

Abstract
Effects of tromethamine (Tris), polyvinylpyrrolidone (PVP-K25), and low molecular weight chitosan (LM-CH) on dissolution and therapeutic efficacy of glimepiride (Gmp) were investigated using physical mixtures (PMs), coground mixtures, coprecipitates (Coppts) or kneaded mixtures (KMs), and compared with drug alone. Fourier transform infrared spectroscopy, differential scanning colorimetry, and X-ray diffractometry were performed to identify any physicochemical interaction with Gmp. Surface morphology was examined via scanning electron microscopy. The results of Gmp in vitro dissolution revealed that it was greatly enhanced by Coppt with Tris or PVP-K25 and KM with LM-CH at a drug to carrier ratio of 1:8. Gmp amorphization by PVP-K25 and LM-CH was a major factor in increasing Gmp dissolution. Being basic, Tris might increase the pH of the microdiffusion layer around Gmp particles improving its dissolution. Formation of water-soluble complexes suggested by solubility study may also explain the enhanced dissolution. Capsules were prepared from Coppts and KM 1:8 drug to carrier binary systems and also with Tris PMs. In vivo, the hypoglycemic efficacy of Gmp capsules in rabbits increased by 1.63-, 1.50-, and 1.46-fold for 1:8 Coppts with Tris or PVP-K25 and KM with LM-CH respectively, compared with Gmp alone. Surprisingly, the response to Tris PM 1:20 capsules was 1.52-fold revealing statistically insignificant difference to that of Tris Coppt 1:8 (1.63 fold). As a conclusion, dissolution enhancement and hypoglycemic potentiation by 1:20 PM of Gmp/Tris, being simple and easy to prepare, may enable development of a reduced-dose and fast-release oral dosage form of Gmp.
Key words: dissolution enhancement, glimepiride, low molecular weight chitosan, polyvinylpyrrolidone, tromethamine
INTRODUCTION
On therapeutic application, poorly water-soluble agents encounter a number of serious proplems such as, poor absorption and bioavailability upon oral administration and formation of aggregates following intravenous administration that can cause embolization of blood vessels resulting in serious respiratory system failure, local toxicity, and/or lowered systemic bioavailability (1). Hence, about 40% of potentially valuable drug candidates identified by high throughput screening are rejected and never enter a formulation development stage due to their poor water solubility (2). Consequently, various techniques such as micronization, solubilization, salt formation, complexation, change in physical form, use of prodrugs, drug derivitization, alteration in pH, addition of surfactants, and others have been employed in order to improve the dissolution and bioavailability of sparingly soluble drugs (3,4). Hydrophilic carriers, such as Tris, LM-CH, and PVP (Fig. 1b, c, and d respectively) have been widely used for this purpose. For example, different solid binary systems as physical mixtures (PMs) and coprecipitates (Coppts) with Tris and PVP were successful in increasing the dissolution and the analgesic effect of nimesulide (5). Dissolution rates of benzoic acid (6), hydrochlorothiazide (7), and furosemide (8) were markedly enhanced by use of Tris as a carrier. Enhancement of naproxen dissolution by PMs, coground mixtures (GMs), and kneaded mixtures (KMs) with chitosan has been reported (9). The ability of chitosan to improve the solubility of some drugs as aceclofenac has been illustrated (10). Dissolution improvement of poorly water-soluble sulfonylureas was reported in purpose of increasing their therapeutic efficacy. For example, glibenclamide solubility was enhanced by PVP (11) and Tris (12). That of repaglinide was increased by ultra-rapid freezing with Tris (13). The effects of cyclodextrin and chitosan on the solubility of glyburide have been investigated (14).
Fig. 1.
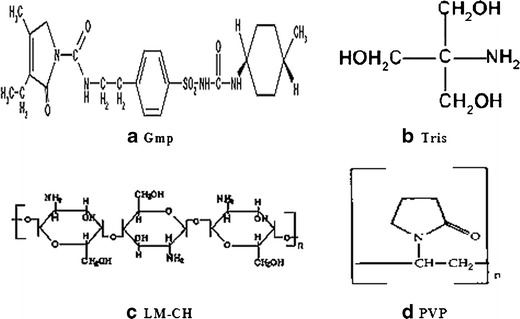
Chemical structures of the drug and the tested carriers (Gmp glimepiride (21), Tris tromethamine (5), LM-CH low molecular weight chitosan (24), PVP polyvinylpyrrolidone (30))
Gmp, Fig. 1a, is one of the third generation sulfonylurea drugs used for treatment of diabetis mellitus type 2 (15). Several investigations reported that this hypoglycemic drug showed number of advantages over the other sulfonylureas including lower dosage, rapid onset, and longer duration of action (16,17). However, its poor water solubility and slow dissolution may result in irreproducible clinical response or therapeutic failure because of subtherapeutic plasma levels (18). Recently, dissolution improvement of the widely used sulfonylurea, Gmp, was obtained utilizing complexes with cyclodextrin and its derivatives (19), solid dispersion with polyethylene glycol 6000 (20) or hyperbranched poly(esteramides) (21), cosolvents (22), and microencapsulation by spray congealing technology using hydrophilic meltable carriers (23). Therefore, it was worthy of trying to prepare different Gmp binary systems with each of Tris, LM-CH, and PVP-K25, investigate and compare the effects of these carriers in the prepared systems on dissolution and hypoglycemic efficacy of Gmp.
MATERIAL AND METHODS
Materials
Gmp (Batch # RM0700075) was kindly supplied by Delta Pharma Co., The 10th of Ramadan City, Egypt. Tris and PVP-K25 were obtained from Merck Co., Darmstadt, Germany. LM-CH was purchased from Sigma Chem. Co., St Louis, MO, USA. Dicalcium phosphate was obtained from Mendell, UK. Glucose oxidase peroxidase (GOD/POD) kit was purchased from Spinreact, S. A. U., Spain. Amaryl 3 mg tablets manufactured by Aventis, Cairo, Egypt, were used as a commercial product for comparison. All other chemicals were of reagent grade.
Methods
Preparation of the Different Solid Binary Systems and Capsules
Gmp with each of Tris, LM-CH, and PVP-K25 as PMs, GMs, and Coppts in different drug/carrier ratios were prepared (Table I). With LM-CH, however, the Coppts were replaced with KMs.
Table I.
Dissolution Efficiency (DE) and Relative Dissolution Rate (RDR), from Different Binary Systems and Capsules
| Drug:carrier | Binary systema | Capsulesb | Fold increasec | |||
|---|---|---|---|---|---|---|
| DEd30 | RDRe30 | DEf60 | RDRg60 | DEd30 | DEf60 | |
| Gmp alone | 6.19 | _ | 6.01 | _ | _ | _ |
| Tris PM 1:8 | 13.06 | 2.10 | _ | 2.11 | _ | |
| Tris PM 1:10 | 15.58 | 2.89 | 21.70 | 3.19 | 2.52 | 3.61 |
| Tris PM 1:15 | 19.72 | 3.75 | 26.72 | 4.13 | 3.19 | 4.44 |
| Tris PM 1:20 | 26.99 | 3.99 | 33.48 | 5.21 | 4.36 | 5.57 |
| Tris GM 1:8 | 38.43 | 5.85 | _ | 6.21 | _ | |
| Tris GM 1:20 | 46.77 | 6.95 | _ | 7.56 | _ | |
| Tris Coppt 1:8 | 65.17 | 9.24 | 49.45 | 8.65 | 10.53 | 8.23 |
| LM-CH PM 1:8 | 13.76 | 1.87 | _ | 2.22 | ||
| LM-CH PM 1:20 | 16.52 | 2.45 | _ | 2.67 | ||
| LM-CH GM 1:8 | 38.37 | 5.70 | _ | 6.2 | ||
| LM-CH GM 1:20 | 46.10 | 7.04 | _ | 7.45 | ||
| LM-CH KM 1:8 | 59.80 | 9.28 | 44.45 | 8.29 | 9.66 | 7.39 |
| PVP-K25 PM 1:8 | 18.93 | 2.79 | _ | 3.06 | ||
| PVP-K25 PM 1:20 | 9.73 | 1.52 | _ | 1.57 | ||
| PVP-K25 GM 1:8 | 37.70 | 5.80 | _ | 6.09 | ||
| PVP-K25 GM 1:20 11:20 | 32.05 | 5.02 | _ | 5.18 | ||
| PVP-K25 Coppt 1:8 | 67.33 | 9.81 | 54.78 | 9.11 | 10.88 | 9.11 |
a PM indicates physical mixture; GM ground mixture; Coppt coprecipitate; and KM kneaded mixture
bEach capsule contains 3 mg Gmp and amount of carrier specified by its ratio. The final weight was 50 mg by dilution with dicalcium phosphate excipient when needed
cCompared with Gmp alone
dThe dissolution efficiencies at 30 min for the powders and capsules
eThe relative dissolution rate at 30 min for the powders and capsules
fThe dissolution efficiencies at 60 min for the powders and capsules
gThe relative dissolution rate at 60 min for the powders and capsules
PMs were prepared by geometric proportion technique. GMs were prepared by grinding the corresponding PM in vibrational mill for 1 h (Microne Micronising Mill, England). The Coppts were prepared by the solvent evaporation technique (5,20). The solvent was methanol and evaporation was under vacuum at 40°C. To prepare KMs of LM-CH, the corresponding PM was kneaded for 0.5 h with distilled water in an amount equal to 1.2 times of total weight of the PM using mortar and pestle (24). The resulting KM was dried at room temperature in a desiccator containing anhydrous calcium chloride for 48 h. The resulting binary systems were ground if necessary and passed through a 250-μm sieve.
Weights of the prepared systems, each equivalent to 3 mg Gmp, was diluted with dicalcium phosphate when needed, as excipient, to 50 mg total weight and dispensed in hard gelatin capsules (Table I).
Estimation of Drug Content
An accurately weighed amount of each of the binary systems equivalent to 3 mg of Gmp was dissolved in minimum amount of methanol with sonication. The volume was adjusted with distilled water to 100 ml in a volumetric flask filtered to remove undissolved material if any. The solution was assayed for drug content spectrophotometrically at 226 nm using blank containing an equivalent amount of the corresponding carrier. Three replicates were prepared and the average drug contents were estimated.
Characterization of the Prepared Solid Binary Systems
Fourier Transform Infrared Spectroscopy
Fourier transform infrared (FTIR) spectra of all carriers and binary systems were obtained using Mattson 5000 FTIR Spectrophotometer (Madison Instruments, Middleton, Wisconsin, USA). KBr discs were prepared by means of hydrostatic press. The scanning range was 500 to 3900 cm−1
Differential Scanning Colorimetry (DSC)
Differential scanning colorimetry (DSC) data were performed using a Perkin-Elmer differential scanning colorimeter model DSC-4 (New York, USA). It was calibrated with indium (99.99% purity, m.p. 156.6°C), at heating rates of 10°C/min. One- to 5-mg samples were heated in aluminum crimped pans under nitrogen gas flow, in the 30–450°C range.
X-Ray Diffractometry
X-ray diffractometry (XRD) patterns were obtained using a Diano X-ray diffractometer equipped with Co Kα (USA). The tube operated at 45 kv, 9 mA.
Scanning Electron Microscopy
The surface morphology of Gmp 1:8 Coppt with each of Tris and PVP-K25 were examined by means of a scanning electron microscope (JEOL, Japan), compared with Gmp alone and both the 1:8 and 1:20 PMs. The powders were fixed on a brass stub using double-sided adhesive tape and then made electrically conductive by coating, in a vacuum, with a thin layer of gold (approximately 150 Å) for 30 s. The pictures were taken at an excitation voltage of 25 kV.
Solubility Studies
Solubility measurements were carried out according to the method of (25). An excess amount of Gmp in either aqueous or phosphate buffer (pH = 6.8) solutions containing different concentrations of each carrier equivalent to drug to carrier ratios of 1:1, 1:2, 1:4, 1:8, 1:10, 1:20, 1:40, and 1:60. The suspensions were shaken at 37 ± 0.5°C for 72 h and then filtered through a millipore filter (0.45 μ). An aliquot portion of the filtrate was properly diluted when necessary and analyzed for its drug content by measuring its ultraviolet absorbance at 226 nm against blank solution containing the same concentration of the studied carrier. The solubility phase diagrams were constructed and the apparent stability constants (Ks) were calculated according to the following equation (26):
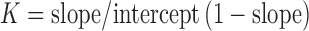 |
Where, the intercept is the solubility of Gmp alone in the respective medium.
In Vitro Dissolution Study
Dissolution of Gmp from all prepared binary systems was assessed by USP apparatus II (paddle method) at 37°C using 500 ml of phosphate buffer (pH 6.8) (19) and stirred at 100 rpm. With respect to capsules, the same procedure was employed except that USP apparatus I (basket method) was used. Aliquots from the dissolution medium were withdrawn at time intervals of 15, 30, 60, 90, 120 and 180 min and the medium was replenished by equal volumes of fresh dissolution medium. The samples were filtered through a millipore filter (pore size 45 μm) and analyzed for Gmp content by measuring its absorbance at 226 nm. Buffer containing an equivalent amount of the tested carrier (Tris, LM-CH, or PVP-K25) and/or excipient was used as a blank after filtration. Each experiment was done in triplicate and the average % dissolved was calculated at each time interval. Values of dissolution efficiency (DE) were calculated as the percent ratio of area under the dissolution curve up to the time, t, to that of the area of the rectangle described by 100% dissolution at the same time (26,27). The value of relative dissolution rate (RDR) was estimated as the ratio of the percentage dissolved Gmp from each binary system or capsule to that of Gmp alone at the same time (5).
Therapeutic Efficacy Assessment
Male rabbits weighing 1.75–2 kg were kept on a standard diet. The animals were fasted overnight prior to the experiment with water ad libitum. Food was withheld during the experiment. The rabbits were randomized into eight groups each of six animals. Animal use protocol was approved by the research ethical committee at Mansoura University in accordance with “principles of laboratory animal care” (NIH publication No. 85-23, revised 1985). One group of animals served for the control capsule and another for the marketed one (amaryl 3 mg). The other groups were for capsules of the selected binary systems as shown in Table II. Blood samples were collected from the marginal ear vein of the animal at time intervals 1, 2, 3, 4, 5, 6, 8, 10, and 24 h after drug administration (BGa). Each animal served as its own control by taking a sample of blood before drug administration (BGb). Blood glucose level was assessed by glucose oxidase peroxidase method using GOD/POD kit (28). The hypoglycemic response was evaluated as percentage reduction in blood glucose level (%RBGL) and the data were plotted as mean values versus time. The area under these response curves (AUC0–24 h) were calculated using Prism 4 software (19,29). The values of %RBGL were calculated as follows:
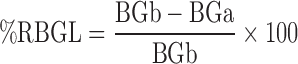 |
Table II.
Solubility Parameters of Gmp with The Tested Carriers at 37°C
| Carrier | In buffer pH 6.8 | In water | ||
|---|---|---|---|---|
| Slope | Stability constant (K s; mg−1 ml) | Slope | Stability constant (K s; mg−1 ml) | |
| Tris | 0.0026 | 0.965 | 0.0024 | 1.002 |
| LM-CH | 0.0009 | 0.334 | 0.0008 | 0.334 |
| PVP-K25 | 0.0019 | 0.705 | 0.0018 | 0.751 |
Statistical Analysis
In vitro, the dissolution data were represented as average of triplicate and standard deviation. The values of the maximum %RBGL and AUC0–24 h of the different groups were statistically analyzed based on one-way analysis of variance test followed by Tukey–Kramer test to compare between pairs. This statistical analysis was computed with GraphPad InStat version 3.0 (GraphPad Software, Inc., CA, USA) at different levels of P values.
RESULTS AND DISCUSSION
Drug Content of the Prepared Solid Binary Systems
Gmp content in different binary systems ranged from 92.07% to 99.3%. PMs with the three carriers showed higher average drug contents than the corresponding GMs, Coppts and KMs may be due to the simpler method of preparation that minimized drug loss.
Characterization of the Prepared Solid Binary Systems
FTIR
FTIR spectra of the 1:8 drug/carrier ratio are shown in Fig. 2. The 1:2 ratio was added for comparison. The spectrum of Gmp reveals its characteristic peaks of N–H stretch at wavenumbers of 3369 and 3288 cm−1 (19). Spectra of Gmp with each of 8 Tris and LM-CH either as PM, GM and Coppt or KM appeared as combined spectra of both drug and the corresponding carrier (Fig. 2a and b, respectively). These results may indicate absence of identified interaction between Gmp and either of Tris or LM-CH. On the other hand, Gmp with PVP-K25 1:8 either as GM or Coppt showed a broadening or complete disappearance of Gmp N–H stretching peaks contrary to the corresponding PM. This may be due to an interaction between Gmp N–H groups and PVP-K25 carbonyl groups through hydrogen bonding on cogrinding and coprecipitation. A similar reaction of PVP carbonyl groups with hydroxyl groups of anionic indomethacin through hydrogen bonding has been reported (30).
Fig. 2.
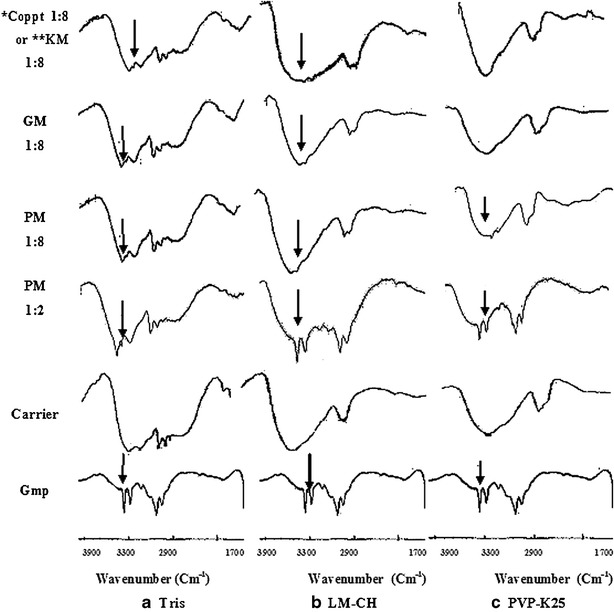
FTIR spectra of some prepared binary systems compared with Gmp and carriers, each alone. The arrows indicate Gmp N–H stretching peaks (PM physical mixture, GM coground mixture; Coppt coprecipitate, KM kneaded mixture, *Coppt with Tris or PVP-K25, **KM with LM-CH)
DSC
Figure 3 illustrates DSC data of the prepared systems with the tested carriers at drug to carrier ratio of 1:8 compared with those of Gmp and carriers each alone. DSC curves of PMs of the three carriers with the drug at a ratio of 1:2 was included in this figure to demonstrate the effect of dilution on the height of Gmp peak. The crystalline anhydrous nature of untreated Gmp was expressed by a sharp endothermic peak (Tpeak = 212°C) corresponding to its melting point (20). Pure Tris experienced two sharp endothermic peaks: at 172.8°C close to its reported melting point range (168–172°C) (31) and at 141°C representing its solid–solid transition (8) (Fig. 3a). The disappearance of the melting point peak of Gmp (Tpeak = 212°C) in the PMs (1:2 and 1:8) with Tris might be due to the thermal energy supplied during DSC scan which brought both Gmp and Tris to a highly dispersed state. Thus, more contact may be allowed resulting in an interaction that could not occur in their solid PMs and was not seen in their FTIR results. The possibilty of conversion of some crystalline compounds into highly dispersed state due to the thermal energy supplied during DSC scan has been reported in other studies (5,32). Excess Tris without interaction during DSC scan may account for the presence of the two peaks (141°C and 172°C). Both GM and Coppt with Tris experienced only one peak for Tris (140°C) and absence of the peak at 171°C. This could be explained by change of the carrier crystals with these harsh treatments as has been suggested by others (33). The broad endotherm of LM-CH confirmed its amorphous hydrated nature (Fig. 3b; 9). This figure shows that Gmp peaks existed in all LM-CH preparations (PM1:2, PM 1:8, GM 1:8 and KM 1:8) negating interaction of this carrier with Gmp as evidenced by FTIR data. The peak heights were 3.06, 2.81, and 0.47 mv for PM, GM, and KM in 1:8 drug carrier ratio, respectively, indicating variation in crystalinity of Gmp in the different systems with LM-CH. More amorphization of the drug by kneading took place than by cogrinding with LM-CH. The broad endotherm of PVP-K25 indicated its amorphous nature (Fig. 3c). Coppt in 1:8 of Gmp/PVP-K25 showed a broadening and shift in Gmp endothermic peak. The shift in Gmp peak was small in case of PM compared with Coppt. In accordance with FTIR data, these findings indicate interaction of Gmp and its amorphization on cogrinding and coprecipitation with PVP-K25.
Fig. 3.
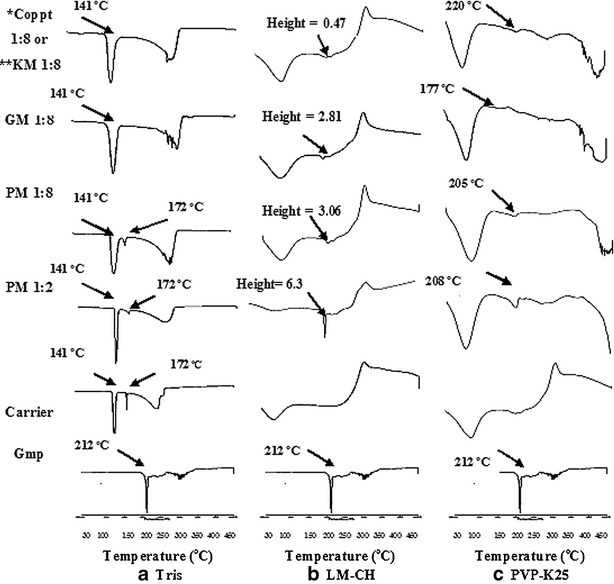
DSC curves of some prepared binary systems with the different carriers compared with Gmp and carriers, each alone (PM physical mixture, GM coground mixture, Coppt coprecipitate, KM kneaded mixture, *Coppt with Tris or PVP-K25, **KM with LM-CH)
XRD
XRD spectra at a diffraction angle of 2-theta for different binary systems with each of Tris, LM-CH, and PVP-K25 are depicted in Fig. 4a, b, and c, respectively. Drug and carriers, each alone, were included for comparison. XRD patterns at a ratio of 1:2 drug/carrier were added to the spectra to show clearly the impact of the carrier ratio and the method of preparation on Gmp crystallinity compared with 1:8.
Fig. 4.
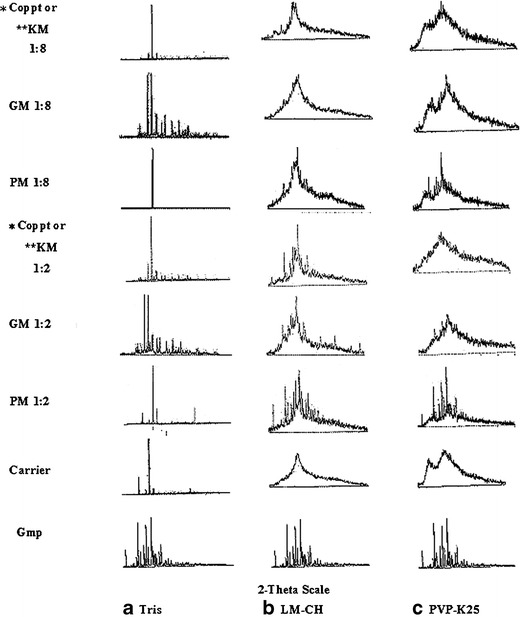
XRD spectra of some prepared binary systems with the different carriers compared with Gmp and carriers, each alone (PM physical mixture, GM coground mixture, Coppt coprecipitate, KM kneaded mixture, *Coppt with Tris or PVP-K25, **KM with LM-CH)
Numerous sharp diffraction peaks of pure Gmp were recorded indicating its crystalline nature (20). Tris alone exhibited a number of short diffraction peaks and one long distinct peak that confirmed its highly crystalline nature (Fig. 4a). The peaks of crystalline Gmp and Tris exist in their binary systems. In case of Tris GMs and Coppts with Gmp, possible change of the crystalline structure of the carrier might account for their different diffraction patterns from those of the corrsponding PMs (33).
LM-CH (9) and PVP-K25 (5,32) amorphous natures were represented by halo diffraction patterns characterized by absence of sharp peaks (Fig. 4b and c, respectively). The ratio of 1:2, GM and KM with LM-CH showed less crystallinity of Gmp than that noticed with corresponding PM. These findings were more clear at a ratio of 1:8 may be due to dilution with higher LM-CH content. GMs and Coppts with PVP-K25, at both ratios 1:2 and 1:8, resulted in a pronounced reduction in Gmp crystallinity compared with the corresponding PMs. The results obtained with scanning electron microscopy (SEM) hereafter together with that seen with XRD would augment each other.
SEM
The photomicrographs of the different drug/carriers ratios are shown in Fig. 5. The morphology of the PM 1:20 and Coppt 1:8 with Tris appeared as broken crystals with irregular shape and size similar to that of drug alone. Factors other than amorphization helped in increasing the dissolution rate of the drug in this case, possibly the pH of the microdiffusion layer around the drug particles. With LM-CH and PVP-K25, however, the PMs showed crystals of the drug dispersed in amorphous matrix of either polymer. On the other hand, the photomicrographs of the KM 1:8 with LM-CH or Coppt 1:8 with PVP-K25 reveal amorphous-looking lumps. The closer contact between the hydrophilic carriers and drug in addition to the amorphization were influential in increasing the dissolution rate of Gmp.
Fig. 5.

SEM of Gmp alone (a), Gmp/Tris PM 1:20 (b), Gmp/Tris Coppt 1:8 (c), Gmp/LM-CH PM 1:8 (d), Gmp/LM-CH KM 1:8 (e), Gmp/PVP-K25 PM 1:8 (f), and Gmp/PVP-K25 Coppt 1:8 (g)
Solubility Studies
The solubilities of Gmp in phosphate buffer 6.8 and water were 2.66 and 2.41 μg/ml, respectively, indicating its poor solubility in both media. A linear increase in Gmp solubility in the following order: Tris > PVP-K25 > LM-CH has been recorded with increased carrier levels (Fig. 6). Formation of water-soluble complexes between Gmp and these carriers can be suggested. Water-soluble complexes of Tris with nimesulide (5), PVP with acetaminophen (34), and chitosan with naproxen (9) have been reported. Some authors used the slopes of the linear solubility curves (AL) and the apparent stability constants (Ks) as indicatives of the solubilizing efficiency and the binding affinity of the carriers towards examined drugs, respectively (26). Tris showed highest solubilizing efficiency and binding affinity followed by PVP-K25 then LM-CH as clarified by the slope and Ks values, respectively, of the corresponding linear curves (Table II). Being compratively strong basic, Tris may be expected to form more stabilized complexes with Gmp, hence higher solubility of this drug was observed (5,7,13). The hydrogen bonding of PVP with Gmp as suggested by FTIR results might stabilize the complex with this drug (Fig. 2c).
Fig. 6.
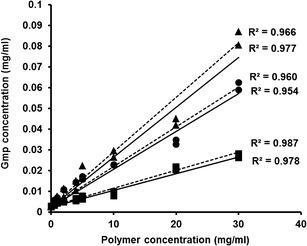
Phase-solubility diagrams of Gmp in phosphate buffer pH 6.8 (dotted lines) and distilled water (solid lines) in the presence of increasing concentrations of tromethamine (Tris, triangles), low molecular weight chitosan (LM-CH, squares), and polyvinylpyrrolidone-K25 (PVP-K25, circles)
In Vitro Dissolution
The tested binary systems were prepared at different drug to carrier ratios, however, a noticeable enhancement in Gmp dissolution was observed at a drug to 12 carrier ratio of 1:8. PMs and GMs at a drug to carrier ratio of 1:20 were formed to investigate the possibility of optimal dissolution using these techniques that are simpler than kneading and coprecipitation. Hence, the dissolution results in phosphate buffer pH 6.8 at only these two ratios were illustrated for binary systems with Tris, LM-CH, and PVP-K25 in Fig. 7a, b, and c respectively. Table I represents the values of DE and RDR of Gmp and its binary systems with the three carriers, as well as those of the prepared capsules. Generally the ratio of 1:8, Coppt with Tris or PVP-K25 and KM with LM-CH showed almost complete dissolution after 30 min (burst effect) with 10.53-, 10.88-, and 9.66-fold increase in DE30 respectively, compared with drug alone (Table I). In accordance, the values of RDR30 of the above mentioned binary systems were 9.24, 9.81, and 9.28, respectively. With the three carriers, the increase in Gmp dissolution at a drug to carrier ratio 1:8 was in the following order: Coppt or KM > GM > PM (Fig. 7). The DE30 and RDR30 values for these systems, illustrated in Table I, confirm these orders.
Fig. 7.
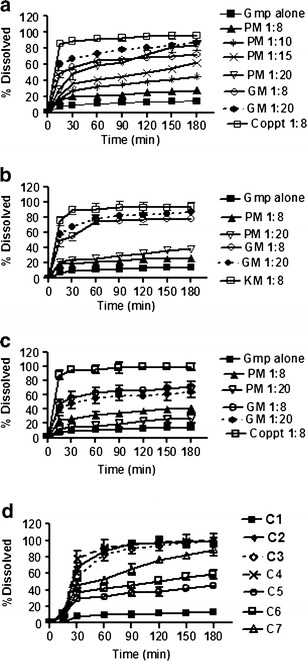
Dissolution of Gmp in phosphate buffer pH 6.8 from some binary systems with a Tris, b LM-CH, c PVP-K25 and d the prepared capsules; all compared with Gmp alone; (see Table III for capsules composition; PM physical mixture, GM coground mixture, Coppt coprecipitate, KM kneaded mixture)
The superiority of dissolution in case of coprecipitation with PVP-K25 and kneading with LM-CH can be attributed to amorphization of Gmp as confirmed with XRD and SEM results. An amorphous drug would be expected to dissolve faster than a crystalline material because of its high energy state (5). The increased dissolution of some drugs by cogrinding and kneading with chitosan was represented in the literature (9,35). Coppts with each of Tris and PVP potentiated the dissolution and the analgesic effect of nimesulide (5).
With LM-CH and PVP-K25, the less crystalline structure of Gmp confirmed by XRD data on cogrinding may explain the higher dissolution observed with GMs than the corresponding PMs (Fig. 4b and c, respectively). A pronounced increase in the viscosity of the diffusion layer due to higher PVP concentration may account for the lower Gmp dissolution from PMs and GMs with this carrier at a drug to carrier ratio of 1:20 than the corresponding 1:8 binary systems. Similar behavior with PVP was documented in the literature (5). A unique factor pertinent to Tris; being basic could increase the pH of the microenvironment around Gmp particles augmenting dissolution. To ascertain this, PM of Gmp/Tris in 1:10, 1:15 and 1:20 were prepared and dissolution rates were assessed. A gradual increase in dissolution rate of Gmp was noticed with the increase in Tris ratio. The increase in DE30 for Gmp/Tris PMs 1:10, 1:15 and 1:20 was 2.52-, 3.19- and 4.36- fold of Gmp alone, respectively (Table I). Nevertheless, these values were lower than that of Gmp/Tris 1:8 Coppt (10.53-fold, Table I). Higher dissolution of Gmp from GMs and Coppts with Tris than the corresponding PMs was noted probably due to more intimate contact between Gmp and this carrier.
The Coppts and KM in 1:8 ratio were selected to prepare capsules of Gmp (Table I). Moreover, capsules of Gmp physically mixed with different Tris ratios (1:10, 1:15, and 1:20) were also included for comparison (Table I). Gmp dissolution from the prepared capsules is described in Fig. 7d together with that of the drug alone. Generally, in vitro dissolution of Gmp from capsules of the different systems was higher than that observed with the drug alone and parallel to that of the corresponding powders. Complete dissolution was within 60 min with lag time of 30 min, for the gelatin shell to swell and dissolve. The highest Gmp dissolution observed with capsules of Tris PMs was that of 1:20 ratio (DE60 33.48). However, it was lower than that experienced with capsules containing the Coppt of Gmp/Tris in 1:8 binary system (DE60 49.45; Table I).
Therapeutic Efficacy Assessment
Capsules containing Coppts or KM in 1:8 Gmp/carrier ratio as well as PMs with different ratios of Tris were selected for in vivo experiment in rabbits (Table III). Figures 8 and 9 elucidate the average %RBGL in normal rabbits after treatment with capsules of the selected binary systems as in Table III. The area under the curve of %RBGL-time was used as a reflection of the insulin level by some authors (29). The results as %RBGL and AUC0–24 h were statistically compared with those capsules of Gmp alone and marketed tablet (amaryl 3 mg; Table III) (19). The data of the two Coppts and KM, were statistically insignificant between themselves, P > 0.05, while each was significantly different from capsules of Gmp alone and the marketed tablet, P < 0.05 or <0.01 (Table III). The hypoglycemic efficacy (AUC0–24 h) of Tris and PVP-K25 Coppts and LM-CH KM were higher by 1.63-, 1.50-, and 1.46-fold respectively, compared with that of Gmp alone (Table III).
Table III.
Statistical Analysis of The Hypoglycemic Response Parameters
| Capsulesa | T max (h) | Mean max. %RBGL ± S.E. | AUC0–24 h ± S.E. | P value | |
|---|---|---|---|---|---|
| Mean Max. %RBGL | AUC0–24 h | ||||
| C1 | 4 | 33.3 ± 1.5 | 311.7 ± 15.2 | _ | _ |
| Control | |||||
| Gmp alone | |||||
| Marketed tablet | 4 | 34.0 ± 0.6 | 342.9 ± 13.8 | >0.05b | >0.05b |
| C2 | 4 | 44.7 ± 2.6 | 509.3 ± 27.6 | <0.01b,c | <0.001b |
| Coppt Gmp/Tris 1:8 | >0.05d,e,f | <0.01c | |||
| >0.05d,e,f | |||||
| C3 KM | 4 | 41.7 ± 2.2 | 456.3 ± 16.7 | <0.05b | <0.01b |
| Gmp/LM-CH 1:8 | >0.05c | <0.05c | |||
| C4 Coppt | 4 | 42.2 ± 1.8 | 466.5 ± 45.2 | <0.05b,c | <0.01b |
| Gmp/PVP-K25 1:8 | <0.05c | ||||
| C5 PM Gmp/Tris 1:10 | 4 | 35.2 ± 1.5 | 354.2 ± 14.9 | >0.05b,c | >0.05b,c |
| C6 PM Gmp/Tris 1:15 | 4 | 36.5 ± 1.3 | 404.4 ± 33.6 | >0.05b,c | >0.05b,c |
| C7 PM Gmp/Tris 1:20 | 4 | 41.2 ± 1.3 | 474.8 ± 41.1 | <0.01b,c | <0.01b |
| >0.05d,e,f | <0.05c | ||||
| >0.05d,e,f | |||||
aEach capsule contains 3 mg Gmp
bCompared with C1
cCompared with marketed tablet
dCompared with C3
eCompared with C4
fCompared with C7
Fig. 8.
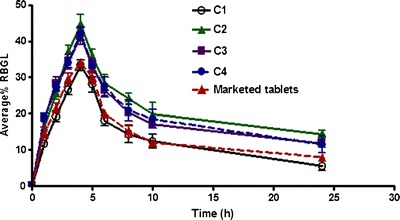
Hypoglycemic efficacy of Gmp from capsules with different systems compared with the drug alone and marketed tablet (%RBGL percentage reduction in blood glucose level, see Table III for capsules composition)
Fig. 9.
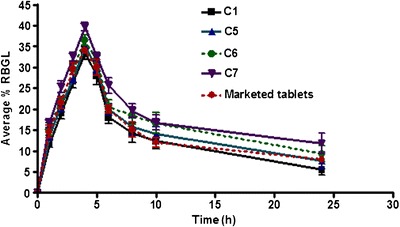
Effect of capsules containing different ratios of physically mixed Tris on the hypoglycemic efficacy of Gmp compared with that containing Gmp alone and marketed tablet (%RBGL percentage reduction in blood glucose level, see Table III for capsules composition)
Conversely from dissolution studies, the 1:20 PM with Tris showed a statistically similar response (P > 0.05) to Coppt Gmp/Tris and to that with PVP-K25 (Table III) despite the amorphizing power of the later revealed by solid state analysis. This might be because of the basic effect of Tris on the microenvironmental pH around Gmp particles in the GIT resulting in higher dissolution rate. This result makes the PM of Gmp/Tris in 1:20 ratio a promising system of high Gmp bioavailability. Repaglinide an antidiabetic drug with slight solubility has been ultra-rapid freezed with Tris (13). The 1:10 and 1:15 PMs with Tris, C5 and C6, respectively, exhibited statistically insignificant difference from the control (C1) and marketed tablet (Table III).
In Vitro–In Vivo Correlation
In vitro and in vivo correlation was examined using DE60 of the tested capsules as the in vitro parameter and mean maximum %RBGL as the in vivo one.
After some trials to get the highest correlation coefficient squared (r2) value possible, it was found that with LM-CH as KM 1:8 (C3), PVP-K25 as Coppt 1:8 (C4) and Gmp alone (C1; Table III), the value of r2 was 0.9770 (Fig. 10a). A slightly lower r2 value, 0.9404, was obtained with Tris PMs 1:10, 1:15 and 1:20 (C5, C6 and C7, respectively), and Coppt 1:8 (C2) together with Gmp alone (C1; Fig. 10b). Surprisingly, these correlations came in accordance with the suggestion of the mechanisms involved in increasing the dissolution rate of Gmp. In the first case (Fig. 10a), amorphization of Gmp with the carriers whether LM-CH or PVP-K25 was the common mechanism. In the second case (Fig. 10b) however, alkalization of the diffusion layer around Gmp particles by Tris could play a major role.
Fig. 10.
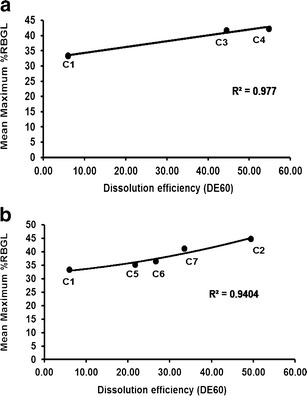
a In vitro–in vivo correlation of capsules containing a KM 1:8 with LM-CH (C3), Coppt 1:8 with PVP-K25 (C4) and that of Gmp alone (C1). b Coppt 1:8 (C2), PM 1:10, PM 1:15, and PM 1:20 with Tris (C5, C6, and C7, respectively) and that of Gmp alone (C1; see Table III for capsules composition and Table I for DE60)
CONCLUSIONS
Gmp/carrier at a ratio of 1:8, Coppt with each of Tris and PVP-K25 and KM with LM-CH showed highly enhanced dissolution of the drug by 9.24-, 9.81-, and 9.28-fold respectively, compared with the drug alone. Gmp amorphization by PVP-K25 and LM-CH was a major factor in increasing Gmp dissolution rate. Being basic, Tris possibly increased the pH of the microdiffusion layer around Gmp particles improving its dissolution. As well, formation of water-soluble complexes may account for the enhanced dissolution. Capsules of PM 1:20 with Tris resulted in an improvement in the therapeutic effect statistically similar to that obtained with Coppt 1:8 drug/Tris possibly due to the increase in the microenvironmental pH of the drug particles in the GIT. Being simple and easy to prepare, the PM 1:20 Gmp/Tris might be promising as a soluble form with a reduced dose of the drug.
REFERENCES
- 1.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56:1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 2.Thompson D, Chaubal MV. Cyclodextrins (CDS)—excipients by definition, drug delivery systems by function (part I: injectable applications) Drug Deliv Technol. 2000;2:34–38. [Google Scholar]
- 3.Garad SD. How to improve the bioavailability of poorly soluble drugs. Am Pharm Rev. 2004;7:80–85. [Google Scholar]
- 4.Nokhodchi A, Javadzadeh Y, Siahi-Shadbad MR, Barzegar Jalali M. The effect of type and concentration of vehicles on the dissolution rates of a poorly water soluble drug (indomethacin) from liquisolid compacts. J Pharm Pharm Sci. 2005;8:18–25. [PubMed] [Google Scholar]
- 5.Abdelkader H, Abdallah OY, Salem HS. Comparison of the effect of tromethamine and polyvinylpyrrolidone on dissolution properties and analgesic effect of nimesulide. AAPS PharmSciTech. 2007;8:E1–E8. doi: 10.1208/pt0804100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGloughlin RM, Corrigan OI. Dissolution characteristics of benzoic acid–TRIS mixtures. Int J Pharm. 1992;82:135–143. doi: 10.1016/0378-5173(92)90082-D. [DOI] [Google Scholar]
- 7.Gabr KE, Borg TM. Characterization of hydrochlorothiazide trometamol mixtures: formulation of fast release and soluble tablets. Pharm Ind. 1999;61:281–285. [Google Scholar]
- 8.Magda AE. Physicochemical characterisation of coprecipitates of furesemide with tromethamine. Alex J Pharm Sci. 2005;19:1–8. [Google Scholar]
- 9.Mura P, Zerouk N, Mennini N, Maestrelli F, Chemtob C. Development and characterization of naproxen–chitosan solid systems with improved drug dissolution properties. Europ J Pharm Sci. 2003;19:67–75. doi: 10.1016/S0928-0987(03)00068-X. [DOI] [PubMed] [Google Scholar]
- 10.Mutalik S, Anju P, Manoj K, Usha AN. Enhancement of dissolution rate and bioavailability of aceclofenac: a chitosan-based solvent change approach. Int J Pharm. 2008;350:279–290. doi: 10.1016/j.ijpharm.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 11.Iwata M, Ueda H. Dissolution properties of glibenclamide in combinations with polyvinylpyrrolidone. Drug Dev Ind Pharm. 1996;22:1161–1165. doi: 10.3109/03639049609065953. [DOI] [Google Scholar]
- 12.El Sayed GM. Role of tromethamine as a dissolution and bioavailability enhancer of oral glibenclamide. STP Pharma Sci. 1998;8:169–173. [Google Scholar]
- 13.Purvis T, Mattucci ME, Crisp MT, Johnston KP. Rapidly dissolving repaglinide powders produced by ultra-rapid freezing process. AAPS PharmSciTech. 2007;8:E52–E60. doi: 10.1208/pt0803058. [DOI] [PubMed] [Google Scholar]
- 14.Zerrouk N, Corti G, Ancillotti S, Maestrelli F, Mura P. Influence of cyclodextrins and chitosan, separately or in combination, on glyburide solubility and permeability. Eur J Pharm Biopharm. 2006;62:241–246. doi: 10.1016/j.ejpb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 15.Kouichi I, Masaki W, Youhei N, Takahiro S, Nobuki T, Masahiko T, Hideyuki K, Kensuke Y, Masao S, Susumu K, Takuya A, Shigehiro K. Efficacy of glimepiride in Japanese type 2 diabetic subjects. Diab Res Clin Pract. 2005;68:250–257. doi: 10.1016/j.diabres.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 16.Geinsen K. Special pharmacology of the new sulfonylurea glimepiride. Drug Res. 1988;38:1120–1130. [PubMed] [Google Scholar]
- 17.Muller G, Wied S, Wetekam E, Crecelius A, Unkelbach A, Punter J. Stimulation of glucose utilization in 3 T3 adipocytes and rat diaphragm in vitro by the sulfonylureas, glimepiride and glibenclamide, is correlated with modulations of the cAMP regulatory cascade. Biochem Pharmacol. 1994;48:985–996. doi: 10.1016/0006-2952(94)90369-7. [DOI] [PubMed] [Google Scholar]
- 18.Massimo MB. Glimepiride in type 2 diabetes mellitus: a review of the worldwide therapeutic experience. Clin Ther. 2003;25:799–816. doi: 10.1016/S0149-2918(03)80109-1. [DOI] [PubMed] [Google Scholar]
- 19.Ammar HO, Salama HA, Ghorab M, Mahmoud AA. Formulation and biological evaluation of glimepiride-cyclodextrin-compound systems. Int J Pharm. 2006;309:129–138. doi: 10.1016/j.ijpharm.2005.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Abhinav M, Vasanti S, Tyangi R, Shukla A. Formulation and evaluation of solid dispersions of an anti-diabetic drug. Curr Trends Biotechnol and Pharm. 2009;3:76–84. [Google Scholar]
- 21.Reven S, Grdadolnik J, Kristl J, Žagar E. Hyperbranched poly(esteramides) as solubility enhancers for poorly water-soluble drug glimepiride. Int J Pharm. 2010;396:119–126. doi: 10.1016/j.ijpharm.2010.06.033. [DOI] [PubMed] [Google Scholar]
- 22.Seedher N, Kanojia M. Co-solvent solubilization of some poorly soluble antidiabetic drugs. Pharm Dev Technol. 2009;14:185–192. doi: 10.1080/10837450802498894. [DOI] [PubMed] [Google Scholar]
- 23.Ilić I, Dreu R, Burjak M, Homar M, Kerč J, Srčič S. Microparticle size control and glimepiride microencapsulation using spray congealing technology. Int J Pharm. 2009;381:176–183. doi: 10.1016/j.ijpharm.2009.05.011. [DOI] [PubMed] [Google Scholar]
- 24.Shiraishi S, Arahira M, Imai T, Otagiri M. Enhancement of dissolution rates of several drugs by low-molecular chitosan and alginate. Chem Pharm Bull. 1990;38:185–187. doi: 10.1248/cpb.38.185. [DOI] [Google Scholar]
- 25.Higuchi T, Connors KA. Phase-solubility techniques. Adv Anal Chem Instr. 1965;7:117–212. [Google Scholar]
- 26.Ahuja N, Katare OP, Singh B. Studies on dissolution enhancement and mathematical modeling of drug release of a poorly water-soluble drug using water-soluble carriers. Eur J Pharm Biopharm. 2007;65:26–38. doi: 10.1016/j.ejpb.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 27.Khan K. The concept of dissolution efficiency. J Pharm Pharmac. 1975;27:48–49. doi: 10.1111/j.2042-7158.1975.tb09378.x. [DOI] [PubMed] [Google Scholar]
- 28.Kulkarni JS, Mehta AA, Santani DD, Goyal RK. Effect of chronic treatment with cromakalim and glibenclamide in alloxan-induced diabetic rats. Pharmacol Res. 2002;46:101–105. doi: 10.1016/S1043-6618(02)00078-6. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Mitra AK. Effect of phospholipid chain length, concentration, charge and vesicle size on pulmonary insulin absorption. Pharm Res. 1996;13:76–79. doi: 10.1023/A:1016029317299. [DOI] [PubMed] [Google Scholar]
- 30.Taylor LS, Zografi G. Spectroscopic characterization of interaction between PVP and indomethacin in amorphous dispersion. Pharm Res. 1997;14:1691–1698. doi: 10.1023/A:1012167410376. [DOI] [PubMed] [Google Scholar]
- 31.The United States Pharmacopoeia 28 and NF23. Rockville, MD: United States Pharmacopoeial Convention; 2005.
- 32.Zerrouk N, Mennini N, Francesca M, Chemtob C, Mura P. Comparison of the effect of chitosan and polyvinylpyrrolidone on dissolution properties and analgesic effect of naproxen. Eur J Pharm Biopharm. 2004;57:93–99. doi: 10.1016/S0939-6411(03)00112-7. [DOI] [PubMed] [Google Scholar]
- 33.Lee JW, Kim SY, Kim SS, Lee YM, Lee KH, Kim SJ. Synthesis and characteristics of interpenetrating polymer network hydrogel composed of chitosan and poly (acrylic acid) J Appl Polym Sci. 1999;73:113–120. doi: 10.1002/(SICI)1097-4628(19990705)73:1<113::AID-APP13>3.0.CO;2-D. [DOI] [Google Scholar]
- 34.Garekani HA, Sadeghi F, Ghazi A. Increasing the aqueous solubility of acetaminophen in the presence of polyvinylpyrrolidone and investigation of the mechanisms involved. Drug Dev Ind Pharm. 2003;29:173–179. doi: 10.1081/DDC-120016725. [DOI] [PubMed] [Google Scholar]
- 35.Portero A, Remunan-Lopez C, Vila-Jato JL. Effect of chitosan and chitosan glutamate enhancing the dissolution properties of the poorly water soluble drug nifedipine. Int J Pharm. 1998;175:75–84. doi: 10.1016/S0378-5173(98)00245-2. [DOI] [Google Scholar]