5173(99)00256-2. [DOI] [PubMed] [Google Scholar]
- 32.Maeda H. The enhanced permeability and retention (EPR) effect in tumor vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzym Regul. 2000;41:189–207. doi: 10.1016/S0065-2571(00)00013-3. [DOI] [PubMed] [Google Scholar]
- 33.Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. J Control Release. 2000;65:271–284. doi: 10.1016/S0168-3659(99)00248-5. [DOI] [PubMed] [Google Scholar]
- 34.Ramsay EC, Anantha M, Zastre J, Meijs M, Zonderhuis J, Strutt D, Webb MS, Waterhouse D, Bally MB. Irinophore C: a liposome formulation of irinotecan with substantially improved therapeutic efficacy against a panel of human xenograft tumors. Clin Cancer Res. 2008;14:1208–1217. doi: 10.1158/1078-0432.CCR-07-0780. [DOI] [PubMed] [Google Scholar]
- 35.Wen X, Wen J. Research on the clinical pharmacokinetics of irinotecan. Chin J Mod Drug Appl. 2008;2:42–44. [Google Scholar]
- 36.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid–anchored folic acid–PEG conjugates. Adv Drug Deliv Rev. 2004;56:1177–1192. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]